

Summary
Citrullination of joint proteins by the protein arginine deiminase (PAD) family of enzymes is recognized increasingly as a key process in the pathogenesis of rheumatoid arthritis. This present study was undertaken to explore the efficacy of a novel PAD4‐selective inhibitor, GSK199, in the murine collagen‐induced arthritis model of rheumatoid arthritis. Mice were dosed daily from the time of collagen immunization with GSK199. Efficacy was assessed against a wide range of end‐points, including clinical disease scores, joint histology and immunohistochemistry, serum and joint citrulline levels and quantification of synovial autoantibodies using a proteomic array containing joint peptides. Administration of GSK199 at 30 mg/kg led to significant effects on arthritis, assessed both by global clinical disease activity and by histological analyses of synovial inflammation, pannus formation and damage to cartilage and bone. In addition, significant decreases in complement C3 deposition in both synovium and cartilage were observed robustly with GSK199 at 10 mg/kg. Neither the total levels of citrulline measurable in joint and serum, nor levels of circulating collagen antibodies, were affected significantly by treatment with GSK199 at any dose level. In contrast, a subset of serum antibodies reactive against citrullinated and non‐citrullinated joint peptides were reduced with GSK199 treatment. These data extend our previous demonstration of efficacy with the pan‐PAD inhibitor Cl‐amidine and demonstrate robustly that PAD4 inhibition alone is sufficient to block murine arthritis clinical and histopathological end‐points.
Keywords: ACPA, complement, citrullination, PAD4, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease that affects synovial tissue in multiple joints. Symptoms include pain and swelling of the small peripheral joints, and debilitating joint destruction resulting from bone erosion and pannus formation is characteristic of the disease. Approximately 60% of patients with RA test positive for antibodies to citrullinated protein antigens (ACPA) 1, which can manifest for years before onset of clinical disease 2, 3, 4, 5. Such autoantibodies could therefore contribute to the early development and progression of disease.
The enzymes responsible for generating citrullinated protein antigens are protein arginine deiminases (PADs), of which there are five in mammals 6, 7, 8. The different PADs demonstrate variable tissue expression and localization, with PAD2 and PAD4 being particularly relevant to RA because of their expression in macrophages and neutrophils in the RA synovium, respectively 9. In addition, variable evidence for and against an underlying genetic association for PAD4 with RA exists across multiple studies 10, 11, 12, and may be suggestive of interactions with concomitant environmental factors such as smoking in distinct (Caucasian versus Asian) patient populations. PADs catalyze the post‐translational modification of arginine residues to citrulline, a phenomenon termed deimination, which has been implicated in cell differentiation 13, stem cell pluripotency 14, apoptosis 15, neutrophil extracellular trap (NET) formation 16, transcriptional regulation 17, 18, antigen processing in autophagy 19, 20, inflammation 21, the cornification of skin 22, demyelination in multiple sclerosis 23, chemokine regulation 24, 25, 26, spinal cord injury repair 27 and various normal cellular processes. The proposed role in neutrophil extracellular trap (NET)osis is also pertinent for RA, as NETs are deficient in the absence of PAD4 28 and PAD4 is released extracellularly in RA joints, due probably to the pathological status of RA neutrophils 29. In addition, a known single nucleotide polymorphism (SNP) associated with RA (C1858T in PTPN22) has been shown recently to reduce direct interaction between PTPN22 and PAD4, leading to de ‐repression of both hypercitrullination and NETosis 30.
While citrullination is a normal, if ill‐understood, physiological process and citrullinated proteins are found in many organs associated with tissue injury 31, the immune response to citrullinated proteins is commonly reported to be specific for RA.
In order to assess the role of citrullination on the pathogenesis of inflammatory arthritis, we previously tested the effects of Cl‐amidine (an irreversible, peptidomimetic, pan‐PAD inhibitor 32, 33, 34) in the murine collagen‐induced arthritis (CIA) model of RA 35. In these studies, pan‐PAD inhibition decreased inflammation and joint destruction, as well as having a modest effect on epitope spreading to both citrullinated and non‐citrullinated autoantigens. However, as Cl‐amidine is not selective for PAD4 over other PAD family members, the precise role of PAD4 inhibition in treating CIA is difficult to infer. GSK199 was described recently as a selective, reversible, small‐molecule inhibitor of PAD4 and also of NET production 36. It operates via a novel mechanism of action (binding preferentially to an inactive conformation of the enzyme), and crystallographic studies have identified key conformational changes at the enzyme's active site, accounting for both its potency and selectivity. Herein we demonstrate that treatment with GSK199 is sufficient to prevent CIA and results in decreased paw inflammation, joint destruction and a substantially reduced deposition of complement C3 in the joints as evaluated by histological assessments, with more modest effects on antibody responses against a subset of citrullinated proteins and other autoantigens. These results demonstrate that PAD4 inhibition is sufficient for control of murine arthritis and strengthen PAD4 as a promising therapeutic target to address unmet clinical need in inflammatory arthritis.
Materials and methods
Collagen‐induced arthritis
CIA was induced in 6–8‐week‐old dark brown Agouti (DBA)/1J mice (from The Jackson Laboratory, Bar Harbor, ME, USA) by intradermal injection on day 0 with 100 µl of Incomplete Freund's adjuvant (IFA) containing 200 µg of bovine CII (Collagen Type II; Elastin Products, Owensville, MO, USA) along with 200 µg of inactivated Mycobacterium tuberculosis (H37Ra; Difco, Detroit, MI, USA) 37. A boost injection with the same reagents was also performed on day 21. All animal experiments were conducted in accordance with the GSK policy on the Care, Welfare and Treatment of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of Colorado School of Medicine.
Treatment groups
All groups of mice (n = 7) were dosed daily throughout the study, i.e. from the first injection on day 0 to termination on day 35. Mice were divided into six groups to receive six distinct interventions: no injection, 0·9% NaCl (vehicle control), 10 mg/kg GSK199, 30 mg/kg q.d. (once a day) GSK199, 30 mg/kg b.i.d. (twice a day) GSK199 (with a 6‐h interval between doses) or 0·25 mg/kg dexamethasone. All injections were subcutaneous (s.c.) apart from dexamethasone, which was administered intraperitonally (i.p.). After a booster injection on day 21, two trained laboratory personnel blinded to treatment status evaluated clinical disease activity independently. The maximum score of 12 per animal reflected the sum of a three‐point scale per paw. The scoring guidelines were: 0 = normal joint; 1 = modest inflammation with redness; 2 = severe swelling and erythema over the entire paw; and 3 = deformed paw/joint with ankylosis, joint rigidity and functional impairment.
