

Abstract
Neuronal and neuroendocrine L-type calcium channels (Cav1.2, Cav1.3) open readily at relatively low membrane potentials and allow Ca2+ to enter the cells near resting potentials. In this way, Cav1.2 and Cav1.3 shape the action potential waveform, contribute to gene expression, synaptic plasticity, neuronal differentiation, hormone secretion and pacemaker activity. In the chromaffin cells (CCs) of the adrenal medulla, Cav1.3 is highly expressed and is shown to support most of the pacemaking current that sustains action potential (AP) firings and part of the catecholamine secretion. Cav1.3 forms Ca2+-nanodomains with the fast inactivating BK channels and drives the resting SK currents. These latter set the inter-spike interval duration between consecutive spikes during spontaneous firing and the rate of spike adaptation during sustained depolarizations. Cav1.3 plays also a primary role in the switch from “tonic” to “burst” firing that occurs in mouse CCs when either the availability of voltage-gated Na channels (Nav) is reduced or the β2 subunit featuring the fast inactivating BK channels is deleted. Here, we discuss the functional role of these “neuron-like” firing modes in CCs and how Cav1.3 contributes to them. The open issue is to understand how these novel firing patterns are adapted to regulate the quantity of circulating catecholamines during resting condition or in response to acute and chronic stress.
Keywords: BK and SK currents, exocytosis, L-type channels, Nav1.3 and Nav1.7 channels, tonic and burst firing
Introduction
The chromaffin cells (CCs) of the adrenal medulla are the major source of circulating adrenaline that mobilize the body response to situations of fear, stress and danger [1]. In adult animals the release of adrenaline from CCs is strictly controlled by the sympathetic nervous system (neurogenic control) [2]. Upon splanchnic nerve discharge, repeated quantal release of acetylcholine (ACh) leads to the activation of nicotinic and muscarinic receptors, sustained CC depolarizations and increased firing frequency [3-5]. This causes voltage-gated Ca2+ channel openings, increased Ca2+-entry and robust exocytosis of catecholamine-containing vesicles [5-8]. Adrenaline release is finely regulated by all types of the voltage-gated Ca2+ channels (Cav) that are expressed in CCs (Cav1, Cav2 and Cav3; L-, N-, P/Q-, R- and T-type). These channels open readily during membrane depolarization and warrant rapid Ca2+-fluxes of variable size and time course in response to the broad range of membrane potentials (-50 to +40 mV) spanned during an action potential (AP) waveform.
Given the strong Ca2+-dependence of catecholamines secretion and the effective voltage-sensitivity of Cav channels it is obvious that CCs activity is strictly conditioned by the effective AP firing patterns driving the cell. CCs fire at frequencies that change from 0.2-1 Hz at rest to 20-30 Hz during stress-mimicking conditions. In order to do so, CCs are endowed with a broad array of Na+, Ca2+ and K+ channels that warrant suitably shaped APs and carefully adjusted Ca2+ influx depending on the cell needs [9]. Although neurogenically controlled, a variable fraction of bovine, rat, mouse, and human adrenal chromaffin cells fire spontaneously when cultured in vitro [10-20] or prepared in slices of the adrenal medulla [21-23]. Firing occurs either in form of “tonic irregular” sequences of single action potentials [9] or in form of burst firings where a series of APs ride on top of a depolarizing slow-wave plateau potential [16, 21, 22, 24, 25]. In this latter case, slightly depolarized plateau potentials of 100-300 ms can drive large Ca2+ influx. This in turn produces enhanced catecholamine release, as proved in isolated rat CCs (RCCs) where secretion induced by repeated bursts is remarkably enhanced if compared to the release induced by single APs of increasing frequency [26]. In this view, there is strong parallelism between stimulus-secretion coupling of CCs and neurons possessing comparable slow firing rates (0.2–2 Hz) such as midbrain dopaminergic neurons [9, 27, 28]. In these neurons burst firing is twice as potent in triggering secretion as compared to trains of regularly spaced APs, possessing the same average frequency [29]. Switching from tonic to burst firing is thus a common mechanism that neurons and neuroendocrine cells adopt to boost stimulus-secretion coupling when required.
The strong dependence of neurotransmitter release on AP-shape, frequency and firing patterns (tonic vs. bursts) demands for a detailed analysis of the gating properties of the different ion channel types that contribute to the size and time course of pacemaking currents. Past and recent studies in mouse and rat CCs have highlighted the orchestrated role that Ca2+, Na+ and K+ channels play in shaping the pacemaker current and sustaining the firing modes of CCs under different physiological conditions. Variations of one or more of these components may explain the firing changes that occur under extreme physiological conditions when, for instance, the levels of released neurotransmitters, extracellular pH, extracellular K+ concentration, NO and hormones are significantly altered. This would be even more critical in neonatal CCs where the control of catecholamine release is mainly “non-neurogenic” and sensitive to hypoxia [30, 31].
In this review of the special issue celebrating the 50th anniversary of Ca2+ channel discovery by Harald Reuter [32], we will focus on the role that L-type Cav1.3 channels play in regulating spontaneous and electrically-evoked AP firing in CCs. We will review the peculiar way in which Cav1.3 regulates the pacemaking of CCs in concert with SK [20] and “fast-inactivating” BK channels [9, 19] that shape APs and set the frequency of firing at rest and during stimulation. We will also review the newly uncovered burst firing patterns of CCs, and how this firing mode may be triggered by reducing Nav channels and “fast-inactivating” BK channel availability [22, 24]. All this appears uniquely linked to the degree of expression and gating properties of Cav1.3 channels that regulate both the neuron-like firing modes and the Ca2+-dependent release of catecholamines from the adrenal medulla.
Tonic versus bursts firing mode in chromaffin cells: basic requirements
In neurons and neuroendocrine cells, spontaneous firing takes place when sufficient inward current is driven by small voltage perturbations near resting potential. Critical for triggering spontaneous AP oscillations is the net balance between inward and outward currents (leak, Na+, Ca2+ and K+) near resting potential. When the inward depolarizing current outweighs the outward current, a more positive unstable potential is reached at the start of the “negative conductance” region of the IV characteristics [33, 34]. At this unstable potential, any small depolarization causes a tiny inward current that further depolarizes the cell and triggers an AP train. Low-threshold spontaneous membrane potential oscillations occur easily if the cell possesses high input resistance (2-5 GΩ) and is equipped with sufficiently large densities of Nav and Cav channels that activate readily at low voltages like chromaffin cells [11, 35, 36]. An additional fundamental requirement for driving spontaneous firing in slow spiking cells is the presence of a slowly inactivating current that sustains the slow pacemaker potential.
Spontaneous firing is rather variable in CCs regardless of whether the cells are isolated [9-11, 16, 18-20, 24] or in slices of the adrenal gland [21-23]. In many CCs the firing is “tonic irregular”, i.e., spikes occur at variable frequency (Fig. 1a). In mouse CCs (MCCs), the degree of regularity is negatively correlated with spike frequencies recorded at rest. Fast spiking cells are typically more regular and display “tonic regular” firings (Fig. 1b).
Fig. (1).
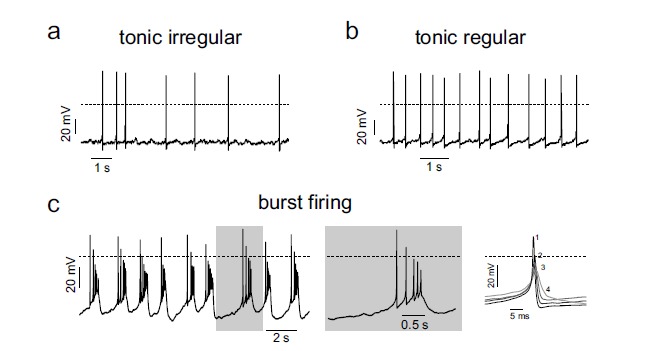
Different firing patterns of spontaneously active RCCs and MCCs. a, b) Spontaneous AP traces recorded from two different RCCs displaying typical tonic irregular and tonic regular firing patterns, respectively. c) Representative trace of spontaneous AP trains fired in a burst-like mode recorded from a MCC. To the right are shown a single burst at an expanded time scale (grey window) and the overlap of consecutive APs within a burst. Numbers indicate the position/sequence in the burst (adapted from ref. [24]).
As in many neurons, a fraction of CCs can undergo also spontaneous bursts at normal physiological conditions [22, 24]. Burst firing of APs occurs as a train of APs on top of a sustained plateau potential of variable duration (Fig. 1c). The burst terminates with a marked afterhypolarization (AHP) whose deepness sets the duration of the inter-bursts interval (IBI): deeper AHPs generate longer IBIs. The origin of this “neuron-like” bursting mode in CCs is completely unknown but one can speculate that it derives from accumulated K+ channel inactivation and from the presence of sizeable slowly inactivating inward currents (Ca2+ or Na+) that sustain the plateau potential and part of the spikes amplitude. Any reduction of the K+ currents activated during the first AP attenuates the AHP to more positive values (plateau potential) where Cav and Nav channels can be activated to sustain the quickly repeated APs of the bursts. As several ion channels contribute to set the plateau potentials and spike amplitudes, the key issue is then to uncover how their degree of expression, gating properties and sensitivity to endogenous and exogenous modulators may alter the equilibrium between tonic and burst firing modes in CCs. Below we will review the biophysical features of Cav1.3, BK, SK and Nav channels that in concert set the firing of CCs.
