

Summary
The thyrotrophin receptor (TSHR) A‐subunit is the autoantigen targeted by pathogenic autoantibodies that cause Graves' hyperthyroidism, a common autoimmune disease in humans. Previously, we reported that pathogenic TSHR antibodies develop spontaneously in thyroiditis‐susceptible non‐obese diabetic (NOD).H2h4 mice bearing a human TSHR A‐subunit transgene, which is expressed at low levels in both the thyroid and thymus (Lo‐expressor transgene). The present study tested recent evidence that high intrathymic TSHR expression protects against the development of pathogenic TSHR antibodies in humans. By successive back‐crossing, we transferred to the NOD.H2h4 background a human TSHR A‐subunit transgene expressed at high levels in the thyroid and thymus (Hi‐expressor transgene). In the sixth back‐cross generation (> 98% NOD.H2h4 genome), only transgenic offspring produced spontaneously immunoglobulin (Ig)G class non‐pathogenic human TSHR A‐subunit antibodies. In contrast, both transgenic and non‐transgenic offspring developed antibodies to thyroglobulin and thyroid peroxidase. However, non‐pathogenic human TSHR antibody levels in Hi‐expressor offspring were lower than in Lo‐expressor transgenic mice. Moreover, pathogenic TSHR antibodies, detected by inhibition of TSH binding to the TSHR, only developed in back‐cross offspring bearing the Lo‐expressor, but not the Hi‐expressor, transgene. High versus low expression human TSHR A‐subunit in the NOD.H2h4 thymus was not explained by the transgene locations, namely chromosome 2 (127–147 Mb; Hi‐expressor) and chromosome 1 (22.9–39.3 Mb; low expressor). Nevertheless, using thyroiditis‐prone NOD.H2h4 mice and two transgenic lines, our data support the association from human studies that low intrathymic TSHR expression is associated with susceptibility to developing pathogenic TSHR antibodies, while high intrathymic TSHR expression is protective.
Keywords: autoimmunity, Graves' disease, intrathymic transcription, thyroid autoantigens, tolerance, TSHR autoantibodies
Introduction
Graves' hyperthyroidism, a common autoimmune disease in humans, is caused by autoantibodies that stimulate the thyrotrophin receptor (TSHR) 1, 2. Genes conferring susceptibility to Graves' disease include single nucleotide polymorphorphisms (SNPs) in non‐coding regions of the TSHR (reviewed in 3). TSHR genotypes responsible for low intrathymic TSHR transcription levels are associated with susceptibility to Graves' disease and, conversely, TSHR genotypes responsible for high intrathymic TSHR expression are protective 4. Moreover, a transcriptional repressor of interferon (IFN)‐α, a cytokine that can trigger thyroid autoimmunity, interacts with a Graves' disease‐associated TSHR SNP to reduce intrathymic TSHR expression 5.
These observations provide insight into the breakdown in self‐tolerance to the TSHR and TSHR autoantibody production in patients with Graves' disease. Central tolerance is determined by ‘T cell education’: immature T cells that bind with high affinity to peptides derived from self‐proteins expressed in the thymus undergo apoptosis and are deleted 6. Defects in central tolerance contribute to autoimmune susceptibility. For example, in autoimmune‐prone non‐obese diabetic (NOD) mice, the altered process of thymic education leads to an increased proportion of autoreactive T cells in peripheral lymphoid organs 7, 8, 9, 10. Moreover, manipulating the magnitude of thymic autoantigen expression in mice controls autoreactive T cell deletion and affects the development of autoimmune disease 11. T cells play a critical role in providing help to B cells for antibody production. In particular, defective central tolerance in mice lacking the autoimmune regulator gene is associated with the development of autoantibodies to a variety of autoantigens 12, 13. Indeed, in studying the association between TSHR SNPs and Graves' hyperthyroidism, the selection criteria of individuals included, in addition to clinical parameters, the presence of TSHR antibodies in patients but not in controls 4.
Previously, we generated two transgenic lines of BALB/c mice with the human TSHR A‐subunit targeted to the thyroid gland 14. One line expresses very high, and the other much lower, intrathyroidal levels of the human TSHR A‐subunit 15. Consistent with the intrathyroidal differences, intrathymic human TSHR A‐subunit transcripts were extremely high in the ‘Hi‐expressor’ line but similar to the levels of the endogenous mouse TSHR in the ‘Lo‐expressor’ line 16. As might be expected, TSHR autoantibodies were induced readily in the Lo‐expressors by immunization with moderate titres of human TSHR A‐subunit adenovirus. In contrast, inducing TSHR antibodies in Hi‐expressor mice required injection of extremely high‐titre human TSHR A‐subunit adenovirus without or with TSHR A‐subunit protein in adjuvant 16, 17.
BALB/c mice do not develop pathogenic TSHR antibodies spontaneously and (like the TSHR human A‐subunit transgenic BALB/c mice) only do so after immunization with plasmid or adenovirus vectors expressing the TSHR or its A‐subunit (reviewed in 18). Conversely, NOD.H2h4 mice develop autoantibodies spontaneously to the thyroid autoantigen thyroglobulin (Tg) 19, 20, 21 and later to thyroid peroxidase (TPO) 22. Because of their predisposition to thyroid autoimmunity, we hypothesized that NOD.H2h4 mice would break tolerance spontaneously to a protein expressed transgenically in the thyroid. Therefore, we transferred the Lo‐expressor human TSHR A‐subunit transgene 15 to the NOD.H2h4 background by back‐crossing and selection for the transgene. Indeed, hTSHR/NOD.H2h4 mice develop non‐pathogenic TSHR antibodies detectable by enzyme‐linked immunosorbent assay (ELISA) and, more importantly, pathogenic TSHR‐stimulating antibodies resembling those in patients with Graves' disease 23.
The availability of Hi‐expressor TSHR A‐subunit BALB/c mice provided the opportunity to test the evidence from human studies that self‐tolerance to the TSHR is not broken when this antigen is expressed highly in the thymus. We therefore transferred the Hi‐expressor TSHR A‐subunit transgene from the BALB/c line to the NOD.H2h4 background. We observed spontaneous loss of tolerance to the hTSHR A‐subunit as measured by the emergence of non‐pathogenic, but not (as in the Lo‐expressor animals) pathogenic TSHR antibodies.