GSK199 pharmacokinetic measurements
Mice were bled prior to receiving treatments on days 2 and 15, and 2 h post‐treatment on days 1, 7, 14, 21, 28 and 35. Blood samples were analysed for the free base of GSK199 using protein precipitation and high performance liquid chromatography‐mass spectrometry (HPLC‐MS/MS) analysis. Acetonitrile (350 µl) containing an internal standard was added to blood samples (20 µl blood diluted previously with 20 µl water). Samples were mixed by mechanical shaking for 20 min, and then centrifuged (2988 g for 20 min at room temperature). A 150 µl volume of supernatant was transferred to another 96‐well plate and evaporated to dryness under a steady stream of nitrogen at 40°C. The plate was reconstituted in a suitable volume of 0·1% formic acid in acetonitrile: water, 5 : 95 (v/v) and analysed for GSK199 by reverse‐phase HPLC‐MS/MS using a heat‐assisted electrospray interface in positive ion mode (Sciex API 4000) and a Kinetix C18 column (50 × 2·1 mm ID, 2.7 μm; Phenomenex, Torrance, CA, USA). The mobile phase was delivered as a linear gradient of 5–95% acetonitrile (containing 0·1% v/v formic acid) : water (containing 0·1% formic acid) for 1·1 min, held for 0·05 min and returned to starting conditions. The overall run‐time was 1·4 min. Nominal MRM (multiple reaction monitoring) transition for GSK199 was 433–333. Concentration range for the assay was: 0·005–10 µg/ml (0·012–23 µM) with a lower limit of quantification (LLQ) of 0·005 µg/ml (0·012 µM).
Histological examination
At the end of the dosing period, the forepaws and right hind limb (including the paw, ankle and knee) were removed from each mouse and fixed immediately in 10% buffered formalin (Fisher Scientific (Kalamazoo, MI)). After embedding in paraffin, the tissue was sectioned and stained for further analysis. Haematoxylin and eosin stains were used for global assessment of histopathology and toluidine blue staining enabled evaluation of cartilage changes 38. An assessor (blinded to treatment status) then scored the joint sections (on a scale of 0–5) for presence and magnitude of synovial inflammation, pannus development and damage to bone and cartilage. Composite data from five joints per animal were then expressed as mean ± standard error of the mean (s.e.m.). Mouse C3 deposition in synovium and cartilage was also scored quantitatively on a scale of 0–3 following immunohistological processing 39.
Measurement of total citrulline content
At day 35 left hind knees were excised and flash‐frozen in liquid nitrogen. After enrichment of synovial tissue by further dissection, homogenates were prepared as described by Moscarello 40, 41. In brief, individual samples were resuspended at a final concentration of 20 mg tissue per 0·1 ml buffer in 50 mM HEPES pH 7·6, 1·0 mM ethylenediamine tetraacetic acid (EDTA), 0·5 mM DTT (dithiothreitol) and 0·43 mM PMSF (phenylmethylsulphonyl fluoride). Cell extracts were homogenized and debris was pelleted by centrifugation. Synovial lysate (60 μl) or serum lysate (20 μl) was added to a reaction buffer containing 50 mM NaCl, 10 mM CaCl2, 2 mM DTT and 100 mM Tris pH 7·6 for analysis of total citrulline content. This involved the addition of 200 µl of a colour development solution specific for citrullinated proteins (COLDER) 42. After vortexing, the sample was incubated for 20 min at 95°C. The absorbance at 540 nm was then interpolated off a standard curve of known citrulline concentrations. Duplicate measurements were employed, and the citrulline levels were normalized for the total protein concentration of each sample.
Anti‐collagen antibody determination
Specific enzyme‐linked immunosorbent assays (ELISAs) for murine and bovine CII antibodies [both immunoglobulin (Ig)G2a and IgG1] were employed, with triplicate measurements, as described previously 37. After coating overnight at 4°C with 5 µg/ml of CII (Chondrex, Seattle, WA, USA), 96‐well plates were washed three times with phosphate‐buffered saline (PBS) containing 0·05% Tween and 1% bovine serum albumin (BSA), and blocked with PBS and 1% BSA for 4 h at 4°C. After removal of the blocking solution, 50 µl of each serum sample (diluted 1 : 2000 in PBS) were added (in triplicate) and incubated overnight at 4°C. The plates were washed in PBS with 0·05% Tween and incubated with horseradish peroxidase (HRP)‐conjugated anti‐mouse IgG1 or IgG2a antibodies (1 : 1000 dilution in PBS; Southern Biotechnology Associates, Birmingham, AL, USA) for 4 h at 4°C to detect bound IgG1 and IgG2a antibodies. After washing a further three times with PBS/0·05% Tween, reactivity was visualized by adding 100 µL of 1‐step TMB reagent (3,3',5,5'‐tetramethylbenzidine; ThermoFisher Scientific, Waltham, MA, USA). Absorbance was read at 450 nm. A standard curve for each IgG isotype was generated using mixed and serially diluted serum from mice with severe disease. These data were expressed as mean optical density (OD) values ± s.e.m.
Synovial proteome microarray
Sera from mice treated with GSK199 or vehicle were analysed for autoantibody responses in multiplex format against synovial proteome arrays. These contained proteins and peptides (191 in total) representing candidate autoantigens in RA 43, 44 and have been validated extensively with both monoclonal antibody panels and reference sera specific for many of the target proteins. Significant differences in autoantibody reactivities between GSK199 and vehicle treatments were identified by SAM analysis (significance analysis of microarrays; version 3.08). Data were arranged using hierarchical cluster analysis (Cluster version 3.0 software) and displayed as heatmaps [Java TreeView software version 1.1.3 created by Alok (alokito@users.sourceforge.net)] 45, 46. An Accession number (GSE69680) for the complete microarray data is approved and available in the GEO public database at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE69680.
Statistical analysis
Analysis of variance (anova) with one‐sided comparisons to saline treatment was used to examine the CIA data (clinical disease activity scores, histology, anti‐collagen antibody levels and synovial/serum citrulline content). Pearson's correlation coefficient was employed to compare histology and clinical disease scoring. Data were expressed as arithmetic or geometric means (for pharmacokinetic data) and P < 0·05 was considered statistically significant.
Results
CIA clinical disease activity is reduced by treatment with GSK199
To test if inhibition of PAD4 activity was sufficient for efficacy against CIA, arthritis was induced in DBA1/J mice by immunization with bovine CII on days 0 and 21, and clinical disease activity was monitored during days 21–35 (Fig. 1a). From day 0 mice began receiving either 10 or 30 mg/kg (q.d. or b.i.d.) GSK199 s.c., 0·25 mg/kg dexamethasone i.p., 0·9% saline s.c. or no treatment. Mice continued to receive these treatments daily throughout the course of the experiment. As predicted, mice receiving 0·25 mg/kg dexamethasone developed very little clinical disease. Of note, mice receiving the vehicle control (0·9% saline) showed a 36% decrease in disease severity [measured by area under the curve (AUC), Fig. 1b] compared to control mice receiving no treatment, due possibly to placebo effects from regular handling. However, mice receiving daily s.c. injections of GSK199 in vehicle showed clinical disease activity reductions of 25% (10 mg/kg) and 54% (30 mg/kg q.d., P < 0·05) relative to saline‐treated control, and reductions of 52% (10 mg/kg, P < 0·05) and 71% (30 mg/kg q.d., P < 0·05) compared to mice receiving no treatment. Therefore, treatment with GSK199 significantly reduces clinical disease activity globally in CIA.