The CAV channels of chromaffin cells
Mammalian adrenal chromaffin cells express all types of Cav channels (L, N, P/Q, R and T-type) [5, 8, 9, 37, 38]. Their expression density changes remarkably among animal species, cell conditions and development. In adult animals L, N, P/Q and R-type channels are the dominant species [5, 8, 10, 39]. They shape action potential waveforms, control catecholamine secretion and vesicle retrieval. Despite the many reports, convincing proof of a specific co-localization of either one of the expressed Cav channel types with the secretory apparatus is still lacking. Each channel type control catecholamine release with the same Ca2+-dependence [6, 40-48]. The question is thus, why chromaffin cells which are spherically shaped cells and do not possess neuron-like morphology and specialized pre-post microdomains need so many different Ca2+ channel types for their function? A possibility is that depending on their different sensitivity to external signaling and intracellular second messengers, the many Cav channel types warrant compensatory paracrine and autocrine responses to different CCs functioning conditions. Along this view, Cav2.1 and Cav2.2 channels can be effectively down-regulated by released ATP and opioids that are co-released with adrenaline and noradrenaline [49-54]. Conversely, Cav1.2 and Cav1.3 L-type channels could be both strongly up-regulated by elevating the cAMP/PKA levels [55-57] or drastically down-regulated by cGMP/PKG mediated pathways [57]. Cav channels can also undergo drastic up-regulation during cell re-modeling, as in the case of Cav3.2 T-type channels [6, 39]. Cav3 channels are usually not expressed in adult CCs but are functionally active in embryonic CCs when innervation of the adrenal medulla has not reached complete maturation [30, 58, 59]. At this developmental stage, CCs respond only to non-neurogenic stimuli. Cav3.2 channels are de-novo recruited in adult CCs to sustain “low-threshold” exocytosis during stress mimicking conditions such as during β-adrenergic receptor stimulation [60] or chronic hypoxia [61]. Recent evidence indicates that exogenous expression of Cav3.2 channels in a chromaffin cell line induces “low-threshold” release of catecholamines, which can be blocked by coexpressing a peptide corresponding to the syntaxin-1A interaction domain of the channel [62].
CAV1.3 as pacemaker channel: a complex growing concept
CCs express mainly the two neuronal Cav1.2 and Cav1.3 channel isoforms, that have strong amino acid homologies. They both contribute to about 50% of the total current in cat, rat and mouse CCs [5, 7, 11, 56]. In humans, L-type channels are reported to contribute to 30% of the total [63], but this is most likely an underestimate since the DHP blocking assay was done with cells clamped at rather negative Vh (-80 mV) using low doses of nifedipine (1 µM).
Cav1.2 and Cav1.3 are also directly involved in the control of vesicle endocytosis [64, 65] and pacemaking of spontaneous [10, 11] and evoked AP firings [9]. The last observation is a relatively new concept that gains increasing evidence. Many reports on neurons, cardiac myocytes and neuroendocrine cells suggest the involvements of L-type channels in pacemaking [9]. The question is thus, are both channels suitable for pacemaking or one of them is optimal for triggering auto-rythmicity? Looking at their gating properties, it is evident that Cav1.3 possesses the characteristics to sustain subthreshold currents that may contribute to sustain spontaneous low frequency firing patterns (0,2 to 2 Hz) typical of chromaffin cells [10, 11, 19], substantia nigra pars compact neurons [28, 66-68] and sino-atrial node (SAN) “pacemaker cells” [69].
Data derived from heterologously expressed channels indicate that: i) Cav1.3 has similar single-channel conductance to Cav1.2 [70] but activates with steep voltage-dependence at more negative voltages [71, 72] and, ii) Cav1.3 has faster activation but slower and less complete voltage-dependent inactivation as compared to Cav1.2 [71]. In addition, Cav1.3 is less sensitive to DHPs as compared with Cav1.2, requiring about 10-fold more DHPs for full block at normal Vh (-70 mV) [73]. Cav1.3 sensitivity toward DHPs increases markedly at less negative Vh (-50 mV) reaching comparable IC50 values of Cav1.2 [71]. Thus, at -50 mV saturating concentrations of DHPs that block Cav1.2 ensure also full block of Cav1.3. This is crucial when assaying the role of Cav1.3 during spontaneous AP recordings in firing cells that spend most of their time at the interspike potential around -50 mV (see ref. [36]).
Despite a recent report indicates selective block of reconstituted Cav1.3 channels [74], contradicting findings [75, 76] suggest there are not yet available specific blockers for this Cav1 isoform. Due to this, the exact role of Cav1.3 in cellular auto-rythmicity can be assayed only indirectly using the Ca1.3 deficient mouse (Cav1.3–/–) [77]. This mouse model has a strong phenotype characterized by deafness and bradycardia. Deafness derives from the complete absence of Cav1.3 L-type currents in cochlear inner hair cells and degeneration of outer and inner hair cells [77]. Bradycardia derives from the abolition of the major component of the Cav1.3 L-type current activating at low voltages in the SAN “pacemaker cells” controlling the heart rhythm and rate under physiological conditions [69].
A specific role of L-type channels in CC pacemaking was first reported in RCCs [10]. Increasing doses of nifedipine could either decrease the firing frequency or block the spontaneous firing. In these experiments, a tight coupling of L-type channels to the fast-inactivating BK channels of RCCs was also noticed, in good agreement with previous reports by Chris Lingle’s lab [78, 79]. An L-type pacemaker current of 10 to 20 pA amplitude was first reported in AP-clamp experiments in WT MCCs [11]. A subthreshold inward Ca2+ current of this size passing through the high input resistance of chromaffin cells (2-5 GΩ) [11, 19, 35] is sufficient to generate pacemaker potentials of 10-20 mV that drive the cell from inter-pulse potentials of -50 mV to the threshold of the AP up-stroke (-30 mV) [11]. Deletion of Cav1.3 reduces drastically the amplitude of this pacemaker current and the fraction of firing MCCs [19]. An example is shown in Fig. 2 where the inward Ca2+ currents of WT and Ca1.3–/– MCCs flowing during the inter-pulse interval (ISI) are compared. Loss of Cav1.3: i) decreases markedly the level of the nifedipine-sensitive currents driving spike generation, ii) raises the rheobase (from 4 to 6.6 pA) and iii) decreases the extent of spike frequency adaptation during sustained current injections [20].
Fig. (2).
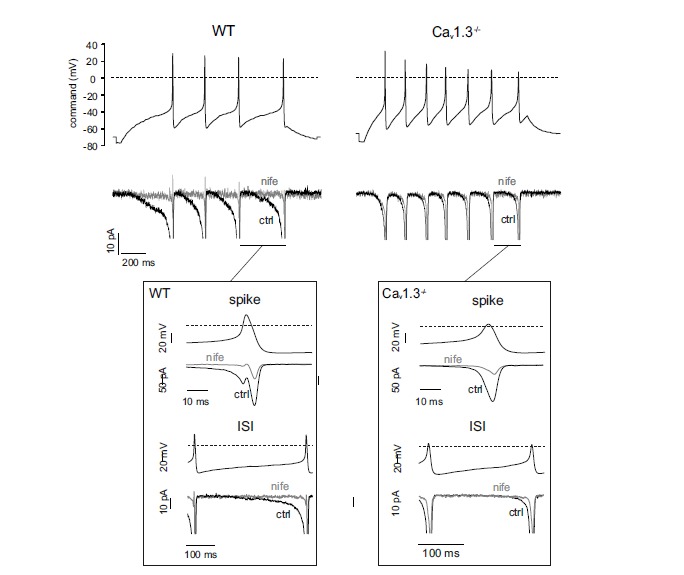
Deletion fo Cav1.3 causes a marked reduction of the pacemaking L-type current flowing during the interspike interval. Top, representative AP trains from a WT (left) and Cav1.3–/– MCC (right) recorded after a 15 pA (700 ms) current step starting from Vh= -70 mV. The voltage traces were used as voltage-clamp command to reveal the interspike specific apamin-sensitive currents. Middle, Ca2+ currents of WT (left) and Cav1.3–/– MCCs (right) recorded in control condition (black trace, ctrl) and in the presence of 3 µM nifedipine (grey, nife). External solution contained 135 mM TEA, 300 nM TTX and 2 mM Ca2+. Bottom insets, Ca2+ currents flowing during the spike and the interspike interval of WT and Cav1.3-/- MCCs shown at an expanded time scale (adapted from ref. [20]).
As pacemaker channel, Cav1.3 appears quite unique if compared with other neuronal channels that generate spike firings and carry mostly Na+ currents, like the hyperpolarization-activated cation channels (HCN) [80], the persistent [81] and resurgent [82] Na+ channels and background channels [83]. The inward Ca2+ current generated by Cav1.3 is always accompanied by outward K+ currents associated with Ca2+-activated BK and SK channels that are highly expressed in neurons [84] and neuroendocrine cells [85, 86]. Thus, activation of Cav1.3 generates net currents that might be inward or outward depending on the degree of coupling and expression density of BK and SK channels. In the case of BK, it depends also on membrane potential since these channels are also steeply voltage-dependent [84].
The CAV1.3-BK channel nanodomains regulate AP shape and cell firing modes
As in many brain regions, BK channels are highly expressed in RCCs [10, 86], MCCs [9, 19, 22] and bovine CCs (BCCs) [85, 87], To activate during physiological depolarization, BK channels need an intracellular Ca2+ concentration of at least 10 µM [84]. Such high concentrations occur only within “Ca2+-nanodomains” in the close vicinity of the Ca2+ source [88]. For this purpose, the neuronal BK channels are often co-localized to either the P/Q- [89], N- [90] or L-type [91] channels. RCCs and MCCs express two different BK channels that are predominantly coupled to L-type Cav1 channels [9, 79] and can be distinguished according to their inactivation kinetics: a fast-inactivating (BKi) and a slowly inactivating channel type (BKs) [9, 19, 78]. The BKi channel is typically expressed in CCs and gives rise to slowly adapting cell firing. The BKs channel has gating properties similar to the central neurons and smooth muscle BK channels and causes fast adapting cell firings [78]. Due to their strong voltage-dependence, BK channels contribute to the repolarization phase of the AP (AHP) [84, 86], influence the refractory period and regulate the firing rate of CCs. In MCCs, block of BK channels by paxilline significantly augments the firing frequency by delaying AP repolarization and slightly reducing the early phase of the AHP [19]. Notice that due to the tight Cav1-BK coupling also nifedipine, like paxilline, delays AP repolarization and reduces the early phase of the AHP in MCCs [9, 19] and RCCs [10, 78] (see Fig. 4 in ref [9]). However, due to the block of Cav1.3, the DHP either blocks or slows down the firing rate.