Methods
Generating NOD.H2h4 mice expressing high levels of the human TSHR A‐subunit
NOD.H2h4 mice (originally from The Jackson Laboratory, Bar Harbor, ME, USA; strain NOD.Cg‐H2h4/DilTacUmmJ) and transgenic BALB/c mice expressing high (Hi) intrathyroidal and intrathymic levels of the human TSHR A‐subunit (line 50.6; subsequently referred to as TSHR‐Hi) 15, 16 were bred at Cedars‐Sinai Medical Center. The Hi‐expressor transgene was transferred to NOD.H2h4 as follows (Fig. 1a): male Hi‐expressor TSHR‐A‐subunit BALB/c mice were crossed to female non‐transgenic NOD.H2h4 mice (Hi‐Tgic‐BALB/c × non‐Tgic‐NOD.H2h4) to generate N1 progeny. Expression of the transgene was determined by polymerase chain reaction (PCR) 14. Transgenic male N1‐Hi pups were bred to wild‐type NOD.H2h4 females to generate ‘N2‐Hi A‐subunit’ mice and the same procedure was repeated to produce the N3‐Hi, N4‐Hi and N5‐Hi A‐subunit generations. At this stage, to introduce the NOD.H2h4 Y chromosome, wild‐type NOD.H2h4 males were crossed to female N5 Hi‐Tgic‐NOD.H2h4 mice to produce the N6 generation. With each back‐cross, the percentage of NOD.H2h4 genes increases from 0% in BALB/c to 98·4% in N6 offspring (Fig. 1b, based on data in 24).
Figure 1.
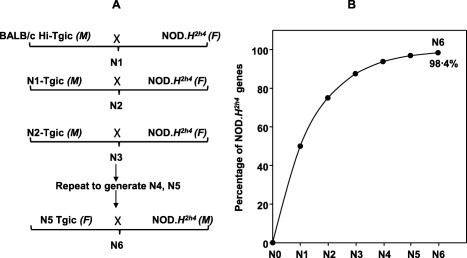
(a) Transferring the Hi‐expressor thyrotrophin receptor (TSHR) A‐subunit transgene from BALB/c mice to non‐obese diabetic (NOD).H2h4 mice by repeated back‐crossing. (b). Increasing percentage of NOD.H2h4 background genes from 0% in BALB/c (N0) to 98.4% in N6 offspring (adapted from data in 24).
From 8 weeks of age, the drinking water in non‐breeding mice was supplemented with 0·05% sodium iodide (NaI; Sigma‐Aldrich, St Louis, MO, USA) for 16 weeks, at which time (age 24 weeks) TSHR‐Hi transgenic (Tgic) NOD.H2h4 and non‐Tgic NOD.H2h4 offspring (N1–N6), as well as parental strains, were euthanized to harvest blood and thyroid tissue. All mouse studies were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at Cedars‐Sinai Medical Center.
TSHR antibody assays
TSHR antibodies were measured using two assays:
-
(a)
ELISA: The ELISA for IgG class TSHR antibodies was performed as reported previously 2. Recombinant TSHR A‐subunit protein secreted by Chinese hamster ovary cells (CHO) with an amplified transgenome 25 was purified from culture supernatants by affinity chromatography 26. ELISA wells (Immulon 4HBX; Thermo Scientific, Rochester, NY, USA) were coated with A‐subunit protein (5 μg/ml) and incubated with test sera (1 : 100 dilution; duplicate aliquots). The positive control used was serum from BALB/c mice immunized with TSHR A‐subunit adenovirus. Antibody binding was detected with horseradish peroxidase‐conjugated mouse anti‐IgG (A 3673; Sigma Aldrich) and the signal was developed with o‐phenylenediamine and H2O2 and the reaction stopped with 20% H2SO4. Data are reported as the optical density (OD) at 490 nm.
-
(b)
TSH binding inhibition (TBI) assay: TBI levels were measured in 25 μl mouse serum using a clinical assay kit (Kronus Inc, Star, ID, USA). The data are reported as the % inhibition of 125I‐TSH binding to the membrane‐bound full length TSHR.
Autoantibodies to thyroglobulin and thyroid peroxidase (TgAb and TPOAb)
Tg was isolated from murine thyroid glands 22. ELISA wells (Immulon 4HBX; Thermo Scientific) were coated with mouse Tg (1·5 μg/ml) and incubated with test sera (duplicate aliquots, 1 : 100 dilution). Antibody binding was detected with horseradish peroxidase‐conjugated goat anti‐mouse IgG (A3673; Sigma Aldrich), the signal developed with o‐phenylenediamine and the reaction stopped using 20% H2SO4. The positive control was serum from BALB/c mice immunized with mouse Tg and complete Freund's adjuvant 17 and the negative control was serum from 8‐week‐old NOD.H2h4 mice on regular water. TgAb data are presented as the optical density (OD) at 490 nm.
TPOAb were measured using CHO cells stably expressing mouse‐TPO 22. Sera (1 : 50 dilution) were incubated with mouse TPO‐CHO cells and binding was detected with fluorescein isothyocyanate‐conjugated affinity purified goat anti‐mouse IgG (M30101; Invitrogen, Carlsbad, CA, USA). Cells stained with propidium iodide (1 μg/ml) were excluded from analysis. The negative control for IgG class antibody binding to mouse TPO‐CHO cells was serum from 8‐week‐old NOD.H2h4 mice. Positive controls were mouse monoclonal antibodies #15 and #64 to human TPO 27, provided to us by Dr J. Ruf (Marseille, France), that recognize mouse TPO 22, 27. Flow cytometry was performed (10 000 events) using a FACSCanto II with cellquest Software (Becton Dickinson, San Jose, CA, USA). Data are reported as the geometric mean (Geo Mean).
Thyroid histology
Thyroid glands were preserved in zinc fixative (BD Pharmingen, San Diego, CA, USA), paraffin‐embedded and serial sections stained with haematoxylin and eosin (IDEXX BioResearch Laboratory Animal and Biological Materials Diagnostic Testing, Columbia, MO, USA).
Genomic insertion of the human TSHR A‐subunit transgene
DNA from Hi and Lo‐expressor BALB/c, non‐transgenic BALB/c and C57BL/6 (the latter used to create the transgenics) were compared using the Illumina Mouse Medium Density panel (Genetic Analysis Facility, The Hospital for Sick Children, Toronto, Canada). Comparison of the genotypes revealed the locations of the transgenes. Where necessary, PCR for polymorphic markers was performed to confirm these locations.