Figure 1.
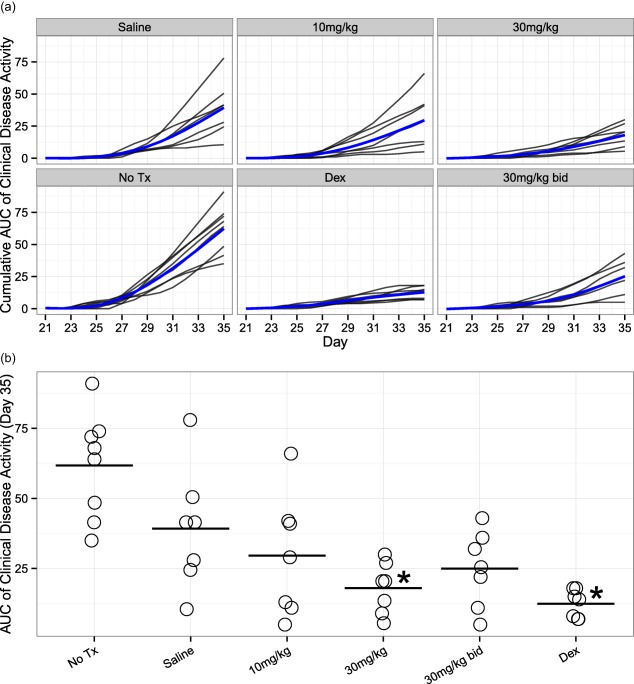
Protein arginine deiminase (PAD4) inhibition decreases clinical disease severity in collagen‐induced arthritis (CIA). Arthritis was induced by two injections, at days 0 and 21, of bovine collagen type II (CII) in Complete Freund's adjuvant (CFA). Mice were treated as described in materials and methods and assessed for clinical disease activity three times each week. (a) Clinical disease activity (CDA) [as cumulative area under the curve (AUC) of CDA] plotted as a function of time. Individual mice are shown as black lines, with the additional blue line representing the mean cumulative activity for each group (estimated from a longitudinal mixed‐model fit). All treatment groups (including the saline vehicle control) showed reduced CDA scores compared to mice receiving no treatment. The plots represent assessments from a single experiment. (b) Plot depicting the AUC of clinical disease activity at day 35 for each mouse per treatment group. AUC was decreased by 36% in the saline group compared to no treatment, while 25, 54 and 36% AUC reductions were observed in 10, 30 and 30 mg/kg b.i.d. groups, respectively, when compared to saline‐treated control mice. Statistically significant changes in AUC compared to saline‐treated mice were observed in 0·25 mg/kg/day dexamethasone‐treated mice and mice receiving GSK199 at 30 mg/kg q.d. (P < 0·05, n = 7 except for the no‐treatment group, where n = 8).
Treatment with GSK199 reduces joint destruction in CIA
To evaluate the effects of treatment with GSK199 on joint destruction in CIA, joints were analysed histologically at the end of the experiment (day 35). As anticipated, histopathology scores for synovial inflammation, pannus invasion and damage to cartilage and bone were reduced markedly (P < 0·05) for the 0·25 mg/kg dexamethasone treatment group compared to vehicle controls (Fig. 2a–d, with representative images in Supporting information, Fig. S1). Scores for all GSK199 treatment groups trended downwards in each category, with the 30 mg/kg and 30 mg/kg b.i.d. treatment groups demonstrating significantly reduced inflammation, pannus and damage scores (P < 0·05) compared to vehicle controls. In addition, GSK199 at 10 mg/kg also affected two of these four histopathology end‐points significantly (synovial inflammation and bone damage). The clinical disease activity and histology scores were correlated significantly with each other (r = 0·80, P < 0·001; Supporting information, Fig. S2). C3 deposition was reduced in the synovium (Fig. 2e) and cartilage (Fig. 2f) of mice treated with GSK199, an effect that is demonstrated clearly in the supporting images (Supporting information, Fig. S3). Remarkably, treatment with 30 mg/kg b.i.d. GSK199 resulted in the elimination of C3 deposition in both compartments (P < 0·05), while treatment with 10 or 30 mg/kg q.d. GSK199 also reduced C3 deposition significantly (P < 0·05). Similar to clinical disease activity, treatment with saline alone resulted in decreased C3 deposition in the synovium and cartilage relative to untreated mice. These results indicate that GSK199 treatment blocks joint destruction and reduces C3 deposition in the joints of mice with CIA.
Figure 2.
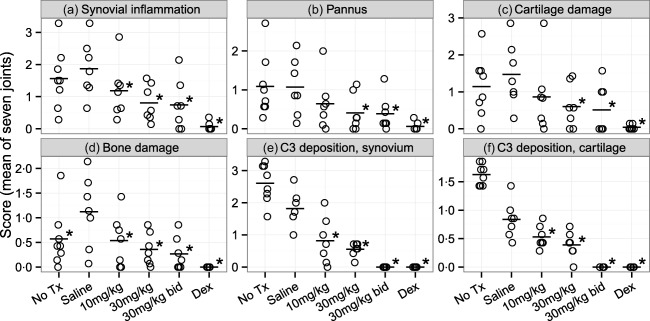
GSK199 treatment reduces histological disease severity. Histological sections were scored on a scale of 0–5 for changes in (a) synovial inflammation, (b) pannus formation, (c) cartilage damage and (d) bone damage. Sections were also scored for complement C3 deposition in both (e) the synovium and (f) cartilage on a scale of 0–3. Data are expressed as the means of seven joints per animal. Treatment with 30 mg/kg (q.d. or b.i.d.) GSK199 resulted in statistically significant, decreased histopathology scores for inflammation, cartilage/bone damage and complement deposition compared to the saline vehicle group (P < 0·05, n = 7 except for no treatment, where n = 8). Treatment with 10 mg/kg GSK199 resulted in statistically significant improvements in synovial inflammation, bone damage and both locations of complement deposition.
Treatment with GSK199 does not alter joint total citrulline levels significantly
To explore whether the effects of GSK199 are reflected by its ability to inhibit PAD4 activity, we assessed synovial citrulline content as a surrogate of PAD activity. Synovial citrulline content (from the knee) showed variability within groups but the total citrulline content of joints from saline‐treated animals was similar to that of arthritic mice not administered with any compound, and both showed markedly higher levels over control mice in which CIA was not induced (labelled as ‘WT’). Citrullination trended downwards in response to all GSK199 doses (Fig. 3a) towards the basal levels seen in the non‐immunized mice, but was less marked than the decrease observed previously with Cl‐amidine 35. It is unlikely, given the attractive profile and pharmacokinetic distribution of GSK199, that inability to access the knee joints would account for this lack of robust effect seen on local citrulline levels, which is probably related to the variability observed. We also measured the citrulline content in serum at day 35. Similar to joint total citrulline levels, serum citrulline content trended downwards with GSK199 treatment (Fig. 3b), but only untreated mice (WT) and mice administered dexamethasone demonstrated a statistically significant decrease in total citrulline levels in the serum (P < 0·05).