Fig. (4).
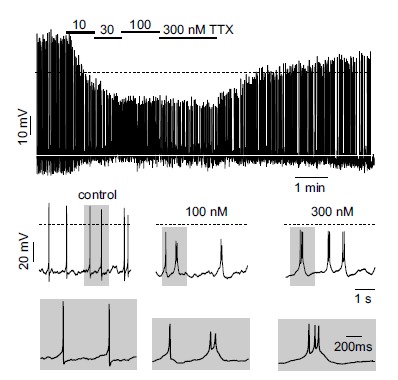
TTX leads to a dose-dependent switch into burst firing of MCCs. Top, representative current-clamp trace showing the action of increasing TTX concentration. Middle and bottom. better view of the effects of increasing TTX concentrations on AP waveforms and firing patterns at slower and faster (grey windows) time scales. Periods of activity were taken from the top panel (adapted from ref. [24]).
Given the critical role of Cav1.3 in pacemaking MCCs, the Cav1-BK channel coupling is a key determinant for setting the AP shape and the firing frequency of CCs. In addition, the coupling is drastically altered by deletion of Cav1.3 in a way that loss of Cav1.3 prevents the expression of BKi channels [19]. This property appears to be an adaptive phenomenon first discovered in cochlear inner hair cells [91]. In MCCs this is evident by comparing the BK channel types expressed in WT and Ca1.3–/– MCCs using standard protocols of Ca2+ pre-loading which maximally activate all available BK channels (Fig. 3a). WT MCCs are shown to express a predominance of fast-inactivating BK currents while Ca1.3–/– MCCs display typically slowly inactivating BK currents. Using fast (BAPTA) and slow (EGTA) Ca2+ buffers it is also possible to prove that the coupling between BKs and Cav1.2 channels in Cav1.3-/- MCCs is BAPTA- and EGTA-sensitive suggesting that the two channels are separated by an average distance of more than 160 nm. Conversely, the coupling between Cav1.3 and BKi channels in WT-MCCs is BAPTA-sensitive but EGTA-insensitive, suggesting an average distance of 50 to 160 nm between the two channels. In other words, BKi and Cav1.3 channels form functional “Ca2+-nanodomains” that regulate the steepness of the AHP phase and thus set the-shape and frequency of AP trains.
Fig. (3).
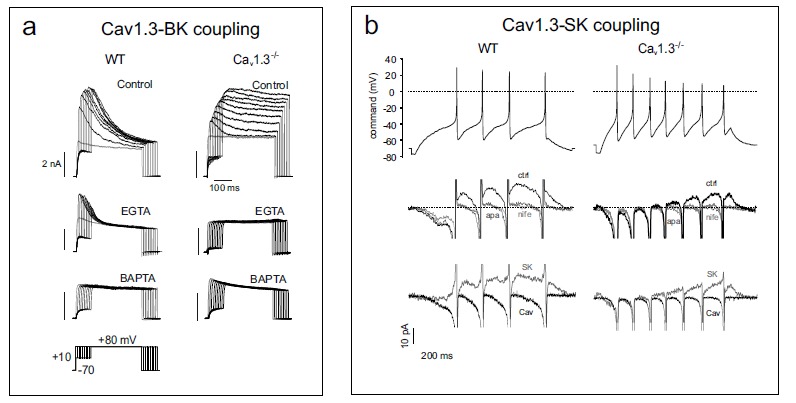
Deletion of Cav1.3 prevents the activation of fast inactivating BK channels and reduces the size of SK currents during spike frequency adaptation. a) The BK channels of Cav1.3−/− MCCs are mainly non-inactivating [19]. Membrane-permeable EGTA-AM is unable to prevent the coupling in WT MCCs but is effective on Cav1.3−/− MCCs. BAPTA-AM is effective on both WT and Cav1.3−/− MCCs (adapted from ref. [9]). The pulse protocol is shown at the bottom left. b). Top, evoked AP trains from a representative WT and Cav1.3-/- MCC used as voltage-clamp command to reveal the Ca2+ and SK currents of the same cells of (Fig. 2). Middle, Ca2+ and SK currents recorded in the presence of a Tyrode standard (2 mM Ca2+, 4 mM K+) containing 300 nM TTX and 1 µM paxilline in control (black traces) and in the presence of either 3 µM nifedipine (light grey s) or 200 nM apamin (dark grey). Notice how nifedipine is equivalent to apamin in blocking the outward SK currents. Bottom, SK currents (grey traces) obtained after subtraction of the apamin trace from the control trace of the middle panel overlapped with the Ca2+ currents of (Fig. 2) (black traces) (adapted from ref. [20]).
Because BKi channel inactivation is associated with the presence of a specific β2 subunit that shifts channel activation toward more negative voltages at a given Ca2+ [92, 93], these findings suggest that loss of Cav1.3 channels may lead to the loss of BK-β2 subunits. This could also indicate that β2 is the critical element regulating the close coupling between Cav1.3 and BKi channels. In the absence of β2 the coupling between BK and Cav1.3 (or Cav1.2) channels is weaker and less critical for cell firing. That β2 may be a key element of BK channels that helps regulating MCCs firing is suggested by recent findings showing that deletion of the β2 subunit produces rather drastic changes to MCCs firing patterns [22]. The knock-out of BK-β2 subunits in fact slows AP repolarization and, during constant current injection, decreases AP firing. This clearly supports the idea that the β2-mediated shift of the BK channel activation range affects repetitive firing and AP properties. Unexpectedly, CCs from BK-β2 deficient mice show an increased tendency toward spontaneous slow-wave burst firings, suggesting that the peculiar activation properties of BK channels in the absence of β2 subunits may predispose CCs to burst firing and, possibly, to an associated increased secretion of catecholamines. It is worth noticing that burst firing occurs also in BK deficient (BK–/–) MCCs during L-type current activation by BayK 8644 [9, 19] and in WT MCCs when Nav channel availability is reduced by either slowly inactivating or blocking them (see below).
The “Functional” CAV1.3-SK Channel Coupling Regulate Cell Firing Frequency and Firing Adaptation
SK channels (KCa2.1, KCa2.2, KCa2.3) are widely expressed throughout the central and peripheral nervous system where they act as key modulators of neuronal excitability [94, 95]. They control intrinsic excitability and pacemaking, dendritic integration, postsynaptic responses as well as synaptic plasticity [84]. Besides affecting firing frequencies, SK channels are involved in switches from single spiking into burst firing patterns and can drive spike frequency adaptation and accommodation [96-98]. SK currents contribute to the AHP phase of single or burst of APs, slowing-down the pacemaker cycle. The high Ca2+-sensitivity of SK channels is at the basis of the prevailing current view that SK channels do not have to be necessarily close to a specific Ca2+ source to get activated [84]. Several studies nevertheless demonstrated a specific coupling of SK channels to Cav channels or neurotransmitter receptors [90, 99-102].
In chromaffin cells, SK channels were first biophysically identified in BCCs [85] and RCCs [86]. Experiments on cat adrenal glands showed that secretion of catecholamines induced by electrical stimulation or acetylcholine application could be potently stimulated by SK block with apamin [103]. In RCCs, the SK channels mediate the biphasic effect of muscarine on AP firing, which consists in a transient hyperpolarization followed by a sustained depolarization [4, 104]. The hyperpolarization is induced by an IP3-mediated increase of intracellular Ca2+ via M1 receptors activation [4] while the depolarization is associated with the block of an M-type K+ channel [12].
SK channels are of extreme importance for the control of MCCs firing [20]. By comparing WT and Ca1.3–/– MCCs firing it has been possible to uncover a “functional” coupling between Cav1.3 and SK channels that slows down MCC's basal firing rate and leads to spike frequency adaptation upon sustained current injections [20]. This is not surprising, since in MCCs, as in most neurons, the SK channels are equally driven by L, N, P/Q and R-type channels (see Fig. 9 in ref. [20]). As Cav1.3 drives most of the slow pacemaking current, it is evident that the SK currents at rest are mainly driven by these L-type channels. Thus, during spontaneous firing the “Cav1.3-SK coupling” builds-up robust subthreshold SK currents that slows-down the pacemaking and guarantees the effective recovery of NaV channels required to maintain stable the AP firings. A critical role of SK currents on MCC firings is evident by observing that: i) the MCC firings is slow and irregular at control and becomes faster and more regular by blocking the SK channels with apamin, ii) the frequency of spontaneously firing MCCs increases nearly 3-fold upon addition of apamin and, iii) exists a negative correlation between the basal firing frequency and the percentage of frequency increase induced by apamin. A corollary of this is that the presence of SK channels decreases the precision of firing in MCCs, which is in contrast to most central neurons where robust SK currents increase the regularity and precision of firing [97, 98, 105, 106]. The increased precision of firing is due to an enhanced NaV channel availability sustained by SK channels that is required for pacemaking these cells.
The “functional” Cav1.3-SK channel coupling has a similar critical role on the response to sustained depolariza- tion. It allows the fast adaptation of the initial high-frequency to a low sustained frequency during prolonged stimulations. Loss of Cav1.3 induces a nearly two-fold reduction of the total Ca2+ charge entering the cell during pacemaking with consequent reduction of SK currents (Fig. 3b). In this case, the induced AP-firing adapts more slowly and generates AP trains of higher frequency and lower amplitude. The “functional” and “not localized” coupling to SK channels thus accounts for the inability of Ca1.3–/– MCCs to adapt their firing frequency. As a consequence, the Cav1.3-SK coupling confers to Cav1.3 the dual role of “drive” and “brake” of MCC's excitability that is crucial when controlling sustained catecholamine release from the adrenal gland during stressful stimuli [5]. The braking action of SK channels is particularly crucial to prevent over-excitation that could damage cells that fire slowly (~1 Hz) and rely on “Ca2+-dependent pacemakers” such as chromaffin cells [19], dopaminergic and histaminergic neurons [67, 107].