Intrathymic expression of the human TSHR A‐subunit in NOD.H2h4 mice measured by quantitative real‐time PCR (qPCR)
Thymuses from 32‐day‐old mice were stored in RNAlater (Life Technologies, Carlsbad, CA, USA) for the following strains: Lo‐expressor hTSHR/NOD.H2h4 (N13, n = 3), Hi‐expressor hTSHR/NOD.H2h4 (N7, n = 3) and non‐transgenic NOD.H2h4 mice (n = 3). Thyroid tissue, obtained at euthanasia, was stored in RNAlater for the following strains: Hi‐expressor hTSHR/NOD.H2h4 (N6, n = 6), Lo‐expressor hTSHR/NOD.H2h4 (N8, n = 4) and non‐transgenic NOD.H2h4 mice (n = 8). Tissues were homogenized with QIAshredder columns (Qiagen, Valencia, CA, USA). Total RNA was prepared using the RNeasy Plus Mini kit (Qiagen). The mRNA samples were treated with TURBO DNase (Life Technologies) to remove genomic DNA. Reverse transcription was performed with the AffinityScript qPCR cDNA synthesis kit (Agilent Technologies, Cedar Creek, TX, USA) using oligo(dT) and random primers. Real‐time PCR was performed using the FastStart SYBR Green Master mix (Roche, Basel, Switzerland) with 2·5% of cDNA (20 μl final volume). Reactions were run on an iCycler Thermal Cycler with iQ5 real‐time PCR detection system module (BioRad Laboratories, Hercules, CA, USA). An initial denaturation step at 95°C (10 min) was followed by denaturation at 95°C (30 s) and annealing and extension at 55°C (30 s) for 40 cycles. Relative gene expression levels were calculated using the comparative Ct method (ΔΔCt), according to the Pfaffl model 28, using Bio‐Rad iQ5 version 2.0 software. Samples were tested in triplicate; parallel controls lacked reverse transcriptase. Data were normalized to mouse β−actin for the thymus and for β‐actin and glyceraldehyde 3‐phosphate dehydrogenase (GADPH) for the thyroid. Primers for the hTSHR A‐subunit were as follows: sense 5'‐GCAAGAAACACCTGGACTCTTAA‐3', hTSHR A‐subunit anti‐sense 5‐GGTGGTGATGGCTAGTCTGA‐3' (Eurofins Genomics, Louisville, KY, USA); these primers were designed to avoid overlap with mouse TSHR. For thymus, the mouse TSHR primer was RT2 qPCR Primer Assay for Mouse Tshr (Qiagen); for thyroid tissue, the mouse TSHR primer was PrimePCR™ SYBR® Green Assay: Tshr, Mouse (Bio‐Rad). For both tissues, we used RT2 qPrimer Assay for mouse actin β and GADPH (from Qiagen).
Results
TSHR ELISA‐type antibodies develop in NOD.H2h4 offspring expressing the Hi‐TSHR A‐subunit transgene
TSHR antibodies of IgG class, measured by ELISA using plates coated with recombinant human TSHR A‐subunit protein, were monitored at each back‐cross generation in the process of transferring the Hi‐TSHR A‐subunit transgene from the BALB/c transgenic to the NOD.H2h4 strain. As expected, ELISA‐type TSHR antibodies were not detected in NOD.H2h4, BALB/c or Hi‐expressor BALB/c transgenic mice. In contrast, TSHR ELISA antibodies developed in a small number of Hi‐TSHR A‐subunit transgenic offspring from the N2–N6 generations of back‐crossing to non‐transgenic NOD.H2h4 parents (Fig. 2a). These studies show that the NOD.H2h4 genetic background, unlike the non‐autoimmune BALB/c strain, permits spontaneous development of antibodies to the intrathyroidal product of the Hi‐expressor TSHR A‐subunit transgene. However, the levels of TSHR A‐subunit ELISA antibodies were consistently lower in offspring from the Hi‐expressor back‐crosses than in comparable numbers of back‐cross offspring generated previously from the Lo‐expressor BALB/c parent (Fig. 2b). The previously reported values for N1–N8 offspring 23 are shown for comparison. Because conditions in the animal facility may vary at different times, we also include unpublished data for mice housed at the same time as the Hi‐expressors, namely N9–N12 non‐transgenic and transgenic offspring.
Figure 2.
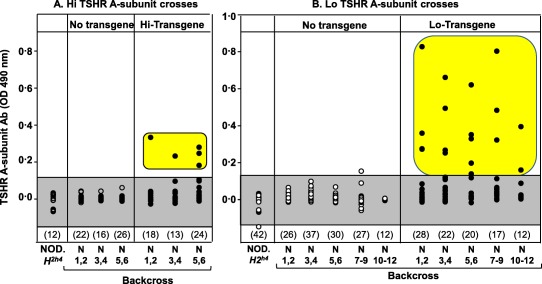
Thyrotrophin receptor (TSHR) A‐subunit antibody (TSHR antibody) detected by enzyme‐linked immunosorbent assay (ELISA) develop in offspring generated by transferring the Hi‐expressor human TSHR A‐subunit transgene from BALB/c mice to the non‐obese diabetic (NOD).H2h4 background (left panel). However, antibodies in the Hi‐expressor offspring are present at lower levels than in NOD.H2h4 back‐crosses generated from Lo‐expressor human TSHR A‐subunit transgenic BALB/c (right panel; data for generations N1–N8 re‐plotted from Rapoport et al. 23 and for N9–N12 offspring (unpublished). N1–N12 = generations of back‐crossing to NOD.H2h4. Values for TSHR antibody, measured by ELISA [optical density (OD) 490 nm], are shown for individual mice (males and females) at 24 weeks of age (NaI‐supplemented drinking water begun at 8 weeks); the numbers of mice in each group are given in parentheses. The shaded areas represent the mean ± 2 standard deviations (s.d.) for antibody levels in NOD.H2h4 littermates not carrying the human A‐subunit transgene. Positive values are highlighted. [Colour figure can be viewed at wileyonlinelibrary.com]
TgAb, TPOAb and thyroid histology in Hi‐expressor A‐subunit NOD.H2h4 back‐crosses
Unlike TSHR ELISA antibodies that developed only in transgenic offspring (Fig. 2a), both wild‐type and Hi‐expressor transgenic offspring developed IgG class autoantibodies to Tg (Fig. 3a) and TPO (Fig. 3b), like the NOD.H2h4 parental strain but unlike the non‐autoimmune strains BALB/c mice and the Hi‐A subunit expressor BALB/c donor of the transgene. Similar observations were made previously for offspring of the Lo‐expressor back‐crosses 23. These data indicate that neither transgene insertion site (see below) nor possible carry‐over genes of BALB/c origin due to the back‐cross strategy affected the spontaneous autoimmune responses to other thyroid antigens in the NOD.H2h4 mice.
Figure 3.