Figure 3.
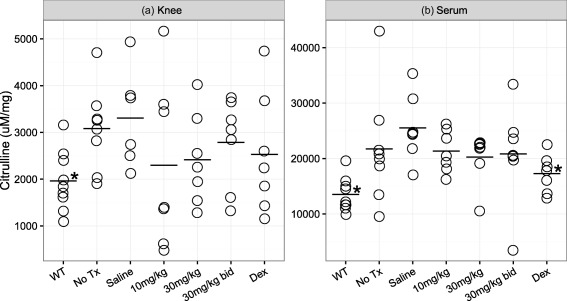
GSK199 treatment does not alter joint and serum total citrulline levels significantly. (a) Synovial citrulline content from the knee. Arthritis was induced in mice by injections of bovine collagen in Complete Freund's adjuvant (CFA) at days 0 and 21. Mice were treated daily, beginning on day 0, with GSK199 (10, 30 or 30 mg/kg b.i.d.) in saline, saline alone, 0·25 mg/kg dexamethasone or were untreated. Mice which received no collagen type II (CII) immunizations (WT) were used as a baseline reference. (b) Serum citrulline content was also measured in the same mice at day 35. GSK199‐treated mice showed slightly decreased levels of total citrulline in the knee and serum, but only wild‐type (WT) mice and dexamethasone‐treated animals had statistically significant decreases in citrulline content compared to saline‐treated controls.
We had anticipated that, as GSK199 was not a pan‐PAD inhibitor, total citrulline levels might not be affected dramatically. To verify that serum GSK199 levels were sufficient to inhibit PAD4 during our study, blood concentrations of GSK199 were determined at regular intervals at expected peak (2 h) and trough concentrations (prior to the next dose) for each group. Sample analysis showed that blood concentrations of GSK199 were consistent between animals and throughout the treatment period, confirming reproducible exposure to GSK199 during the experiment (Supporting information, Fig. S4). Blood concentrations of GSK199 increased in proportion to dose level between the 10 and 30 mg/kg groups dosed q.d. Geometric mean concentrations at 2 h were 4·0 and 11·4 µM for the 10 and 30 mg/kg groups, respectively, and corresponding trough concentrations were 0·02 and 0·10 µM. For the 30 mg/kg b.i.d. group, the geometric mean trough concentration was 0·60 µM, confirming greater exposure and higher trough concentrations compared with the equivalent time‐point in animals dosed q.d. The second dose at 6 h also drives a second peak concentration approximately 1–2 h later. This was confirmed in a satellite pharmacokinetic (PK) study (data not shown), but further blood sampling to generate these data could not be incorporated into the protocol design. As serum levels of GSK199 remained sufficient for PAD inhibition (well above the measured inhibitory concentration 50 (IC50) potencies for binding to and inhibition of the recombinant enzyme 36), these data suggested that systemic levels of GSK199 were sufficiently high throughout the study to inhibit PAD4 and that the lack of significant decrease in total citrulline levels might be attributed to citrullination by uninhibited PAD family members and/or to limitations of the COLDER assay for in‐vivo measurement of citrulline.
Treatment with GSK199 does not alter autoantibody production significantly in CIA
As CIA develops, evolution of both bovine (xeno) and mouse (self) anti‐CII antibodies increases, with the latter reflecting a growing loss of tolerance to self‐epitopes. To determine whether GSK199 treatment recapitulated the effects of Cl‐amidine treatment on autoimmune antibody production, sera from mice from each dosing group were incubated on separate 96‐well plates coated with either bovine or mouse CII and analysed using ELISAs specific for mouse IgG1 and IgG2a antibodies to each CII species.
Unlike Cl‐amidine treatment, none of the GSK199 treatments affected anti‐bovine or anti‐mouse CII IgG1 or IgG2a antibody levels (Supporting information, Fig. S5). These results demonstrated that GSK199 efficacy is not mediated via decreased humoral responses to native or foreign CII.
Treatment with GSK199 reduces epitope spreading in CIA
Synovial antigen array analysis of day 35 sera was performed to assess the effects of GSK199 on auto‐antibody responses. These synovial arrays have been described previously 35, 43, 44. Briefly, these arrays contained 191 peptides or proteins, both with and without citrullination, representative of candidate antigens in RA 47. Following SAM analysis, seven and 14 autoantibody reactivities, respectively, were decreased statistically in CIA mice treated with 30 or 30 mg/kg b.i.d. GSK199 compared to vehicle treatment (Fig. 4). In contrast, more than 150 other epitopes (native and citrullinated) on the arrays did not exhibit significantly different reactivity. Sera from mice treated with 30 mg/kg GSK199 exhibited decreased antibody reactivity to native epitopes from human fibrinogen alpha and beta chains and keratin. The parallel 30 mg/kg b.i.d. sera exhibited decreased antibody responses to native epitopes such as collagen IV, heat shock protein 65, peptides from alpha and beta chains of human fibrinogen (two of which were decreased in the 30 mg/kg q.d. group) and vimentin compared to saline controls; q < 0·1%. Decreased autoantibody responses to citrullinated vimentin and fibromodulin were also observed compared to controls after b.i.d. dosing of GSK199 (q < 0·1%); these epitopes were not affected by GSK199 30 mg/kg q.d. Importantly, there were no differences between treatment with GSK199 or saline alone in the reactivity to 38 other citrullinated epitopes in the array. These data demonstrated that GSK199 treatment decreases autoantibody production in CIA and impairs epitope spreading, to an extent (partial) similar to that observed previously with pan‐PAD inhibition.
Figure 4.
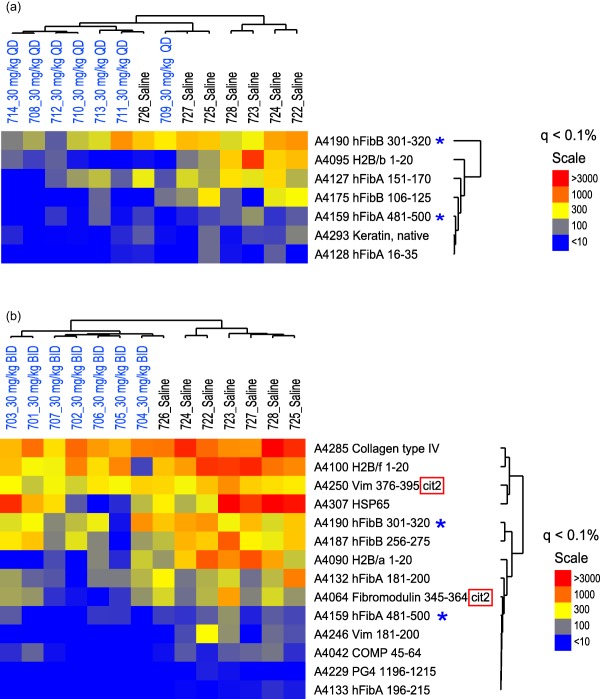
GSK199 treatment reduces epitope spreading in collagen‐induced arthritis (CIA). Sera were collected from (a) 30 mg/kg GSK199 q.d. or (b) 30 mg/kg GSK199 b.i.d. and control‐treated mice. Autoantibodies in these samples were assessed using synovial antigen arrays with a goat anti‐mouse anti‐immunoglobulin (Ig)G/M secondary antibody. Significance analysis of microarrays (SAM) was used to analyse the antigen array data sets and highlighted seven and 14 antigens, respectively, in each treatment group, with significant differences in autoantibody reactivity [false discovery rate (q) < 0·1]. Relationships between the profiles were clarified by hierarchical clustering and the data displayed as heatmaps (with blue representing negative associations, yellow intermediate and red strong positive). GSK199‐treated mice co‐clustered (on the left of the heatmap) while exhibiting lower autoantibody titers against all the seven and 14 antigens (respectively) identified by SAM, including to native epitopes derived from collagen IV and human fibrinogen alpha and beta chains, as well as citrulline‐modified vimentin and fibromodulin peptides (red boxes). Antigens affected by both treatments are also indicated by blue asterisks.