The fast adapting response to sustained depolarization due to the Cav1.3-SK coupling has clear-cut implications on the overall organization of catecholamine release in the adrenal medulla. The first is that increasing the frequency of stimulation does not necessarily lead to a linear increase of catecholamine secretion. The catecholamine release induced by trains of electrical stimulations (0.1 to 30 Hz), saturates between 3 and 10 Hz and then declines in rat and cat chromaffin cells [3, 103, 108]. The second implication is that, similarly to certain neuronal networks [109], the phasic response regulated by the Cav1.3-SK coupling may help the firing synchronization in the adrenal medulla where chromaffin cells are electrically coupled [39]. Synchronization followed by adaptation to low firing frequencies in extended areas of the adrenal medulla may optimize the release of catecholamines during prolonged stressful stimulation, preventing excessive accumulation of undesired levels of circulating catecholamines.
NAV1.3 and NAV1.7 Channels do not Contribute to Pacemaker Current
As in most brain neurons [33, 110], voltage-gated Na+ channels (Nav) are highly expressed in the CCs of all animal species [14, 15, 17, 21, 24, 35, 36, 111-114]. Nav channels generate and shape the AP waveform and thus directly contribute to CCs excitability. Early studies have shown the existence of a background Na+ conductance that could drive Na+-influx at rest in rat and gerbil CCs [13, 14]. TTX application or Na+ removal from the extracellular medium reduces, but does not block rat CCs firing [111]. In MCCs, Nav channel block by TTX preserves AP firing [21, 24, 36], suggesting that Nav channels play a different role in CC excitability than L-type channels [9]. Furthermore, Nav channels in MCCs activate at 24 mV more positive potentials than Cav1.3, suggesting a minor contribution to sub-threshold pacemaker currents [36], even considering that Nav channels are expressed at much higher densities with respect to Cav channels in CCs [35]. Taken all together these observations raise two key questions: do Nav channels contribute to the pacemaking of CCs? How critical Is their availability to trigger CCs firing modes?
Concerning the first issue, the answer is clearly linked to the type of Nav channels expressed in CCs. Presently, Nav1.7 is thought to be the main voltage-gated Nav channel expressed in bovine and human adrenal tissues [115, 116]. In the rat adrenal gland, mRNA encoding Nav1.7 is expressed only in the medulla [117], with a plausible localization in CCs, as also reported in PC12 cells [118]. A recent report by our group shows that cultured MCCs express Nav1.7 (scn9A) and Nav1.3 (scn3A) with a 7.3-fold higher expression level for the scn3A transcript [24]. In conclusion, all the available functional studies on CC Nav channels show convincingly that these channels are TTX-sensitive and fast-inactivating [24, 35, 36, 113, 114]. Fast inactivating Nav channels could partly contribute to pacemaking in fast-spiking neurons [27, 119] with firing frequencies of 40-100 Hz but could hardly sustain pacemaking currents in slow firing cells with inter-pulse intervals of 0.3 to 2 s duration, such as in CCs. All this, however, cannot exclude that CCs could express “persistent” or “resurgent” Na+ currents that partly contribute to slow pacemaking. This possibility has been recently investigated using traditional whole-cell and perforated micro-vesicle single channel recordings in MCCs [24]. It is concluded that the Nav1.3/Nav1.7 fast inactivating channels expressed in MCCs are fully inactivated after step depolarization of 200-1000 ms. The steady-state net currents at these time intervals are <1-2 pA and comparable to the background current noise. It is thus unlikely that Nav1.3 and Nav1.7 contribute to the pacemaker current in MCCs. In addition, MCCs do not possess ”persistent” Nav currents which are typically associated with Nav1.6 (scn8a) [82]. Persistent Na+ currents activate at potentials as negative as -70 mV [120] and sustain tonic firing at elevated frequencies (20-100Hz) in several neurons [121-124]. MCCs do not fire at elevated frequencies and lack scn8a [24]. In addition, the auxiliary β subunit scn4b (NaVβ4) is linked to resurgent Na+ currents [125]. Neither scn4b nor “resurgent” Na+ currents are revealed in MCCs [24].
Nav1.3/Nav1.7 CHANNELS AVAILABILITY REGULATE THE Cav1.3-MEDIATED SWITCH FROM TONIC TO BURST FIRING
Concerning the second question it is worth recalling that as in most neurons, Nav channels availability is critical for shaping the AP waveform in MCCs [20, 36]. Subtle changes in spike shape can lead to drastic changes in Cav channel recruitment and in AP-induced Ca2+ transients [126]. AP broadening is indeed a common way to optimize presynaptic Ca2+ influx at central presynaptic terminals [127]. Neuronal Nav channel availability is drastically reduced by depolarizing the resting membrane potential [128, 129]. The same is true in MCCs where the reduced Nav channel availability drastically alters L-type channel recruitment and, paradoxically, boosts catecholamine secretion [24].
As shown in Vandael et al., 2015 [24], Nav channel availability can be reduced by either blocking Nav1.3/Nav1.7 channels by TTX or by lowering the resting membrane potential from physiological (-50 mV) to steady depolarized potentials (-40 mV). In the latter case, the slow and partial recovery from fast inactivation and the fast entry into the closed-state inactivation at -40 mV, reduces Nav channel availability by nearly 50%. This leads to a reduced AP peak and less KV and BK channel activation that attenuates the fast afterhyperpolarization phase. This in turn slows the return to baseline potentials followed by a switch from net outward to net inward currents after AP termination. When Nav channel availability is reduced up to 20% of total either by TTX block or steady depolarization the conversion from outward to inward current is more drastic and a switch from tonic into burst firing is observed. Figure 4 shows an example of switch from tonic to burst firing in the presence of saturating doses of TTX.
During burst firing, brief periods of high frequency firing are separated by relatively long gaps of no activity. Spikes fire in bursts on top of a slow-wave depolarization plateau that lasts for the whole burst duration. AP-clamp experiments show clearly that while the net current during the AHP of single control spikes is outward, the current during the intraburst interval is net inward and carried by Ca2+ (dashed rectangles in Fig. 5a). Broadening of APs during cell depolarization and bursts of APs in the presence of TTX causes a ten-fold prolongation of Ca2+ currents (bottom inset in Fig. 5b) that, despite the lower amplitudes, carries about four-fold more Ca2+ charges inside the cell [24], demostrating that Ca2+ currents are the main source of the slow depolarization plateau. In conclusion, the incoming Ca2+, together with a lack of sufficient outward K+ current is the triggering event of burst firing when Nav channels availability is strongly attenuated.
Fig. (5).
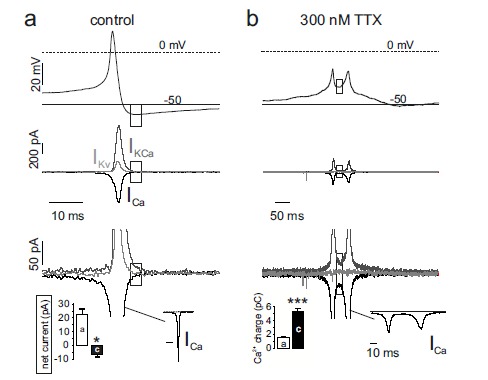
Reducing NaV availability by TTX inverts net current during the AHP from outward to inward. a) Top, AP-clamp experiment showing representative control spike. Middle, Kv currents (ligh grey traces) were measured in a Tyrode standard solution with TTX (300 nM) and Cd2+ (200 μM). Ca2+ currents (black) were measured in the presence of TTX (300 nM) and high extracellular TEA (135 mM). Ca2+-activated K+ currents (dark grey) were obtained by subtracting from a control recording in Tyrode standard with 300 nM TTX the KV and the Ca2+ current. Bottom, close up of the middle panel. The dashed rectangles indicate the AHP phase and the respective currents that sustain it. b) same as for a, using a spike doublet fired after complete block of Nav currents with 300 nM TTX. Bottom-left inset in a: net current amplitudes measured during the AHP phase indicated by the dashed rectangles. Bottom-left inset in b: Ca2+ charge entering the cell during the AHP phase calculated by integrating the corresponding Ca2+ inward current (ICa) shown in full to the right (adapted from ref. [24]).
Spontaneous burst firing is also observed in a minority of MCCs (14%) (Fig. 1c and 6). A train of high frequency spikes emerge on a significantly longer slow-wave depolarization plateau with respect to those induced by TTX (200-500 ms vs. 80-200 ms, respectively). Due to the Nav channel availability, the first spike amplitude is always larger as compared to the subsequent APs that are gradually broader, of lower overshoot and reduced AHP amplitude (Fig. 6). Nav, Kv and BK currents decline rapidly during the burst while Cav currents decline more slowly. Conversely, SK currents build up rapidly to terminate the burst. Thus, burst firings originating from plateau potentials lasting for hundreds of milliseconds give origin to large Ca2+ influxes that may boost the Ca2+-dependent secretion of catecholamines. In this view, burst firing represents a way by which CCs rapidly increase the release of adrenaline and noradrenaline upon requirement.
Fig. (6).
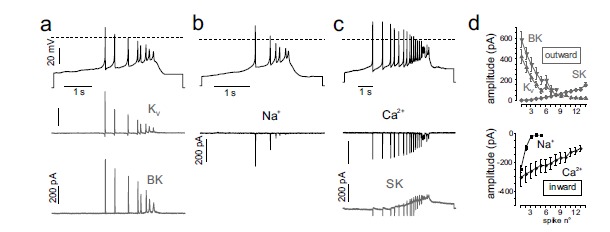
Time courses of K+, Na+ and Ca2+ currents during a slow-wave burst in different MCCs. a) AP-clamp recordings of KV and BK currents. Top shows the burst recorded and used as voltage command (black trace), KV and BK currents are shown in grey. b, c) as for panel a but currents isolated were Ca2+ (black), SK (grey) and Na+ (black). d). Top, BK, SK and Kv outward current amplitudes versus the spike number of the burst. Bottom, same as top but for the Na+ and Ca2+ inward currents (adapted from ref. [24]).