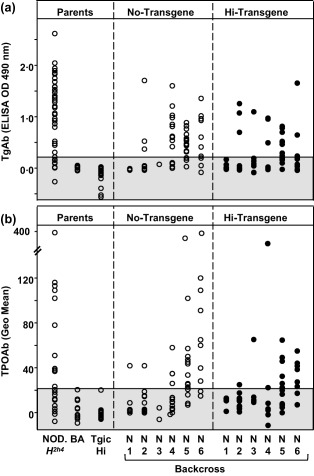
Autoantibodies to thyroglobulin (Tg) and thyroid peroxidase (TPO) develop spontaneously in non‐obese diabetic (NOD).H2h4 back‐cross offspring regardless of whether or not they carried the Hi‐thyrotrophin receptor (TSHR) A‐subunit transgene. Values for individual male and female mice at 24 weeks of age [sodium iodide (NaI)‐supplemented drinking water begun at 8 weeks] are shown for Tg antibody (a, measured by ELISA; OD 490 nm) and TPOAb (b, measured by flow cytometry, geometric mean). The shaded area represents the mean ± 2 standard deviations (s.d.) for antibody levels in parental BALB/c (BA) mice. Tgic Hi = transgenic Hi‐expressor BALB/c mice.
Variable levels of lymphocytic infiltration were observed for thyroid tissue from both non‐transgenic and Hi‐expressor transgenic offspring of the N6 generation (Supporting information, Fig. S1) as observed previously for Lo‐expressor mice of the N5 and N6 generations 23.
Pathogenic TSHR antibodies do not develop in Hi‐expressor hA‐subunit NOD.H2h4 back‐crosses
ELISA‐type TSHR antibodies do not recognize the native hTSHR and are non‐pathogenic. Therefore, we used a clinical assay to test sera for the ability of autoantibodies to inhibit TSH binding to the TSHR, TSH binding inhibition (TBI). This study was restricted to female mice because, as we observed previously, male NOD.H2h4 mice develop very high levels of TSH that cannot be distinguished from TSHR antibodies in the TBI assay 23. Despite the presence of TSHR ELISA antibodies (Fig. 2a), Hi‐expressor back‐cross NOD.H2h4 mice failed to develop TBI activity (Fig. 4a), unlike the Lo‐expressor back‐cross offspring (Fig. 3b) 23. Again, because conditions in the animal facility may vary at different times, we also include unpublished data for mice housed at the same time as the Hi‐expressors, namely N9–N12 non‐transgenic and transgenic offspring. The low levels of TSHR antobodies measured by ELISA in the Hi‐transgenic crosses (all TBI‐negative) are comparable with those of some TBI positive Lo‐transgenic crosses (Supporting information, Fig. S2). Therefore, the absence of TBI activity in the Hi‐expressor transgenics is not due to assay insensitivity.
Figure 4.
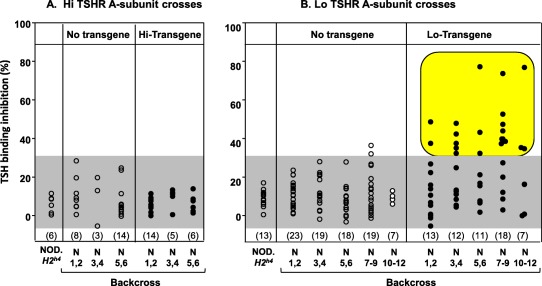
Thyrotrophin receptor (TSHR) antibodies measured by inhibition of TSH binding to the membrane‐bound TSHR (TBI) do not develop in female non‐obese diabetic (NOD).H2h4 back‐cross offspring derived from the BALB/c parent bearing the Hi‐expressor human TSHR A‐subunit transgene (a). In contrast, TSHR antibodies with pathogenic TBI activity develop spontaneously in female Lo‐expressor A‐subunit transgenic NOD.H2h4 back‐cross mice (b; data for generations N1–N7 replotted from Rapoport et al. 23 and for N9–N12 offspring (unpublished). Values are shown as the % binding inhibition of radiolabelled TSH to the TSHR. N1–N12 = generations of back‐crossing to NOD.H2h4. The shaded area represents the mean + 2 standard deviation (s.d.) for all non‐transgenic littermates. Positive values are highlighted. [Colour figure can be viewed at wileyonlinelibrary.com]
Previously, sera from the N8 Lo‐expressor crosses were tested for TBI and for thyroid‐stimulating antibody activity (TSAb) 23. Sera negative for TBI activity are not TSAb‐positive in the spontaneous model (Supporting information, Fig. S3) or in the induced model using TSHR A‐subunit adenovirus (for example 29). For this reason, TSAb activity was not tested in Hi‐expressor A‐subunit transgenic NOD.H2h4 back‐cross mice. Consequently, breaking self‐tolerance to the Hi‐expressor transgene in NOD.H2h4 mice is limited to the generation of non‐functional, non‐pathogenic TSHR antibodies.
Intrathymic and intrathyroidal expression of the TSHR A‐subunit in Hi‐ and Lo‐expressor NOD.H2h4 mice
Using thymic tissue from 32‐week‐old mice, qPCR demonstrated extremely high expression of the human TSHR A‐subunit in Hi‐expressor NOD.H2h4 offspring, almost 200 times as much as the lower levels in Lo‐expressor NOD.H2h4 offspring, and undetectable levels in non‐transgenic mice (Fig. 5a). In contrast, the mouse TSHR was expressed at similar levels in all three mouse strains. Intrathymic expression of the human A‐subunit transgenes in NOD.H2h4 mice is the same as the pattern we observed previously for the Hi‐ and Lo‐transgenes in the BALB/c parents 16 used to generate Hi‐ and Lo‐TSHR A‐subunit crosses.
Figure 5.
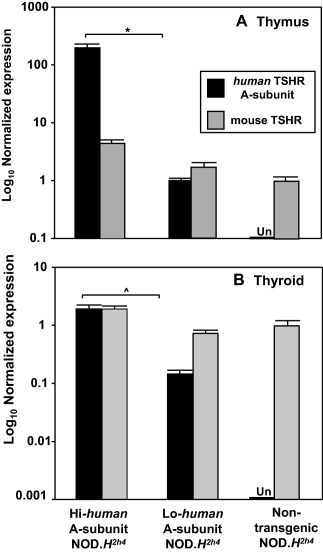
Expression in thymic and thyroid tissue, measured by quantitative polymerase chain reaction (qPCR), of the human thyrotrophin receptor (TSHR) A‐subunit transgene and the endogenous mouse TSHR in Hi‐expressor non‐obese diabetic (NOD).H2h4 offspring, Lo‐expressor NOD.H2h4 offspring and non‐transgenic NOD.H2h4 mice. (a) Thymus; data are shown as log10 relative expression [mean + standard deviation (s.d.) of triplicates] normalized to mouse β‐actin for the human TSHR A‐subunit or the mouse TSHR. (b) Thyroid; data are shown as log10 relative expression (mean + s.d. of triplicates) normalized to mouse β‐actin and glyceraldehyde 3‐phosphate dehydrogenase (GADPH) for the human TSHR A‐subunit or the mouse TSHR. Expression of the human TSHR A‐subunit was undetectable (Un) in both thymus and thyroid of non‐transgenic NOD.H2h4 mice. The thymic data are representative of three comparable experiments. A similar (but separate) study confirmed the thymic expression values for Lo‐expressor transgenics versus non‐transgenic NOD.H2h4 mice 46. Thyroidal mRNA expression levels of the Hi and Lo‐expressor transgenes are similar to previous observations of immunohistochemistry and protein extraction 15. Significantly different human TSHR A‐subunit expression in the Hi‐ versus the Lo‐expressor: *P = 0.009; ^P = 0·001 (t‐tests).