Discussion
Our data indicate that prophylactic treatment with GSK199, a selective small molecule inhibitor of PAD4, with dosing initiated from the same day as the induction of arthritis, resulted in the prevention of clinical and histological disease severity in CIA with effects comparable to those achieved with pan‐PAD inhibition. Epitope spreading, as measured by peptide array analysis, was reduced in a similar fashion to that seen with Cl‐amidine treatment, in that a small subset (< 10%) of humoral responses to some of the peptides were inhibited, but no overlap with the epitope spreading reduction by Cl‐amidine was observed. Notably, PAD4 inhibition did not alter significantly either joint or serum total citrulline levels or autoantibody titres to mouse CII in this study. Finally, we observed significant decreases in C3 deposition in the joints of mice treated with GSK199. These findings suggest that: (1) PAD inhibition, rather than off‐target drug effects, is responsible for blocking disease progression in CIA, (2) PAD4 inhibition is sufficient for this efficacy and (3) PAD inhibition does not appear to affect CIA primarily by blocking either development of citrullinated epitopes or ACPA.
Interestingly, we observed a significant efficacious effect of vehicle (0·9% saline, s.c.) treatment on inflammation severity and C3 deposition in the joints that was not observed consistently in this model with previous Cl‐amidine treatment 35 (PBS, i.p.). Further experiments will be necessary to confirm this effect and elucidate its cause, but it potentially reflects beneficial effects of regular, intensive handling on these vehicle‐dosed mice. However, despite this unexpected beneficial capacity of the vehicle control, GSK199 treatment reduced paw inflammation and C3 deposition to a greater extent than vehicle treatment alone, suggesting strongly that GSK199 treatment confers significant additional benefit over 0·9% saline treatment.
Our observation that a distinct PAD inhibitor utilizing a different mechanism of action, composition and route of delivery to Cl‐amidine affects CIA argues that PAD inhibition is fundamentally responsible for disease efficacy. It is highly unlikely that two structurally and mechanistically unrelated PAD inhibitors could bring about such similar outcomes due to non‐specific effects. That Cl‐amidine, an irreversible peptidomimetic pan‐PAD inhibitor, and GSK199, a reversible PAD4‐selective inhibitor, affect CIA progression similarly in the disease parameters evaluated suggests further that PAD4 inhibition is sufficient for disease control in CIA. Notably, total citrulline levels in the joints and serum of GSK199‐treated mice were not changed significantly compared to vehicle treatment, unlike mice administered Cl‐amidine previously 35. This lack of response may be reflective of the high variability and/or relative insensitivity of this assay (also consistent with significance being reached for Cl‐amidine against this end‐point only by pooling three dosing groups) and is also consistent with stable citrulline levels being observed in serum or lung when PAD4 is depleted in tumour necrosis factor (TNF)‐α‐overexpressing mice, despite marked reduction in parallel arthritis and lung inflammation assessments 48, 49. Additionally, the contribution of free citrulline to the total levels measured is unknown and, as non‐peptidyl citrulline would be detected but unresponsive to PAD4 inhibition, the available window for pharmacological inhibition could be limited. However, PAD2 and PAD4 are both expressed in the inflamed joint 9, so it is also possible that, in the presence of GSK199, uninhibited PAD2 continues to citrullinate joint proteins and could mask some decrease in total citrulline levels by PAD4 inhibition. Despite this, it is of note that treatment with a highly selective PAD4 inhibitor demonstrated such striking efficacy in reducing disease severity in CIA.
Although this efficacy contrasts with an early report that PAD4 knock‐out mice did not show alteration in disease severity in the KBxN passive transfer model of arthritis 50, two further studies 51, 52 (in the GPI‐induced and CIA active induction models, respectively) have since demonstrated clear effects of PAD4 deficiency on arthritis, with increased compensatory expression of PAD2 reported in the latter paper possibly accounting for the partial effects of PAD4 deficiency. Therefore, on balance there is some consistency between genetic and pharmacological effects on PAD4, although future contextualization of variance across different deficient mice/models would be desirable. However, the negative KBxN model data are consistent with observations of parallel pan‐PAD inhibition with Cl‐amidine having no effect on CAIA 35, a similarly passive transfer model of arthritis, and suggest that PAD inhibition cannot alter the effector phase of disease.
In contrast to our experiments with Cl‐amidine, we did not observe significant differences in anti‐mouse CII autoantibody titres in GSK199‐treated mice. This suggests that PAD4 activity does not regulate autoantibody production to collagen epitopes directly. However, GSK199‐treated mice displayed lower autoantibody reactivity to RA peptide arrays. Importantly, mice treated with GSK199 did not show reduced reactivity to citrullinated peptides specifically, but demonstrated reduced reactivity to a subset of both citrullinated and native peptides, as we observed with pan‐PAD inhibition. Of note, a similar modest effect or potential disconnect with synovial peptide arrays is seen when PAD4 is depleted in TNFα over‐expressing mice, despite profound differences in arthritis progression 48. These observations are consistent with a hypothesis that these effects on autoantibody production may be a secondary effect of efficacy, rather than a direct result of PAD inhibition. This protective effect of GSK199 may occur through inhibition of NET formation in arthritis 29. Indeed, a recent paper 53 reproduces the efficacy of Cl‐amidine in CIA and demonstrates elegantly that it is associated with marked decreases in the numbers of neutrophils undergoing NETosis (assessed robustly by quantification of co‐stained images). This report also offers additional evidence for associated immune effects of Cl‐amidine on T helper type 1 (Th1) helper cells in the draining lymph nodes in these mice and demonstrates that NET extracts from diseased animals promote activation of murine and dendritic cells upon co‐incubation.