Figure 7 shows that this indeed is the case if current-clamped MCCs are first hyperpolarized to stop their spontaneous activity and prevent Ca2+ accumulation at the onset of the recording. Control cells and cells pre-treated with TTX are then stimulated for 40 s with a constant depolarizing pulse to induce secretion. The fast quantal release of catecholamines revealed with carbon fibre amperometry [11, 61] (Fig. 7a) shows clearly that the increased AP frequency during bursts in the presence of TTX enhances significantly the quantity of released catecholamines (Fig. 7b, bottom). Thus, block of Nav channels with TTX to induce burst firing paradoxically boosts Ca2+-entry and release of catecholamines. Although not shown, it is likely that most of the exocytosis is sustained by Cav1.3 since is the dominant pacemaking channel supporting prolonged depolarizations in MCCs.
Fig. (7).
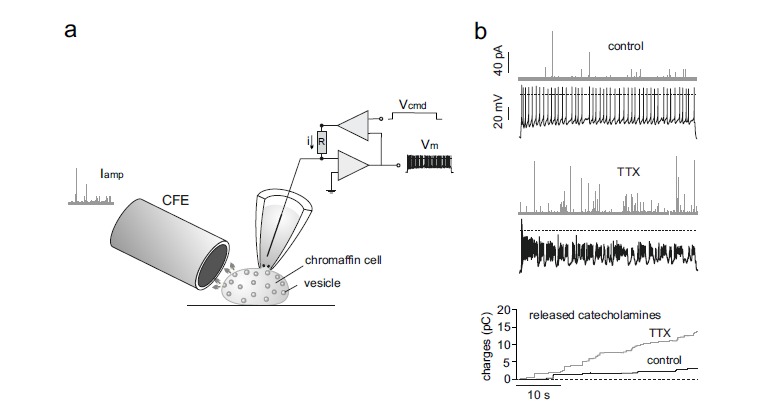
Burst firing associated with Nav channel block boosts MCC exocytosis. a) Experimental arrangement to record catecholamine secretion using a carbon fibre microelectrode (CFE) from a current-clamped MCC. The cell is constantly depolarized to evoke an AP train (Vm) by passing a constant current (i) through the glass pipette using a square pulse voltage command (Vcmd). The quantal release of adrenaline and noradrenaline is detected in forms of amperometric currents by the CFE (Iamp) placed very close to the CC. b) Top, example of combined recording of APs (black traces) and amperometric currents (grey traces) by CFE amperometry in control. Middle, combined recording of AP bursts and amperometric events during TTX application to the same cell of the top. Bottom, overlap of cumulative secretion plots obtained by integrating the amperometric recordings shown in a and b for control (black trace) and TTX application (grey trace) (adapted from ref. [24]).
Tonic Versus Burst Firing in Chromaffin Cells: A “Neuron-Like” Way of Signalling to Boost Exocytosis
The present findings show that CCs possess intrinsic “neuron-like” firing properties typical of some central neurons that undergo burst firing [27]. CCs spontaneously fire at rest in an irregular tonic manner and change into burst firing depending on the expression density and gating conditions of Na+, Ca2+ and K+ channels that control the firing. Recent evidence supports the view that this occurs when the net current passing during the AP repolarization phase turns from outward to inward, i.e., when inward Na+ and Ca2+ currents prevail on outward K+ currents. Thus, burst firing is apparent in MCCs when Kv channels are blocked by TEA [21] or when the β2 subunit controlling the fast inactivation rate of Slo1 BK channels is deleted [22]. Burst firing occurs also in a minority of MCCs under physiological conditions [22, 24], most likely because of a critical balance between functioning Na+, K+ and Ca2+ channels. In RCCs and BCCs, burst firing appears when either the ERG K+ channel is blocked by the antiarrhythmic drug WAY or when an M-current is blocked by an histamine-mediated pathway [16, 18]. In this view, any modulatory mechanism that leads to a depression of K+ outward current or to a potentiation (or maintenance) of Ca2+ currents is potentially able to induce burst firing in CCs. Indeed, burst firing occurs also when the availability of functional fast-inactivating Nav1.3/Nav1.7 channels is strongly attenuated by block with TTX or by accelerating the Nav channel entry into closed-state inactivation at more depolarized resting potentials. This latter, represents a novel modulatory pathway by which “fast inactivating” Nav channels control burst firing in spontaneously firing cells [24]. A role typically played by “persistent” and “resurgent” neuronal Nav channels.
Burst firing in CCs is in all cases sustained by the L-type Cav1.3 currents which activate at relatively low voltages and inactivate slowly at potentials near rest. Cav1.3 sets and regulates the firing cycle by sustaining the slow pacemaking current and by triggering BK and SK currents. In CCs the slow-wave bursts enhance the quantity of Ca2+-entry inside the cell and thus boost exocytosis of catecholamines. In altered physiological conditions, this may occur in various ways: i) by any mechanism that dampens Kv and BK channel gating, ii) by regulatory pathways which up-regulate L-type channels [56, 57] or, iii) by modulatory pathways that cause steady CC depolarizations and drastically reduce Nav1.3/Nav1.7 channel availability. This latter might be applicable to conditions such as plasma hyperkalemia [36], acidosis [130] or increased histamine levels that induce sustained depolarizations, burst firing [18] and enhanced circulating catecholamines [131]. Of particular interest in CC biology is the potential role of muscarinic receptors, whose activation causes prolonged membrane depolarizations, increased cell firing and sustained catecholamines release [4, 21]. The action is mediated by the M1 muscarinic receptor [132] and proceeds through the inhibition of TASK1-like channels [130]. As muscarine activates the PKC pathway in RCCs [4, 133], it is likely that Nav channels may undergo the well-known PKC-dependent down-regulation mediated by muscarinic receptors [134]. This, together with the 30% reduced Nav channels availability at depolarized Vh, would critically increase the probability of MCCs to switch into burst firing patterns, boosting catecholamine release during ACh-driven acute stress responses.
An open question is what could be the clinical impact of CC burst firing and increased catecholamine release following Nav channel block. Nav channel blockers are widely used in therapy and thus, it would be interesting, for instance, to establish whether increased levels of catecholamines may occur in patients with ventricular tachycardia treated with Nav1.5 channel blockers [135]. Obviously, an increased level of circulating catecholamines will reverse the beneficial effects of Nav1.5 blockers. To this specific issue we can observe that cardiac Nav1.5 channels are structurally and functionally different from neuronal Nav1.3/Nav1.7 channels [110]. Nav1.5 channels are TTX-insensitive and carry large “late” sodium currents (INaL) in myocardial pathologies [136] while Nav1.3 and Nav1.7 are TTX-sensitive and fast-inactivating [24, 36]. In addition, several used Nav1.5 blockers are many-fold more potent in inhibiting INaL than the peak INa [137]. For instance, the piperazine derivative ranolazine exerts its therapeutic effects at ~10 µM with minimal or no effect on peak INa. Given this, it seems unlikely that a Nav1.5 blocker could reduce the peak of fast-inactivating Nav1.3/Nav1.7 currents of CCs to less than 20% to induce burst firing and increased circulating catecholamines that would enhance cardiac arrhythmias in hearts treated with INaL blockers [136].
We conclude by observing that in physiological, pathological or therapeutical conditions, the density of functional Nav1 channels is a key element for driving Cav1.3 channels availability and CCs function. For this, is very important to identify all possible causes that down-regulate Nav1.3/Nav1.7 channels availability and shift CCs firing from tonic to bursts, including potential Nav channel blockers used in therapy [138].