Thyroid expression of the transgenes was studied in tissues obtained at euthanasia. Expression of the human A‐subunit was high in the Hi‐expressor mice, lower in the Lo‐expressor offspring and absent from non‐transgenic NOD.H2h4 mice (Fig. 5b). The mouse TSHR was expressed at similar levels in all three mouse strains. We had not studied intrathyroidal expression previously of the endogenous mouse TSHR in BALB/c mice transgenic for the human A‐subunit. However, human A‐subunit transgene expression measured by real‐time PCR in NOD.H2h4 mice was comparable to our previous observations in BALB/c mice using immunohistochemistry or protein extraction 15. Of interest, relative to the mouse TSHR, the human A‐subunit is expresssed more highly in the thymus than in the thyroid.
Genomic insertion of the human TSHR A‐subunit transgene
The genomic insertions of the human A‐subunit transgenes were determined in the BALB/c parents from which the transgenes were transferred to NOD.H2h4 mice. The human A‐subunit transgenic mice were generated using F2 oocytes from the F1 of [C57BL/6 × BALB/c] mice 14. Subsequently, transgenic offspring were back‐crossed to the BALB/c background for at least 10 generations. We performed a medium density single nucleotide polymorphism analysis on DNA from BALB/c, C57BL/6 and the Hi‐ and Lo‐A‐subunit transgenic mice on the BALB/c background. We found that both the Hi‐ and Lo‐expressor strains were more than 99% pure BALB/c.
The Hi‐expressor transgene is located on chromosome (Chr) 2 (127–147Mb) and the Lo‐expressor transgene on Chr 1 (22·9–39·3 Mb). Polymorphic markers for D2Mit280 confirmed the Hi‐expressor A‐subunit transgene to be near 146 Mb on Chr 2 and D1Mit411 markers confirmed the Lo‐expressor transgene to be near 33 Mb on Chr 1 (Table 1). The insertion sites do not overlap with genes that play a role in thymic expression of tissue restricted antigens, including autoimmune regulator (Aire) (for example, 12, 13, 30, 31, E26 avian leukaemia oncogene (Ets1) 32 and FEZ family zinc finger 2 (Fezf2) 33, with the genes encoding the thyroid autoantigens (TSHR or Tg) or with other genes encoding immune molecules associated with susceptibility to thyroid autoimmunity 3, 34, 35, 36, 37, with the possible exception of AFF3 AF4/FMR2 family, member 3 (Aff3) 34, 36. The difference in the locations of Aire and the transgenes is consistent with our previous observation that the absence of Aire had little effect on Graves' disease induced in BALB/c mice by immunization with A‐subunit adenovirus 16.
Table 1.
Chromosomal locations of the Lo‐ and Hi‐expressor transgenes do not overlap with genes responsible for thymic expression of tissue restricted antigens or with murine genes associated with susceptibility to human thyroid autoimmunity (except for AFF3)
| Gene | Name | Chr | Megabase | Ref |
|---|---|---|---|---|
| Lo‐TSHR | Human TSHR A‐subunit Lo‐expressor | 1 | 22·9–39·3 (near 33 Mb) | Present |
| Hi‐TSHR | Human TSHR A‐subunit Hi‐expressor | 2 | 127–147 (near 146 Mb) | Present |
| Genes controlling intrathymic tissue restricted antigens | ||||
| Aire | Autoimmune regulator | 10 | 78·030022–78·043610 | 12, 30 |
| Fezf2 | Fez family zinc finger 2 | 14 | 12·341892–12·345865 | 33 |
| Ets1 | E26 avian leukaemia oncogene | 9 | 32·636210–32·757820 | 32 |
| Genes associated with thyroid autoimmunity | ||||
| Aff3 | AFF3 AF4/FMR2 family, member 3 | 1 | 38·175991– 38·627837 | 34, 36 |
| ICOS | Inducible T cell co‐stimulator | 1 | 60·960773–61·000322 | 3 |
| CD28 | CD28 antigen | 1 | 60·746388–60·773359 | 3 |
| CTLA‐4 | Cytotoxic lymphocyte‐associated protein 4 | 1 | 61·212315–61·219120 | 3, 34, 36 |
| Fcgr4 | Fcgr4 Fc receptor, IgG, low affinity (Fcrl3) | 1 | 171·018926–171029761 | 34, 36 |
| IL2ra | Interleukin 2 receptor, alpha chain/Cd25 | 2 | 11·642792–11·693194 | 3, 34 |
| 1f1h1 | Interferon induced with helicase C domain1 | 2 | 62·595612–62·646255 | 34, 36 |
| CD40 | CD40 antigen | 2 | 165·055604–165·071654 | 3, 34, 36 |
| Ptpn22 | Protein tyrosine phosphatase 22 | 3 | 103·856804– 03·912252 | 37 |
| Magi3 | Membrane associated guanylate kinase‐ | 3 | 104·013259–104·220406 | 37 |
| WW and PDZ domain containing 3 pdf | ||||
| Bach2 | BTB and CNC homology 2 | 4 | 32·238574–32·586108 | 37 |
| Tshr | Thyrotrophin receptor | 12 | 91·400993–91·540509 | 3, 34 |
| Tg | Thyroglobulin | 15 | 66·670756–66·850721 | 3 |
| H2‐antibody | Histocompatibility 2, class 2 | 17 | 34·263227–34·269418 | 3, 34 |
| CD226 | CD226 antigen | 18 | 89·176954–89·272232 | 34, 36 |
| Scgb3A2 | Secretoglobin, family 3A, member 2 | 18 | 43·764281–43·767399 | 34, 36 |
| FoxP3 | Forkhead box P3 | X | 7·579676–7·595243 | 3, 34 |
Discussion
Of two transgenic BALB/c mouse lines generated by targeting the human TSHR A‐subunit to the thyroid using a thyroid‐specific (thyroglobulin) promoter, in one line the TSHR A‐subunit transgene is expressed at a high level in both the thyroid and thymus (‘Hi‐expressor’), whereas in the second line the same transgene is expressed at a low level in both the thyroid and thymus (‘Lo‐expressor’) 15, 16. Previously, we introduced (by back‐crossing) the Lo‐expressor transgene from non‐autoimmune BALB/c mice to the thyroiditis‐prone NOD.H2h4 strain 23. In the present study, using the Hi‐expressor human TSHR A‐subunit transgene, we repeated this back‐cross from BALB/c to NOD.H2h4 mice. This experiment, together with our previous findings 23, allowed us to test in mice the evidence that has arisen in humans 4, 5 that TSHR mRNA transcripts expressed at low levels in the thymus are associated with susceptibility to Graves' disease while high intrathymic TSHR mRNA levels are protective 4, 5.