In such absence of profound effects on multiple mechanistic end‐points in our model, it is likely that additional processes contribute to the striking clinical activity and histopathological efficacy. We propose two complementary mechanisms. First, the reduction in C3 deposition in the joints of animals treated all doses of GSK199 was the most sensitive outcome of this study (i.e. also observed in the 10 mg/kg dosing group) and suggests that PAD4 inhibition may alter the complement response, an important system in both murine and human arthritis. Notably, complement inhibition of the alternative pathway amplification loop, as well as a decrease in the levels of C5a, C3a and membrane attack complex generation, have all been shown previously to result in decreases in joint damage in CIA and/or CAIA 39, 54, 55, 56, 57, 58. Secondly, PAD4 is known to affect gene transcription via epigenetic regulation of the histone arginine methylation/citrullination balance (reviewed by 59) and it is likely that PAD4 may be regulating multiple genes (including key complement pathway members) at the transcript level. Further experiments are needed to address this and other hypotheses in order to understand any epigenetic manifestations of PAD inhibition on multiple facets of arthritis and complement biology.
During the preparation of this paper, a complementary report has been published 60, demonstrating impressive efficacy in a model of mouse CIA with the second‐generation peptidomimetic PAD family inhibitor BB‐Cl‐amidine, when dosed therapeutically. Consistent with our data, minimal effects on ACPA responses versus a subset of citrullinated epitopes are reported. Efficacy was proposed instead to be linked to profound effects on the modulation of T helper subsets from Th1 and Th17 to Th2. Given the increasing evidence for epigenetic effects driving immune mechanisms, such changes are likely to involve transcriptional mechanisms. In addition, as BB‐Cl‐amidine is biased towards PAD2 over PAD4, maintaining the potency against PAD4 from Cl‐amidine while inhibiting PAD2 ∼10 times more potently 61, the authors propose that PAD4 may be the wrong family member to target. This possibility would need detailed understanding of the in‐vivo target engagement for PAD2 over PAD4 at the dose used, but is not consistent with the profound efficacy reported here with GSK199. The potential for agents targeting either enzyme (or both) in arthritis still seems highly justified.
In conclusion, GSK199 treatment decreases epitope spreading in a largely ACPA‐independent manner and substantially affects the clinical and histological disease severity of CIA. However, GSK199 treatment does not appear to reduce total citrulline levels or anti‐mouse CII autoantibody titres. Considered with our results using the pan‐PAD inhibitor Cl‐amidine, these findings suggest that PAD inhibition controls disease by exerting broader effects on the immune system than simply by altering the generation of citrullinated epitopes. In concert with the growing literature in this field, these potential mechanisms are varied, but may include the regulation of complement protein activation and/or deposition in the joint (described herein), T helper cell activity 60, dendritic cell maturation 53 or other proinflammatory pathways known to participate in this disease process 62. Crucially, however, the findings reported in this manuscript confirm definitively that specific inhibition of PAD4 is sufficient to prevent arthritis progression in this CIA model.
Disclosure
H. D. L., D. C. C., S. T., D. L., R. K. P. and P.P.T. are full‐time employees and shareholders of GlaxoSmithKline. W. H. R. was a paid consultant of Padlock Therapeutics, and his laboratory receives funds for research from Gilead Sciences. Authors from the Holers laboratory (V. C. W., N. K. B., K. N. C., G. M. and V. M. H.) were paid to run this study by GSK.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Representative histopathology (toluidine‐blue) images (a–f) from the knee joints of mice with collagen‐induced arthritis (CIA) with and without treatments. All joints were fixed with 10% neutral buffered formalin, paraffin‐embedded, and sectioned at a thickness of 5 µm. The top three panels show staining with T‐blue (blue colour) from the knee joints of CIA mice: (a) no treatment (no Tx), (b) with saline (0.9%) or (c) 10 mg/kg GSK199. The bottom three panels show staining with T‐blue from the knee joints of CIA mice: (d) 30 mg/kg GSK199 b.i.d., (e) 30 mg/kg GSK199 q.d. or (f) dexamethasone (Dex). Areas of synovium (S‐black arrow), area of cartilage (C‐black arrow), bone (B‐black arrow) and meniscus (M) have been identified. The sections were also counterstained with haematoxylin and eosin ((H&E) and photographed under the ×10 objective using the Zeiss Observer D1 (AXIO) microscope. The red scale bar shown at the lower right‐hand corner in all panels from (a–f) is 0·1 mm (100 µm).
Fig. S2. Correlation between total clinical disease activity (expressed as area under the curve) and histology scores (expressed as average of four scores), plotted both by individual treatments and as a composite of all dosing groups.
Fig. S3. Representative immunohistochemistry (C3 deposition) images (a–f) from the knee joints of mice with collagen‐induced arthritis (CIA) with and without treatments. All joints were fixed with 10% neutral buffered formalin, paraffin‐embedded, and sectioned at a thickness of 5 µm. All sections have been stained with anti‐C3 antibody (brown colour) and also counterstained with haematoxylin and eosin (H&E, blue colour). No brown colour indicated the absence of complement protein C3 deposition or below detection limits. The top three panels show staining with C3 (brown colour) from the knee joints of CIA mice: (a) no treatment (no Tx), (b) with saline (0·9%) or (c) 10 mg/kg GSK199. The bottom three panels show staining with C3 antibody (brown color) from the knee joints of CIA mice: (d) 30 mg/kg GSK199 b.i.d., (e) 30 mg/kg GSK199 q.d. or (f) dexamethasone (Dex). Areas of synovium (S‐black arrow), area of cartilage (C‐black arrow), bone (B‐black arrow) and meniscus (M) have been identified. The sections were photographed under the ×10 objective using the Zeiss Observer D1 (AXIO) microscope. The red scale bar shown at the lower right‐hand corner in all panels from (a‐f) is 0·1 mm (100 µm).
Fig. S4. Pharmacokinetic measurement of GSK199 compound levels in blood of mice in the study. Geometric means of peak and trough blood concentrations following subcutaneous (s.c.) administration of GSK199 at 10 and 30 mg/kg q.d. and 30 mg/kg b.i.d. to the dark brown Agouti (DBA)/1J mice; sampled on days 1, 7, 14, 21, 28 and 35 during the study at expected peak (2 h: circles) and trough concentrations (prior to the next dose: triangles) for each group.
Fig. S5. GSK199 treatment does not reduce autoantibody titres in collagen‐induced arthritis (CIA). Sera from individual dosed mice were incubated with bovine (a, b) or mouse (c, d) collagen type II (CII). The presence of either anti‐mouse immunoglobulin IgG1 (a, c) or IgG2a antibody (b, d) was detected by enzyme‐linked immunosorbent assay (ELISA). The 0·25 mg/kg dexamethasone group had significantly reduced titres for bovine CII/IgG1, but no other titres from any group showed statistically significant decreases compared to saline (n = 7 except for the untreated group, where n = 8).
Acknowledgements
V. C. W., N. K. B., S. T., H. D. L. and V. M. H. designed the study. V. C. W., N. K. B., K. N. C. and G. M. performed the in‐vivo experiments and primary analyses. P. C. and W. H. R. delivered the ACPA data and analysis. D. L. and S. T. generated the pharmacokinetic data. D. C. C. performed all the statistical analyses. V. C. W., H. D. L. and V. M. H. wrote and refined the manuscript. P. P. T. and R. K. P. guided aspects of this work and reviewed the manuscript.