ACKNOWLEDGEMENTS
This work was supported by the Italian MIUR (PRIN 2010/2011 project 2010JFYFY2) and the University of Torino.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Cryer P.E. Physiology and pathophysiology of the human sympathoadrenal neuroendocrine system. N. Engl. J. Med. 1980;303(8):436–444. doi: 10.1056/NEJM198008213030806. [DOI] [PubMed] [Google Scholar]
- 2.Shah S.D., Tse T.F., Clutter W.E., Cryer P.E. The human sympathochromaffin system. Am. J. Physiol. Endoc. M. 1984;247(3 Pt. 1):E380–E384. doi: 10.1152/ajpendo.1984.247.3.E380. [DOI] [PubMed] [Google Scholar]
- 3.Wakade A.R. Studies on secretion of catecholamines evoked by acetylcholine or transmural stimulation of the rat adrenal gland. J. Physiol. 1981;313:463–480. doi: 10.1113/jphysiol.1981.sp013676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olivos L., Artalejo A.R. Muscarinic excitation-secretion coupling in chromaffin cells. Acta Physiol. (Oxf.) 2007;192:213–220. doi: 10.1111/j.1748-1716.2007.01816.x. [DOI] [PubMed] [Google Scholar]
- 5.Garcia A.G., Garcia-De-Diego A.M., Gandia L., Borges R., Garcia-Sancho J. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 2006;86:1093–1131. doi: 10.1152/physrev.00039.2005. [DOI] [PubMed] [Google Scholar]
- 6.Mahapatra S., Calorio C., Vandael D.H., Marcantoni A., Carabelli V., Carbone E. Calcium channel types contributing to chromaffin cell excitability, exocytosis and endocytosis. Cell Calcium. 2012;51(3-4):321–330. doi: 10.1016/j.ceca.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Baldelli P., Hernandez-Guijo J.M., Carabelli V., Novara M., Cesetti T., Andres-Mateos E., Montiel C., Carbone E. Direct and remote modulation of L-channels in chromaffin cells - Distinct actions on alpha(1C) and alpha(1D) subunits? Mol. Neurobiol. 2004;29(1):73–96. doi: 10.1385/MN:29:1:73. [DOI] [PubMed] [Google Scholar]
- 8.Marcantoni A., Carabelli V., Comunanza V., Hoddah H., Carbone E. Calcium channels in chromaffin cells: focus on L and T types. Acta Physiol. (Oxf.) 2008;192(2):233–246. doi: 10.1111/j.1748-1716.2007.01815.x. [DOI] [PubMed] [Google Scholar]
- 9.Vandael D.H., Marcantoni A., Mahapatra S., Caro A., Ruth P., Zuccotti A., Knipper M., Carbone E. Ca(v)1.3 and BK Channels for Timing and Regulating Cell Firing. Mol. Neurobiol. 2010;42(3):185–198. doi: 10.1007/s12035-010-8151-3. [DOI] [PubMed] [Google Scholar]
- 10.Marcantoni A., Baldelli P., Hernandez-Guijo J.M., Comunanza V., Carabelli V., Carbone E. L-type calcium channels in adrenal chromaffin cells: Role in pace-making and secretion. Cell Calcium. 2007;42(4-5):397–408. doi: 10.1016/j.ceca.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 11.Marcantoni A., Carabelli V., Vandael D.H., Comunanza V., Carbone E. PDE type-4 inhibition increases L-type Ca2+ currents, action potential firing, and quantal size of exocytosis in mouse chromaffin cells. Pflüg. Arch. Eur. J. Phy. 2009;457(5):1093–1110. doi: 10.1007/s00424-008-0584-4. [DOI] [PubMed] [Google Scholar]
- 12.Akaike A., Mine Y., Sasa M., Takaori S. Voltage and current clamp studies of muscarinic and nicotinic excitation of the rat adrenal chromaffin cells. J. Pharmacol. Exp. Ther. 1990;255:333–339. [PubMed] [Google Scholar]
- 13.Biales B., Dichter M., Tischler A. Electrical excitability of cultured adrenal chromaffin cells. J. Physiol. 1976;262:743–753. doi: 10.1113/jphysiol.1976.sp011618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt B.L., Hagiwara S., Kidokoro Y., Miyazaki S. Action potentials in the rat chromaffin cell and effects of acetylcholine. J. Physiol. 1976;263:417–439. doi: 10.1113/jphysiol.1976.sp011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Diego A.M. Electrophysiological and morphological features underlying neurotransmission efficacy at the splanchnic nerve-chromaffin cell synapse of bovine adrenal medulla. Am. J. Physiol. Cell Physiol. 2009;298:C397–C405. doi: 10.1152/ajpcell.00440.2009. [DOI] [PubMed] [Google Scholar]
- 16.Gullo F., Ales E., Rosati B., Lecchi M., Masi A., Guasti L., Cano-Abad M.F., Arcangeli A., Lopez M.G., Wanke E. ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J. 2002;17:330–332. doi: 10.1096/fj.02-0200fje. [DOI] [PubMed] [Google Scholar]
- 17.Hollins B., Ikeda S.R. Inward currents underlying action potentials in rat adrenal chromaffin cells. J. Neurophysiol. 1996;76:1195–1211. doi: 10.1152/jn.1996.76.2.1195. [DOI] [PubMed] [Google Scholar]
- 18.Wallace D.J., Chen C., Marley P.D. Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. J. Physiol. 2002;540:921–939. doi: 10.1113/jphysiol.2001.013370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcantoni A., Vandael D.H., Mahapatra S., Carabelli V., Sinnegger-Brauns M.J., Striessnig J., Carbone E. Loss of Cav1.3 Channels Reveals the Critical Role of L-Type and BK Channel Coupling in Pacemaking Mouse Adrenal Chromaffin Cells. J. Neurosci. 2010;30(2):491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandael D.H., Zuccotti A., Striessnig J., Carbone E. Ca(V)1.3-Driven SK Channel Activation Regulates Pacemaking and Spike Frequency Adaptation in Mouse Chromaffin Cells. J. Neurosci. 2012;32(46):16345–16359. doi: 10.1523/JNEUROSCI.3715-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nassar-Gentina V., Pollard H.B., Rojas E. Electrical activity in chromaffin cells of intact mouse adrenal gland. Am. J. Physiol. Cell Physiol. 1988;254:C675–C683. doi: 10.1152/ajpcell.1988.254.5.C675. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Espinosa P.L., Yang C., Gonzalez-Perez V., Xia X.M., Lingle C.J. Knockout of the BK beta2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow-wave burst activity. J. Gen. Physiol. 2014;144:275–295. doi: 10.1085/jgp.201411253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colomer C., Lafont C., Guerineau N.C. Stress-induced intercellular communication remodeling in the rat adrenal medulla. Ann. N. Y. Acad. Sci. 2009;1148:106–111. doi: 10.1196/annals.1410.040. [DOI] [PubMed] [Google Scholar]
- 24.Vandael D.H., Ottaviani M.M., Legros C., Lefort C., Guérineau N.C., Allio A., Carabelli V., Carbone E. Reduced availability of Nav channels by depolarization or block by TTX boosts burst firing and catecholamine release in mouse chromaffin cells. J. Physiol. 2015;593:905–927. doi: 10.1113/jphysiol.2014.283374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Micheletti M., Brioschi A., Fesce R., Grohovaz F. A novel pattern of fast calcium oscillations points to calcium and electrical activity cross-talk in rat chromaffin cells. Cell. Mol. Life Sci. 2004;62:95–104. doi: 10.1007/s00018-004-4338-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duan K., Yu X., Zhang C., Zhou Z. Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J. Neurosci. 2003;23:11235–11243. doi: 10.1523/JNEUROSCI.23-35-11235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bean B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 28.Puopolo M., Raviola E., Bean B.P. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonon F.G. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neurosci. 1988;24(1):19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- 30.Levitsky K.L., Lopez-Barneo J. Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J. Physiol. 2009;587:1917–1929. doi: 10.1113/jphysiol.2009.168989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seidler F.J., Slotkin T.A. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. J. Physiol. 1985;358:1–16. doi: 10.1113/jphysiol.1985.sp015536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reuter H. On the effect of adrenaline on the cellular Ca-metabolism in the guinea pig atrium. N.-S. Arch. Ex. Path Ph. 1965;251:401–412. [PubMed] [Google Scholar]
- 33.Catterall W.A., Raman I.M., Robinson H.P., Sejnowski T.J., Paulsen O. The Hodgkin-Huxley heritage: from channels to circuits. J. Neurosci. 2012;32:14064–14073. doi: 10.1523/JNEUROSCI.3403-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamada-Hanff J., Bean B.P. Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J. Neurosci. 2013;33:15011–15021. doi: 10.1523/JNEUROSCI.0577-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fenwick E.M., Marty A., Neher E. Sodium and calcium channels in bovine chromaffin cells. J. Physiol. 1982;331:599–635. doi: 10.1113/jphysiol.1982.sp014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahapatra S., Marcantoni A., Vandael D.H., Striessnig J., Carbone E. Are Ca(v)1.3 pacemaker channels in chromaffin cells? Possible bias from resting cell conditions and DHP blockers usage. Channels. 2011;5(3):219–224. doi: 10.4161/chan.5.3.15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carbone E., Carabelli V., Cesetti T., Baldelli P., Hernandez-Guijo J.M., Giusta L. G-protein- and cAMP-dependent L-channel gating modulation: a manyfold system to control calcium entry in neurosecretory cells. Pflüg. Arch. Eur. J. Physiol. 2001;442(6):801–813. doi: 10.1007/s004240100607. [DOI] [PubMed] [Google Scholar]
- 38.Carbone E., Marcantoni A., Giancippoli A., Guido D., Carabelli V. T-type channels-secretion coupling: evidence for a fast low-threshold exocytosis. Pflüg. Arch. Eur. J. Physiol. 2006;453(3):373–383. doi: 10.1007/s00424-006-0100-7. [DOI] [PubMed] [Google Scholar]
- 39.Guerineau N.C., Desarmenien M.G., Carabelli V., Carbone E. Functional Chromaffin Cell Plasticity in Response to Stress: Focus on Nicotinic, Gap Junction, and Voltage-Gated Ca2+ Channels. J. Mol. Neurosci. 2012;48(2):368–386. doi: 10.1007/s12031-012-9707-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Artalejo C.R., Adams M.E., Fox A.P. Three types of Ca2+ channel trigger secretion with different efficacies in chromaffin cells. Nature. 1994;367:72–76. doi: 10.1038/367072a0. [DOI] [PubMed] [Google Scholar]
- 41.Carabelli V., Giancippoli A., Baldelli P., Carbone E., Artalejo A.R. Distinct potentiation of L-type currents and secretion by cAMP in rat chromaffin cells. Biophys. J. 2003;85(2):1326–1337. doi: 10.1016/S0006-3495(03)74567-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carabelli V., Marcantoni A., Comunanza V., Carbone E. Fast exocytosis mediated by T- and L-type channels in chromaffin cells: distinct voltage-dependence but similar Ca2+-dependence. Eur. Biophys. J. 2007;36(7):753–762. doi: 10.1007/s00249-007-0138-2. [DOI] [PubMed] [Google Scholar]