In mice, loss of tolerance to the human TSHR A‐subunit (autologous in the transgenics) is assessed most readily using ELISA to measure TSHR antibodies. The spontaneous emergence of TSHR antibodies of IgG class (reflecting a role for T cells) clearly indicates a loss of tolerance to this self‐antigen. By the sixth back‐cross stage, transgenic mice have more than 98% of the NOD.H2h4 genome 24. At this stage, back‐cross offspring from both Hi‐expressor transgenic parents (present study) and Lo‐expressor transgenic parents (previous report 23) produced ELISA‐type antibodies to the human TSHR A‐subunit, albeit at lower levels in the Hi‐expressor offspring.
ELISA‐type TSHR antibodies are non‐pathogenic and are not associated with human Graves' disease 38. We therefore measured TSH binding inhibition (TBI), an assay for pathogenic TSHR autoantibodies. This assay is restricted to sera from female mice because male NOD.H2h4 mice develop high TSH levels which cannot be distinguished from TBI antibody activity 23. Previously, we observed that pathogenic TSHR antibodies measured by TBI developed in transgenic back‐cross offspring from the Lo‐expressor parent 23 and, as we show here, TBI also develop in subsequent back‐crosses. In contrast, in transgenic offspring from the Hi‐expressor parent, TBI activity was conspicuously absent.
In the Lo‐expressor human TSHR A‐subunit NOD.H2h4 transgenics, the spontaneous development of pathogenic TSHR antibodies can also be detected in a bioassay for thyroid stimulating antibody (TSAb) activity. However, transgenic NOD.H2h4 mice do not become hyperthyroid 23, for two reasons. First, extensive data indicate that antibodies induced to the human TSHR cross‐react very poorly with the endogenous mouse TSHR expressed on the surface of thyrocytes 29, 39, 40. Secondly, although the human TSHR A‐subunit transgene is targeted to, and expressed by, the mouse thyrocytes, only the holoreceptor, not the isolated A‐subunit, can transduce a signal and increase thyroid hormone secretion. In the present study with Hi‐expressor human TSHR A‐subunit NOD.H2h4 transgenics, we did not assay for TSAb activity because of negativity in the TBI assay. In our experience, TSAb activity is detected only when the TBI assay is positive.
Our work 2, as well as that of others (for example 41, 42, 43, 44, has focused upon the TSHR A‐subunit because of evidence for the importance of this component, rather than the holoreceptor, in the induction and/or the affinity maturation of pathogenic TSHR autoantibodies. The holoreceptor is (of course) required for TSHR antibodies to stimulate the thyroid leading to hyperthyroidism. Intramolecular cleavage of the TSH holoreceptor on the cell surface leads to shedding of the A‐subunit (reviewed in 45). Lacking the transmembrane domain, isolated A‐subunits expressed by cells are secreted and not retained in the plasma membrane 25.
The present study, taken together with previous data, suggests that the spontaneous development of TSHR autoantibodies involves not only central tolerance but the amount of TSHR expressed in the thyroid. Compared with the other thyroid autoantigens, namely TPO and even more abundant Tg, the amount of TSHR protein in the thyroid is very low (reviewed in 18). As noted earlier, in wild‐type NOD.H2h4 mice, Tg autoantibodies develop first and are followed later by autoantibodies to the less abundant TPO 22. Our previous observations in NOD.H2h4 recipients of the Lo‐expressor human A‐subunit transgene indicate that spontaneous development of TSHR antibodies (both pathogenic and non‐pathogenic) requires not only an appropriate genetic background but additional TSHR A‐subunit protein in the thyroid to stimulate the immune system. This deduction is made because the thyroidal murine TSHR levels in non‐transgenic NOD.H2h4 are insufficient to induce antibody production, which requires supplementation of human TSHR A‐subunit by the Lo‐expressor transgene. At the opposite extreme, the human transgenic A‐subunit is expressed highly in both the thymus and the thyroid in NOD.H2h4 offspring; these mice develop low levels of TSHR antibodies, but only of the non‐pathogenic type. These relationships indicate that production of TSHR antibodies is more difficult for Hi‐ than for Lo‐expressor NOD.H2h4 offspring, in agreement with our previous observations for induced TSHR antibody responses in BALB/c mice with the same Hi‐ and Lo‐expressor transgenes 16, 17. We suggest a biphasic ‘Goldilocks’ effect for the level of TSHR expression in breaking tolerance: not too little and not too much. Thus, generation of pathogenic TSHR autoantibodies increases with increasing autoantigen expression until, at higher expression levels, increased central tolerance reduces the ability of the immune system to generate such autoantibodies.
Variability in intrathymic TSHR mRNA transcripts in humans involves TSHR SNPs in the non‐coding region of the TSHR receptor 4, 5. Intrathymic expression of the human TSHR A‐subunit was consistent with our previous observations for BALB/c transgenic mice 16; namely, extremely high in NOD.H2h4 recipients of the Hi‐expressor transgene, much lower in recipients of the Lo‐expressor transgene and absent from non‐transgenic mice. We established the insertion sites of the transgenes, namely Chr 1 for the Lo‐transgene and Chr 2 for the Hi‐transgene. These locations do not reveal the basis for high versus low TSHR A‐subunit expression in our mouse strains. Although the mechanisms responsible for high versus low intrathymic expression levels are different in transgenic mice and humans, the availability of mice bearing the Hi and Lo‐expressor human TSHR A‐subunit transgenes allowed us to test the outcome of high versus low TSHR intrathymic expression in a thyroid autoimmune‐susceptible background. High TSHR A‐subunit expression is compatible with development of non‐functional TSHR antibodies, but bioactive antibodies develop only in Lo‐TSHR expressor NOD.H2h4.
In conclusion, using thyroiditis‐prone NOD.H2h4 mice and two transgenic BALB/c lines, our data support the concept, established in humans 4, 5, that low intrathymic expression of the TSHR is associated with susceptibility to developing pathogenic TSHR autoantibodies in Graves' disease, whereas high intrathymic TSHR mRNA levels are protective.