References
- 1. Whiting PF, Smidt N, Sterne JA et al Systematic review: accuracy of anti‐citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–64. [DOI] [PubMed] [Google Scholar]
- 2. Majka DS, Holers VM. Can we accurately predict the development of rheumatoid arthritis in the preclinical phase? Arthritis Rheum 2003; 48:2701–5. [DOI] [PubMed] [Google Scholar]
- 3. Nielen MM, van SD, Reesink HW et al Simultaneous development of acute phase response and autoantibodies in preclinical rheumatoid arthritis. Ann Rheum Dis 2006; 65:535–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rantapaa‐Dahlqvist S, Boman K, Tarkowski A, Hallmans G. Up regulation of monocyte chemoattractant protein‐1 expression in anti‐citrulline antibody and immunoglobulin M rheumatoid factor positive subjects precedes onset of inflammatory response and development of overt rheumatoid arthritis. Ann Rheum Dis 2007; 66:121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Hair MJ, van de Sande MG, Ramwadhdoebe TH et al Features of the synovium of individuals at risk of developing rheumatoid arthritis: implications for understanding preclinical rheumatoid arthritis. Arthritis Rheumatol 2014; 66:513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res 2000; 2:249–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chavanas S, Mechin MC, Takahara H et al Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non‐coding segments and a new human gene, PADI6. Gene 2004; 330:19–27. [DOI] [PubMed] [Google Scholar]
- 8. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis‐specific autoantibodies. J Clin Invest 1998; 101:273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foulquier C, Sebbag M, Clavel C et al Peptidyl arginine deiminase type 2 (PAD‐2) and PAD‐4 but not PAD‐1, PAD‐3, and PAD‐6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum 2007; 56:3541–53. [DOI] [PubMed] [Google Scholar]
- 10. Iwamoto T, Ikari K, Nakamura T et al Association between PADI4 and rheumatoid arthritis: a meta‐analysis. Rheumatology (Oxf) 2006; 45:804–7. [DOI] [PubMed] [Google Scholar]
- 11. Burr ML, Naseem H, Hinks A et al PADI4 genotype is not associated with rheumatoid arthritis in a large UK Caucasian population. Ann Rheum Dis 2010; 69:666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kochi Y, Thabet MM, Suzuki A et al PADI4 polymorphism predisposes male smokers to rheumatoid arthritis. Ann Rheum Dis 2011; 70:512–5. [DOI] [PubMed] [Google Scholar]
- 13. Nakashima K, Hagiwara T, Ishigami A et al Molecular characterization of peptidylarginine deiminase in HL‐60 cells induced by retinoic acid and 1alpha,25‐dihydroxyvitamin D(3). J Biol Chem 1999; 274:27786–92. [DOI] [PubMed] [Google Scholar]
- 14. Christophorou MA, Castelo‐Branco G, Halley‐Stott RP et al Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 2014; 507:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu GY, Liao YF, Chang WH et al Overexpression of peptidylarginine deiminase IV features in apoptosis of haematopoietic cells. Apoptosis 2006; 11:183–96. [DOI] [PubMed] [Google Scholar]
- 16. Wang Y, Li M, Stadler S et al Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009; 184:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li P, Yao H, Zhang Z et al Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol Cell Biol 2008; 28:4745–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yao H, Li P, Venters BJ et al Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J Biol Chem 2008; 283:20060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ireland J, Herzog J, Unanue ER. Cutting edge: unique T cells that recognize citrullinated peptides are a feature of protein immunization. J Immunol 2006; 177:1421–5. [DOI] [PubMed] [Google Scholar]
- 20. Ireland JM, Unanue ER. Autophagy in antigen‐presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med 2011; 208:2625–32.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Makrygiannakis D, af KE, Lundberg IE et al Citrullination is an inflammation‐dependent process. Ann Rheum Dis 2006; 65:1219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005; 6:328–40. [DOI] [PubMed] [Google Scholar]
- 23. Mastronardi FG, Wood DD, Mei J et al Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor‐induced peptidylarginine deiminase 4 translocation. J Neurosci 2006; 26:11387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loos T, Mortier A, Gouwy M et al Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: a naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood 2008; 112:2648–56. [DOI] [PubMed] [Google Scholar]
- 25. Proost P, Loos T, Mortier A et al Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med 2008; 205:2085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Struyf S, Noppen S, Loos T et al Citrullination of CXCL12 differentially reduces CXCR4 and CXCR7 binding with loss of inflammatory and anti‐HIV‐1 activity via CXCR4. J Immunol 2009; 182:666–74. [DOI] [PubMed] [Google Scholar]
- 27. Lange S, Gogel S, Leung KY et al Protein deiminases: new players in the developmentally regulated loss of neural regenerative ability. Dev Biol 2011; 355:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 2010; 207:1853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A et al NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013; 5:178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang HH, Dwivedi N, Nicholas AP, Ho IC. The W620 polymorphism in PTPN22 disrupts its interaction with peptidylarginine deiminase type 4 and enhances citrullination and NETosis. Arthritis Rheumatol 2015; 67:2323–34. [DOI] [PubMed] [Google Scholar]
- 31. Vossenaar ER, Smeets TJ, Kraan MC, Raats JM, van Venrooij WJ, Tak PP. The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum 2004; 50:3485–94. [DOI] [PubMed] [Google Scholar]
- 32. Luo Y, Arita K, Bhatia M et al Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry 2006; 45:11727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knuckley B, Causey CP, Pellechia PJ, Cook PF, Thompson PR. Haloacetamidine‐based inactivators of protein arginine deiminase 4 (PAD4): evidence that general acid catalysis promotes efficient inactivation. Chembiochem 2010; 11:161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knuckley B, Causey CP, Jones JE et al Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 2010; 49:4852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Willis VC, Gizinski AM, Banda NK et al N‐alpha‐benzoyl‐N5‐(2‐chloro‐1‐iminoethyl)‐L‐ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen‐induced arthritis. J Immunol 2011; 186:4396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lewis HD, Liddle J, Coote JE et al Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 2015; 11:189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Banda NK, Kraus D, Vondracek A et al Mechanisms of effects of complement inhibition in murine collagen‐induced arthritis. Arthritis Rheum 2002; 46:3065–75. [DOI] [PubMed] [Google Scholar]
- 38. Bendele AM, Chlipala ES, Scherrer J et al Combination benefit of treatment with the cytokine inhibitors interleukin‐1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum 2000; 43:2648–59. [DOI] [PubMed] [Google Scholar]
- 39. Banda NK, Thurman JM, Kraus D et al Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen‐induced arthritis. J Immunol 2006; 177:1904–12. [DOI] [PubMed] [Google Scholar]
- 40. Moscarello MA, Pritzker L, Mastronardi FG, Wood DD. Peptidylarginine deiminase: a candidate factor in demyelinating disease. J Neurochem 2002; 81:335–43. [DOI] [PubMed] [Google Scholar]