- 43.Giancippoli A., Novara M., de Luca A., Baldelli P., Marcantoni A., Carbone E., Carabelli V. Low-threshold exocytosis induced by cAMP-recruited Ca(V)3.2 (alpha(1H)) channels in rat chromaffin cells. Biophys. J. 2006;90(5):1830–1841. doi: 10.1529/biophysj.105.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engisch K.L., Nowycky M.C. Calcium dependence of large dense-cored vesicle exocytosis evoked by calcium influx in bovine adrenal chromaffin cells. J. Neurosci. 1996;16:1359–1369. doi: 10.1523/JNEUROSCI.16-04-01359.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosa J.M., Torregrosa-Hetland C.J., Colmena I., Gutierrez L.M., Garcia A.G., Gandia L. Calcium entry through slow-inactivating L-type calcium channels preferentially triggers endocytosis rather than exocytosis in bovine chromaffin cells. Am. J. Physiol. Cell Physiol. 2011;301:C86–C98. doi: 10.1152/ajpcell.00440.2010. [DOI] [PubMed] [Google Scholar]
- 46.Kim S.J., Lim W., Kim J. Contribution of L- and N-type calcium currents to exocytosis in rat adrenal medullary chromaffin cells. Brain Res. 1995;675:289–296. doi: 10.1016/0006-8993(95)00085-5. [DOI] [PubMed] [Google Scholar]
- 47.Thiagarajan R., Tewolde T., Li Y., Becker P.L., Rich M.M., Engisch K.L. Rab3A negatively regulates activity-dependent modulation of exocytosis in bovine adrenal chromaffin cells. J. Physiol. 2003;555:439–457. doi: 10.1113/jphysiol.2003.056333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulate G., Scott S.R., Gonzalez J., Gilabert J.A., Artalejo A.R. Extracellular ATP regulates exocytosis in inhibiting multiple Ca2+ channel types in bovine chromaffin cells. Pflüg. Arch. Eur. J. Physiol. 2000;439:304–314. doi: 10.1007/s004249900185. [DOI] [PubMed] [Google Scholar]
- 49.Albillos A., Carbone E., Gandia L., Garcia A.G., Pollo A. Opioid inhibition of Ca2+ channel subtypes in bovine chromaffin cells: Selectivity of action and voltage dependence. Eur. J. Neurosci. 1996;8(8):1561–1570. doi: 10.1111/j.1460-9568.1996.tb01301.x. [DOI] [PubMed] [Google Scholar]
- 50.Albillos A., Gandia L., Michelena P., Gilabert J.A., delValle M., Carbone E., Garcia A.G. The mechanism of calcium channel facilitation in bovine chromaffin cells. J. Physiol. 1996;494(3):687–695. doi: 10.1113/jphysiol.1996.sp021524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carabelli V., Carra I., Carbone E. Localized secretion of ATP and opioids revealed through single Ca2+ channel modulation in bovine chromaffin cells. Neuron. 1998;20(6):1255–1268. doi: 10.1016/s0896-6273(00)80505-x. [DOI] [PubMed] [Google Scholar]
- 52.Carabelli V., Lovallo M., Magnelli V., Zucker H., Carbone E. Voltage-dependent modulation of single N-type Ca2+ channel kinetics by receptor agonists in IMR32 cells. Biophys. J. 1996;70(5):2144–2154. doi: 10.1016/S0006-3495(96)79780-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Currie K.P., Fox A.P. ATP serves as a negative feedback inhibitor of voltage-gated Ca2+ channel currents in cultured bovine adrenal chromaffin cells. Neuron. 1996;16:1027–1036. doi: 10.1016/s0896-6273(00)80126-9. [DOI] [PubMed] [Google Scholar]
- 54.Gandia L., Garcia A.G., Morad M. ATP modulation of calcium channels in chromaffin cells. J. Physiol. 1993;470:55–72. doi: 10.1113/jphysiol.1993.sp019847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carabelli V., Hernandez-Guijo J.M., Baldelli P., Carbone E. Direct autocrine inhibition and cAMP-dependent potentiation of single L-type Ca2+ channels in bovine chromaffin cells. J. Physiol. 2001;532(1):73–90. doi: 10.1111/j.1469-7793.2001.0073g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cesetti T., Hernandez-Guijo J.M., Baldelli P., Carabelli V., Carbone E. Opposite action of beta 1- and beta 2-adrenergic receptors on Ca(V)1 L-channel current in rat adrenal chromaffin cells. J. Neurosci. 2003;23(1):73–83. doi: 10.1523/JNEUROSCI.23-01-00073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahapatra S., Marcantoni A., Zuccotti A., Carabelli V., Carbone E. Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J. Physiol. 2012;590(20):5053–5073. doi: 10.1113/jphysiol.2012.236729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Souvannakitti D., Nanduri J., Yuan G., Kumar G.K., Fox A.P., Prabhakar N.R. NADPH oxidase-dependent regulation of T-type Ca2+ channels and ryanodine receptors mediate the augmented exocytosis of catecholamines from intermittent hypoxia-treated neonatal rat chromaffin cells. J. Neurosci. 2010;30:10763–10772. doi: 10.1523/JNEUROSCI.2307-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bournaud R., Hidalgo J., Yu H., Jaimovich E., Shimahara T. Low threshold T-type calcium current in rat embryonic chromaffin cells. J. Physiol. 2001;537:35–44. doi: 10.1111/j.1469-7793.2001.0035k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Novara M., Baldelli P., Cavallari D., Carabelli V., Giancippoli A., Carbone E. Exposure to cAMP and beta-adrenergic stimulation recruits Ca(V)3 T-type channels in rat chromaffin cells through Epac cAMP-receptor proteins. J. Physiol. 2004;558(2):433–449. doi: 10.1113/jphysiol.2004.061184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carabelli V., Marcantoni A., Comunanza V., De Luca A., Diaz J., Borges R., Carbone E. Chronic hypoxia up-regulates alpha(1H) T-type channels and low-threshold catecholamine secretion in rat chromaffin cells. J. Physiol. 2007;584(1):149–165. doi: 10.1113/jphysiol.2007.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss N., Hameed S., Fernandez-Fernandez J.M., Fablet K., Karmazinova M., Poillot C., Proft J., Chen L.N., Bidaud I., Monteil A., Huc-Brandt S., Lacinova L., Lory P., Zamponi G.W., De Waard M.A. Ca(v)3.2/Syntaxin-1A Signaling Complex Controls T-type Channel Activity and Low-threshold Exocytosis. J. Biol. Chem. 2012;287(4):2810–2818. doi: 10.1074/jbc.M111.290882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandia L., Mayorgas I., Michelena P., Cuchillo I., de Pascual R., Abad F., Novalbos J.M., Larranaga E., Garcia A.G. Human adrenal chromaffin cell calcium channels: drastic current facilitation in cell clusters, but not in isolated cells. Pflüg. Arch. Eur. J. Phy. 1998;436(5):696–704. doi: 10.1007/s004240050691. [DOI] [PubMed] [Google Scholar]
- 64.Rosa J.M., de Diego A.M., Gandia L., Garcia A.G. L-type calcium channels are preferentially coupled to endocytosis in bovine chromaffin cells. Biochem. Biophys. Res. Commun. 2007;357(4):834–839. doi: 10.1016/j.bbrc.2007.03.207. [DOI] [PubMed] [Google Scholar]
- 65.Rosa J.M., Orozco A., Conde M., Nanclares C., Colmena I., de Pascual R., Gandia L. Activation of endocytosis and L calcium channels by beta-amyloid peptide: role of sphingolipids. Basic Clin. Pharmacol. 2011;109:46–46. [Google Scholar]
- 66.Mercuri N.B., Bonci A., Calabresi P., Stratta F., Stefani A., Bernardi G. Effects of dihydropyridine calcium-antagonists on rat midbrain dopaminergic-neurons. Br. J. Pharmacol. 1994;113(3):831–838. doi: 10.1111/j.1476-5381.1994.tb17068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan C.S., Guzman J.N., Ilijic E., Mercer J.N., Rick C., Tkatch T., Meredith G.E., Surmeier D.J. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447(7148):1081–1085. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 68.Putzier I., Kullmann P.H., Horn J.P., Levitan E.S. Ca(v)1.3 Channel Voltage Dependence, Not Ca2+ Selectivity, Drives Pacemaker Activity and Amplifies Bursts in Nigral Dopamine Neurons. J. Neurosci. 2009;29(49):15414–15419. doi: 10.1523/JNEUROSCI.4742-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mangoni M.E., Couette B., Bourinet E., Platzer J., Reimer D., Striessnig J., Nargeot J. Functional role of L-type Ca(v)13Ca(2+) channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. USA. 2003;100(9):5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Catterall W.A., Perez-Reyes E., Snutch T.P., Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005;57(4):411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 71.Koschak A., Reimer D., Huber I., Grabner M., Glossmann H., Engel J., Striessnig J. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 2001;276(25):22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 72.Xu W.F., Lipscombe D. Neuronal Ca(v)1.3 alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 2001;21(16):5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Striessnig J. Pharmacology of L-type calcium channels. Curr. Mol. Pharmacol. doi: 10.2174/1874467208666150507105845. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang S., Cooper G., Dunne S.F., Dusel B., Luan C.H., Surmeier D.J., Silverman R.B. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat. Commun. 2012;3:1146. doi: 10.1038/ncomms2149. [DOI] [PubMed] [Google Scholar]
- 75.Ortner N.J., Bock G., Vandael D.H., Mauersberger R., Draheim H.J., Gust R., Carbone E., Tuluc P., Striessnig J. Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+ channel activators. Nat. Commun. 2014;5:3897–3906. doi: 10.1038/ncomms4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang H., Ng C.Y., Yu D., Zhai J., Lam Y., Soong T.W. Modest CaV1.342-selective inhibition by compound 8 is beta-subunit dependent. Nat. Commun. 2014;5:4481. doi: 10.1038/ncomms5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Platzer J., Engel J., Schrott-Fischer A., Stephan K., Bova S., Chen H., Zheng H., Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102(1):89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 78.Solaro C.R., Prakriya M., Ding J.P., Lingle C.J. Inactivating and noninactivating Ca2+- and voltage-dependent K+ current in rat adrenal chromaffin cells. J. Neurosci. 1995;15(9):6110–6123. doi: 10.1523/JNEUROSCI.15-09-06110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prakriya M., Lingle C.J. BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J. Neurophysiol. 1999;81(5):2267–2278. doi: 10.1152/jn.1999.81.5.2267. [DOI] [PubMed] [Google Scholar]
- 80.Pape H.C. Queer current and pacemaker: The hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 81.Crill W.E. Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 82.Raman I.M., Bean B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu B.X., Su Y.H., Das S., Liu J., Xia J.S., Ren D.J. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129(2):371–383. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 84.Fakler B., Adelman J.P. Control of K-Ca channels by calcium nano/microdomains. Neuron. 2008;59(6):873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 85.Marty A., Neher E. Potassium channels in cultured bovine adrenal chromaffin cells. J. Physiol. 1985;367:117–141. doi: 10.1113/jphysiol.1985.sp015817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neely A., Lingle C.J. 2 Components of calcium-activated potassium current in rat adrenal chromaffin cells. J. Physiol. 1992;453:97–131. doi: 10.1113/jphysiol.1992.sp019220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scott R.S., Bustillo D., Olivos-Oré L.A., Cuchillo-Ibanez I., Barahona M.V., Carbone E., Artalejo A.R. Contribution of BK channels to action potential repolarisation at minimal cytosolic Ca2+ concentration in chromaffin cells. Pflüg. Arch. Eur. J. Phy. 2011;462(4):545–557. doi: 10.1007/s00424-011-0991-9. [DOI] [PubMed] [Google Scholar]