Author contributions
S. M. McL. and B. R. designed the study; H. A., B. B., S. L. and R. C. performed the experiments; S. M. McL., S. L. and B R. wrote the manuscript.
Disclosure
No disclosures exist.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Thyroiditis in N6 Hi‐expressor thyrotrophin receptor (TSHR) A‐subunit transgenic and non‐transgenic non‐obese diabetic (NOD).H2h4 mice. Examples of lymphocytic infiltration varying from moderate (a,c,d) to minimal (b) are shown for thyroid histology (haematoxylin and eosin, ×10 magnification; three transgenic mice; one non‐transgenic mouse).
Fig. S2. Regression of thyrotrophin receptor (TSHR) A‐subunit enzyme‐linked immunosorbent assay (ELISA) [optical density (OD) 490 nm] versus TSH binding inhibition (TBI) (% inhibition of TSH binding). Patterned area highlights positive TBI values for mice, albeit with low ELISA values typical of those seen in non‐obese diabetic (NOD).H2h4 recipients of the Hi‐expressor TSHR A‐subunit transgene. Data not published previously as a regression for Lo‐expressor NOD.H2h4 mice reported in Rapoport et al. [23].
Fig. S3. Regression of TSH binding inhibition (TBI) (% inhibition of TSH binding) versus thyroid stimulating antibody (TSAb) (% control). Dotted lines indicate ‘cut‐off’ points for TBI and TS antibody. The patterned area shows that sera negative for TBI also lack TS antibody activity. Data not published previously as a regression for Lo‐expressor non‐obese diabetic (NOD).H2h4 mice reported in Rapoport et al. [23].
Acknowledgements
We thank Dr Jean Ruf (INSERM‐URA, Faculté de Médecine, Marseille, France) for generously providing us with mouse monoclonal antibodies to human TPO. This work was supported by the National Institutes of Health Grants DK 54684 (SMM) and 19289 (BR) and the Canadian Diabetes Association OG‐3–13–4018 (SL).
References
- 1. Chazenbalk GD, Pichurin P, Chen CR et al Thyroid‐stimulating autoantibodies in Graves disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J Clin Invest 2002; 110:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen C‐R, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves' disease is the culprit as well as the victim. J Clin Invest 2003; 111:1897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol 2014; 9:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colobran R, Armengol MP, Faner R et al Association of an SNP with intrathymic transcription of TSHR and Graves' disease: a role for defective thymic tolerance. Hum Mol Genet 2011; 20:3415–23. [DOI] [PubMed] [Google Scholar]
- 5. Stefan M, Wei C, Lombardi A et al Genetic–epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc Natl Acad Sci USA 2014; 111:12562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell 1987; 49:273–80. [DOI] [PubMed] [Google Scholar]
- 7. Lesage S, Hartley SB, Akkaraju S, Wilson J, Townsend M, Goodnow CC. Failure to censor forbidden clones of CD4 T cells in autoimmune diabetes. J Exp Med 2002; 196:1175–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liston A, Lesage S, Gray DH et al Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity 2004; 21:817–30. [DOI] [PubMed] [Google Scholar]
- 9. Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity 2005; 22:385–96. [DOI] [PubMed] [Google Scholar]
- 10. Mingueneau M, Jiang W, Feuerer M, Mathis D, Benoist C. Thymic negative selection is functional in NOD mice. J Exp Med 2012; 209:623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liston A, Gray DH, Lesage S et al Gene dosage–limiting role of Aire in thymic expression, clonal deletion, and organ‐specific autoimmunity. J Exp Med 2004; 200:1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramsey C, Winqvist O, Puhakka L et al Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet 2002; 11:397–409. [DOI] [PubMed] [Google Scholar]
- 13. Jiang W, Anderson MS, Bronson R, Mathis D, Benoist C. Modifier loci condition autoimmunity provoked by Aire deficiency. J Exp Med 2005; 202:805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pichurin PN, Chen C‐R, Chazenbalk GD et al Targeted expression of the human thyrotropin receptor A‐subunit to the mouse thyroid: insight into overcoming the lack of response to A‐subunit adenovirus immunization. J Immunol 2006; 176:668–76. [DOI] [PubMed] [Google Scholar]
- 15. McLachlan SM, Nagayama Y, Pichurin PN et al The link between Graves' disease and Hashimoto's thyroiditis: a role for regulatory T cells. Endocrinol 2007; 148:5724–33. [DOI] [PubMed] [Google Scholar]
- 16. Misharin AV, Nagayama Y, Aliesky HA, Rapoport B, McLachlan SM. Studies in mice deficient for the autoimmune regulator (Aire) and transgenic for the thyrotropin receptor reveal a role for Aire in tolerance for thyroid autoantigens. Endocrinol 2009; 150:2948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McLachlan SM, Aliesky HA, Chen CR, Chong G, Rapoport B. Breaking tolerance in transgenic mice expressing the human TSH receptor A‐subunit: thyroiditis, epitope spreading and adjuvant as a ‘double edged sword’. PLOS ONE 2012; 7:e43517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev 2014; 35:59–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rasooly L, Burek CL, Rose NR. Iodine‐induced autoimmune thyroiditis in NOD‐H2h4 mice. Clin Immunol Immunopathol 1996; 81:287–92. [DOI] [PubMed] [Google Scholar]
- 20. Braley‐Mullen H, Sharp GC, Medling B, Tang H. Spontaneous autoimmune thyroiditis in NOD.H‐2h4 mice. J Autoimmun 1999; 12:157–65. [DOI] [PubMed] [Google Scholar]
- 21. Hutchings PR, Verma S, Phillips JM, Harach SZ, Howlett S, Cooke A. Both CD4(+) T cells and CD8(+) T cells are required for iodine accelerated thyroiditis in NOD mice. Cell Immunol 1999; 192:113–21. [DOI] [PubMed] [Google Scholar]
- 22. Chen CR, Hamidi S, Braley‐Mullen H et al Antibodies to thyroid peroxidase arise spontaneously with age in NOD.H‐2h4 mice and appear after thyroglobulin antibodies. Endocrinol 2010; 151:4583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rapoport B, Aliesky HA, Banuelos B, Chen CR, McLachlan SM. A unique mouse strain that develops spontaneous, iodine‐accelerated, pathogenic antibodies to the human thyrotrophin receptor. J Immunol 2015; 194:4154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Staff of the Jackson Laboratory . In: Fox RR. and Witham BA, eds. Handbook on genetically standardized Jax mice, 5th edn. Bar Harbor, ME: Jackson Laboratory, 1997; 1–143. [Google Scholar]