- 41. Pritzker LB, Nguyen TA, Moscarello MA. The developmental expression and activity of peptidylarginine deiminase in the mouse. Neurosci Lett 1999; 266:161–4. [DOI] [PubMed] [Google Scholar]
- 42. Knipp M, Vasak M. A colorimetric 96‐well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem 2000; 286:257–64. [DOI] [PubMed] [Google Scholar]
- 43. Hueber W, Kidd BA, Tomooka BH et al Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum 2005; 52:2645–55. [DOI] [PubMed] [Google Scholar]
- 44. Robinson WH, DiGennaro C, Hueber W et al Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med 2002; 8:295–301. [DOI] [PubMed] [Google Scholar]
- 45. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98:5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA 1998; 95:14863–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hill JA, Bell DA, Brintnell W et al Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4‐IE transgenic mice. J Exp Med 2008; 205:967–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shelef MA, Sokolove J, Lahey LJ et al Peptidylarginine deiminase 4 contributes to tumor necrosis factor alpha‐induced inflammatory arthritis. Arthritis Rheumatol 2014; 66:1482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bawadekar M, Gendron‐Fitzpatrick A, Rebernick R et al Tumor necrosis factor alpha, citrullination, and peptidylarginine deiminase 4 in lung and joint inflammation. Arthritis Res Ther 2016; 18:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rohrbach AS, Hemmers S, Arandjelovic S, Corr M, Mowen KA. PAD4 is not essential for disease in the K/BxN murine autoantibody‐mediated model of arthritis. Arthritis Res Ther 2012; 14:R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seri Y, Shoda H, Suzuki A et al Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose‐6‐phosphate isomerase‐induced arthritis model. Sci Rep 2015; 5:13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suzuki A, Kochi Y, Shoda H et al Decreased severity of experimental autoimmune arthritis in peptidylarginine deiminase type 4 knockout mice. BMC Musculoskelet Disord 2016; 17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Papadaki G, Kambas K, Choulaki C et al Neutrophil extracellular traps exacerbate Th1‐mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur J Immunol 2016; 46:2542–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol Immunol 1999; 36:905–14. [DOI] [PubMed] [Google Scholar]
- 55. Monach PA, Benoist C, Mathis D. The role of antibodies in mouse models of rheumatoid arthritis, and relevance to human disease. Adv Immunol 2004; 82:217–48. [DOI] [PubMed] [Google Scholar]
- 56. Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody‐induced arthritis in mice requires amplification by the alternative pathway. J Immunol 2007; 179:4101–9. [DOI] [PubMed] [Google Scholar]
- 57. Banda NK, Takahashi M, Levitt B et al Essential role of complement mannose‐binding lectin‐associated serine proteases‐1/3 in the murine collagen antibody‐induced model of inflammatory arthritis. J Immunol 2010; 185:5598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Banda NK, Hyatt S, Antonioli AH et al Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody‐induced arthritis in mice. J Immunol 2012; 188:1469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fuhrmann J, Thompson PR. Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem Biol 2016; 11:654–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kawalkowska J, Quirke AM, Ghari F et al Abrogation of collagen‐induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell‐mediated immune responses. Sci Rep 2016; 6:26430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Knight JS, Subramanian V, O'Dell AA et al Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus‐prone MRL/lpr mice. Ann Rheum Dis 2015; 74:2199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nandakumar KS, Holmdahl R. Antibody‐induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther 2006; 8:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Representative histopathology (toluidine‐blue) images (a–f) from the knee joints of mice with collagen‐induced arthritis (CIA) with and without treatments. All joints were fixed with 10% neutral buffered formalin, paraffin‐embedded, and sectioned at a thickness of 5 µm. The top three panels show staining with T‐blue (blue colour) from the knee joints of CIA mice: (a) no treatment (no Tx), (b) with saline (0.9%) or (c) 10 mg/kg GSK199. The bottom three panels show staining with T‐blue from the knee joints of CIA mice: (d) 30 mg/kg GSK199 b.i.d., (e) 30 mg/kg GSK199 q.d. or (f) dexamethasone (Dex). Areas of synovium (S‐black arrow), area of cartilage (C‐black arrow), bone (B‐black arrow) and meniscus (M) have been identified. The sections were also counterstained with haematoxylin and eosin ((H&E) and photographed under the ×10 objective using the Zeiss Observer D1 (AXIO) microscope. The red scale bar shown at the lower right‐hand corner in all panels from (a–f) is 0·1 mm (100 µm).
Fig. S2. Correlation between total clinical disease activity (expressed as area under the curve) and histology scores (expressed as average of four scores), plotted both by individual treatments and as a composite of all dosing groups.
Fig. S3. Representative immunohistochemistry (C3 deposition) images (a–f) from the knee joints of mice with collagen‐induced arthritis (CIA) with and without treatments. All joints were fixed with 10% neutral buffered formalin, paraffin‐embedded, and sectioned at a thickness of 5 µm. All sections have been stained with anti‐C3 antibody (brown colour) and also counterstained with haematoxylin and eosin (H&E, blue colour). No brown colour indicated the absence of complement protein C3 deposition or below detection limits. The top three panels show staining with C3 (brown colour) from the knee joints of CIA mice: (a) no treatment (no Tx), (b) with saline (0·9%) or (c) 10 mg/kg GSK199. The bottom three panels show staining with C3 antibody (brown color) from the knee joints of CIA mice: (d) 30 mg/kg GSK199 b.i.d., (e) 30 mg/kg GSK199 q.d. or (f) dexamethasone (Dex). Areas of synovium (S‐black arrow), area of cartilage (C‐black arrow), bone (B‐black arrow) and meniscus (M) have been identified. The sections were photographed under the ×10 objective using the Zeiss Observer D1 (AXIO) microscope. The red scale bar shown at the lower right‐hand corner in all panels from (a‐f) is 0·1 mm (100 µm).
Fig. S4. Pharmacokinetic measurement of GSK199 compound levels in blood of mice in the study. Geometric means of peak and trough blood concentrations following subcutaneous (s.c.) administration of GSK199 at 10 and 30 mg/kg q.d. and 30 mg/kg b.i.d. to the dark brown Agouti (DBA)/1J mice; sampled on days 1, 7, 14, 21, 28 and 35 during the study at expected peak (2 h: circles) and trough concentrations (prior to the next dose: triangles) for each group.
Fig. S5. GSK199 treatment does not reduce autoantibody titres in collagen‐induced arthritis (CIA). Sera from individual dosed mice were incubated with bovine (a, b) or mouse (c, d) collagen type II (CII). The presence of either anti‐mouse immunoglobulin IgG1 (a, c) or IgG2a antibody (b, d) was detected by enzyme‐linked immunosorbent assay (ELISA). The 0·25 mg/kg dexamethasone group had significantly reduced titres for bovine CII/IgG1, but no other titres from any group showed statistically significant decreases compared to saline (n = 7 except for the untreated group, where n = 8).