- 25. Chazenbalk GD, Jaume JC, McLachlan SM, Rapoport B. Engineering the human thyrotropin receptor ectodomain from a non‐secreted form to a secreted, highly immunoreactive glycoprotein that neutralizes autoantibodies in Graves' patients' sera. J Biol Chem 1997; 272:18959–65. [DOI] [PubMed] [Google Scholar]
- 26. Chazenbalk GD, Wang Y, Guo J et al A mouse monoclonal antibody to a thyrotropin receptor ectodomain variant provides insight into the exquisite antigenic conformational requirement, epitopes and in vivo concentration of human autoantibodies. J Clin Endocrinol Metab 1999; 84:702–10. [DOI] [PubMed] [Google Scholar]
- 27. Ruf J, Toubert M, Czarnocka B, Durand‐Gorde J, Ferrand M, Carayon P. Relationship between immunological structure and biochemical properties of human thyroid peroxidase. Endocrinol 1989; 125:1211–8. [DOI] [PubMed] [Google Scholar]
- 28. Pfaffl MW. A new mathematical model for relative quantification in real‐time RT–PCR. Nucleic Acids Res 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McLachlan SM, Braley‐Mullen H, Chen CR, Aliesky H, Pichurin PN, Rapoport B. Dissociation between iodide‐induced thyroiditis and antibody‐mediated hyperthyroidism in NOD.H‐2h4 mice. Endocrinol 2005; 146:294–300. [DOI] [PubMed] [Google Scholar]
- 30. Anderson MS, Venanzi ES, Klein L et al Projection of an immunological self shadow within the thymus by the Aire protein. Science 2002; 298:1395–401. [DOI] [PubMed] [Google Scholar]
- 31. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ‐specific T cells. Nat Immunol 2003; 4:350–4. [DOI] [PubMed] [Google Scholar]
- 32. Russell L, John S, Cullen J, Luo W, Shlomchik MJ, Garrett‐Sinha LA. Requirement for transcription factor Ets1 in B cell tolerance to self‐antigens. J Immunol 2015; 195:3574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takaba H, Morishita Y, Tomofuji Y et al Fezf2 orchestrates a thymic program of self‐antigen expression for immune tolerance. Cell 2015; 163:975–87. [DOI] [PubMed] [Google Scholar]
- 34. Brand OJ, Gough SC. Immunogenetic mechanisms leading to thyroid autoimmunity: recent advances in identifying susceptibility genes and regions. Curr Genomics 2011; 12:526–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Teumer A, Rawal R, Homuth G et al Genome‐wide association study identifies four genetic loci associated with thyroid volume and goiter risk. Am J Hum Genet 2011; 88:664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ploski R, Szymanski K, Bednarczuk T. The genetic basis of Graves' disease. Curr Genomics 2011; 12:542–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Medici M, Porcu E, Pistis G et al Identification of novel genetic loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLOS Genet 2014; 10:e1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rapoport B, Aliesky HA, Chen CR, McLachlan SM. Evidence that TSH receptor A‐subunit multimers, not monomers, drive antibody affinity maturation in Graves' disease. J Clin Endocrinol Metab 2015; 100:E871–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagayama Y, Kita‐Furuyama M, Ando T et al A novel murine model of Graves' hyperthyroidism with intramuscular injection of adenovirus expressing the thyrotropin receptor. J Immunol 2002; 168:2789–94. [DOI] [PubMed] [Google Scholar]
- 40. Rapoport B, Williams RW, Chen CR, McLachlan SM. Immunoglobulin heavy chain variable region genes contribute to the induction of thyroid stimulating antibodies in recombinant inbred mice. Genes Immun 2010; 11:254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mizutori Y, Saitoh O, Eguchi K, Nagayama Y. Adenovirus encoding the thyrotropin receptor A‐subunit improves the efficacy of dendritic cell‐induced Graves' hyperthyroidism in mice. J Autoimmun 2006; 26:32–6. [DOI] [PubMed] [Google Scholar]
- 42. Gilbert JA, Gianoukakis AG, Salehi S et al Monoclonal pathogenic antibodies to the thyroid‐stimulating hormone receptor in Graves' disease with potent thyroid‐stimulating activity but differential blocking activity activate multiple signaling pathways. J Immunol 2006; 176:5084–92. [DOI] [PubMed] [Google Scholar]
- 43. Kaneda T, Honda A, Hakozaki A, Fuse T, Muto A, Yoshida T. An improved Graves' disease model established by using in vivo electroporation exhibited long‐term immunity to hyperthyroidism in BALB/c mice. Endocrinol 2007; 148:2335–44. [DOI] [PubMed] [Google Scholar]
- 44. Wu L, Xun L, Yang J et al Induction of murine neonatal tolerance against Graves' disease using recombinant adenovirus expressing the TSH receptor A‐subunit. Endocrinol 2011; 152:1165–71. [DOI] [PubMed] [Google Scholar]
- 45. Rapoport B, McLachlan SM. TSH receptor cleavage into subunits and shedding of the A‐subunit; a molecular and clinical perspective. Endocr Rev 2016; 37:114–34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46. Rapoport B, Banuelos B, Aliesky HA, Hartwig Trier N, McLachlan SM. Critical differences between induced and spontaneous mouse models of Graves' disease with implications for antigen‐specific immunotherapy in humans. J Immunol 2016; 197:4560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Thyroiditis in N6 Hi‐expressor thyrotrophin receptor (TSHR) A‐subunit transgenic and non‐transgenic non‐obese diabetic (NOD).H2h4 mice. Examples of lymphocytic infiltration varying from moderate (a,c,d) to minimal (b) are shown for thyroid histology (haematoxylin and eosin, ×10 magnification; three transgenic mice; one non‐transgenic mouse).
Fig. S2. Regression of thyrotrophin receptor (TSHR) A‐subunit enzyme‐linked immunosorbent assay (ELISA) [optical density (OD) 490 nm] versus TSH binding inhibition (TBI) (% inhibition of TSH binding). Patterned area highlights positive TBI values for mice, albeit with low ELISA values typical of those seen in non‐obese diabetic (NOD).H2h4 recipients of the Hi‐expressor TSHR A‐subunit transgene. Data not published previously as a regression for Lo‐expressor NOD.H2h4 mice reported in Rapoport et al. [23].
Fig. S3. Regression of TSH binding inhibition (TBI) (% inhibition of TSH binding) versus thyroid stimulating antibody (TSAb) (% control). Dotted lines indicate ‘cut‐off’ points for TBI and TS antibody. The patterned area shows that sera negative for TBI also lack TS antibody activity. Data not published previously as a regression for Lo‐expressor non‐obese diabetic (NOD).H2h4 mice reported in Rapoport et al. [23].