- 88.Augustine G.J., Santamaria F., Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40(2):331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- 89.Edgerton J.R., Reinhart P.H. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J. Physiol. 2003;548:53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marrion N.V., Tavalin S.J. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395(6705):900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 91.Brandt A., Striessnig J., Moser T. Ca(V)1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 2003;23(34):10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun L., Xiong Y., Zeng X.H., Wu Y., Pan N., Lingle C.J., Qu A.L., Ding J.P. Differential regulation of action potentials by inactivating and noninactivating BK channels in rat adrenal chromaffin cells. Biophys. J. 2009;97(10):2863–2863. doi: 10.1016/j.bpj.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xia X.M., Ding J.P., Lingle C.J. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J. Neurosci. 1999;19(13):5255–5264. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Faber E.S. Functions and Modulation of Neuronal SK Channels. Cell Biochem. Biophys. 2009;55(3):127–139. doi: 10.1007/s12013-009-9062-7. [DOI] [PubMed] [Google Scholar]
- 95.Stocker M. Ca(2+)-activated K(+) channels: Molecular determinants and function of the SK family. Nat. Rev. Neurosci. 2004;5(10):758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- 96.Engel J., Schultens H.A., Schild D. Small conductance potassium channels cause an activity-dependent spike frequency adaptation and make the transfer function of neurons logarithmic. Biophys. J. 1999;76(3):1310–1319. doi: 10.1016/S0006-3495(99)77293-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wolfart J., Neuhoff H., Franz O., Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J. Neurosci. 2001;21(10):3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hallworth N.E., Wilson C.J., Bevan M.D. Apamin-sensitive small conductance calcium-activated potassium channels, through their selective coupling to voltage-gated calcium channels, are critical determinants of the precision, pace, and pattern of action potential generation in rat subthalamic nucleus neurons in vitro. J. Neurosci. 2003;23(20):7525–7542. doi: 10.1523/JNEUROSCI.23-20-07525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cueni L., Canepari M., Lujan R., Emmenegger Y., Watanabe M., Bond C.T., Franken P., Adelman J.P., Luthi A. T-type Ca(2+) channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat. Neurosci. 2008;11(6):683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- 100.Engbers J.D., Anderson D., Asmara H., Rehak R., Mehaffey W.H., Hameed S., McKay B.E., Kruskic M., Zamponi G.W., Turner R.W. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. USA. 2012;109(7):2601–2606. doi: 10.1073/pnas.1115024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Oliver D., Klocker N., Schuck J., Baukrowitz T., Ruppersberg J.P., Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron. 2000;26(3):595–601. doi: 10.1016/s0896-6273(00)81197-6. [DOI] [PubMed] [Google Scholar]
- 102.Shah M.M., Haylett D.G. (+) currents generated by NMDA receptor activation in rat hippocampal pyramidal neurons. J. Neurophysiol. 2002;87(6):2983–2989. doi: 10.1152/jn.2002.87.6.2983. [DOI] [PubMed] [Google Scholar]
- 103.Montiel C., Lopez M.G., Sanchezgarcia P., Maroto R., Zapater P., Garcia A.G. Contribution of SK and BK channels in the control of catecholamine release by electrical-stimulation of the cat adrenal-gland. J. Physiol. 1995;486(2):427–437. doi: 10.1113/jphysiol.1995.sp020823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Neely A., Lingle C.J. Effects of muscarine on single-rat adrenal chromaffin cells. J. Physiol. 1992;453:133–166. doi: 10.1113/jphysiol.1992.sp019221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deister C.A., Chan C.S., Surmeier D.J., Wilson C.J., Calcium-Activated S.K. Channels Influence Voltage-Gated Ion Channels to Determine the Precision of Firing in Globus Pallidus Neurons. J. Neurosci. 2009;29(26):8452–8461. doi: 10.1523/JNEUROSCI.0576-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Atherton J.F., Bevan M.D. Ionic mechanisms underlying autonomous action potential generation in the somata and dendrites of GABAergic substantia nigra pars reticulata neurons in vitro. J. Neurosci. 2005;25(36):8272–8281. doi: 10.1523/JNEUROSCI.1475-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Surmeier D.J., Guzman J.N., Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium. 2010;47(2):175–182. doi: 10.1016/j.ceca.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alamo L., Garcia A.G., Borges R. Electrically-evoked catecholamine release from cat adrenals - role of cholinergic receptors. Biochem. Pharmacol. 1991;42(5):973–978. doi: 10.1016/0006-2952(91)90277-c. [DOI] [PubMed] [Google Scholar]
- 109.Ladenbauer J., Augustin M., Shiau L., Obermayer K. Impact of Adaptation Currents on Synchronization of Coupled Exponential Integrate-and-Fire Neurons. PLOS Comput. Biol. 2012;8(4):19. doi: 10.1371/journal.pcbi.1002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Catterall W.A., Goldin A.L., Waxman S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 111.Kidokoro Y., Ritchie A.K. Chromaffin cell action potentials and their possible role in adrenaline secretion from rat adrenal medulla. J. Physiol. 1980;307:199–216. doi: 10.1113/jphysiol.1980.sp013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kobayashi H., Shiraishi S., Yanagita T., Yokoo H., Yamamoto R., Minami S., Saitoh T., Wada A. Regulation of voltage-dependent sodium channel expression in adrenal chromaffin cells - Involvement of multiple calcium signaling pathways. In: Oconnor D.T., Eiden L.E., editors. Chromaffin Cell: Transmitter Biosynthesis, Storage, Release, Actions, and Informatics. Vol. 971. 2002. pp. 127–134. [DOI] [PubMed] [Google Scholar]
- 113.Lou X.L., Yu X., Chen X.K., Duan K.L., He L.M., Qu A.L., Xu T., Zhou Z. Na+ channel inactivation: a comparative study between pancreatic islet beta-cells and adrenal chromaffin cells in rat. J. Physiol. 2003;548:191–202. doi: 10.1113/jphysiol.2002.034405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wada A., Wanke E., Gullo F., Schiavon E. Voltage-dependent Na(v)1.7 sodium channels: multiple roles in adrenal chromaffin cells and peripheral nervous system. Acta Physiol. (Oxf.) 2008;192:221–231. doi: 10.1111/j.1748-1716.2007.01810.x. [DOI] [PubMed] [Google Scholar]
- 115.Sangameswaran L., Fish L.M., Koch B.D., Rabert D.K., Delgado S.G., Ilnicka M., Jakeman L.B., Novakovic S., Wong K., Sze P., Tzoumaka E., Stewart G.R., Herman R.C., Chan H., Eglen R.M., Hunter J.C. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 1997;272:14805–14809. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- 116.Klugbauer N., Lacinova L., Flockerzi V., Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morinville A., Fundin B., Meury L., Jureus A., Sandberg K., Krupp J., Ahmad S., O'Donnell D. Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: expression in the autonomic and endocrine systems. J. Comp. Neurol. 2007;504:680–689. doi: 10.1002/cne.21484. [DOI] [PubMed] [Google Scholar]
- 118.Toledo-Aral J.J., Moss B.L., He Z.J., Koszowski A.G., Whisenand T., Levinson S.R., Wolf J.J., Silos-Santiago I., Halegoua S., Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. USA. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carter B.C., Bean B.P. Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron. 2010;64:898–909. doi: 10.1016/j.neuron.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taddese A., Bean B.P. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- 121.Enomoto A., Han J.M., Hsiao C.F., Chandler S.H. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J. Neurophysiol. 2007;98:710–719. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- 122.Smith M.R., Smith R.D., Plummer N.W., Meisler M.H., Goldin A.L. Functional analysis of the mouse Scn8a sodium channel. J. Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rush A.M., Dib-Hajj S.D., Waxman S.G. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J. Physiol. 2005;564:803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mercer J.N., Chan C.S., Tkatch T., Held J., Surmeier D.J. Nav1.6 sodium channels are critical to pacemaking and fast spiking in globus pallidus neurons. J. Neurosci. 2007;27:13552–13566. doi: 10.1523/JNEUROSCI.3430-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bant J.S., Raman I.M. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. USA. 2010;107:12357–12362. doi: 10.1073/pnas.1005633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li L., Bischofberger J., Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J. Neurosci. 2007;27(49):13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Engel D., Jonas P. Presynaptic action potential amplification by voltage-gated Na+ channels in hippocampal mossy fiber boutons. Neuron. 2005;45:405–417. doi: 10.1016/j.neuron.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 128.Cummins T.R., Aglieco F., Renganathan M., Herzog R.I., Dib-Hajj S.D., Waxman S.G. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 2001;21:5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Madeja M. Do neurons have a reserve of sodium channels for the generation of action potentials? A study on acutely isolated CA1 neurons from the guinea-pig hippocampus. Eur. J. Neurosci. 2000;12:1–7. doi: 10.1046/j.1460-9568.2000.00871.x. [DOI] [PubMed] [Google Scholar]
- 130.Inoue M., Harada K., Matsuoka H., Sata T., Warashina A. Inhibition of TASK1-like channels by muscarinic receptor stimulation in rat adrenal medullary cells. J. Neurochem. 2008;106:1804–1814. doi: 10.1111/j.1471-4159.2008.05521.x. [DOI] [PubMed] [Google Scholar]
- 131.Clausen T. Adrenergic control of Na+-K+-homoeostasis. Acta Med. Scand. Suppl. 1983;672:111–115. doi: 10.1111/j.0954-6820.1983.tb01622.x. [DOI] [PubMed] [Google Scholar]
- 132.Harada K., Matsuoka H., Miyata H., Matsui M., Inoue M. Identification of muscarinic receptor subtypes involved in catecholamine secretion in adrenal medullary chromaffin cells by knockout. Br. J. Pharmacol. 2015;172(5):1348–1359. doi: 10.1111/bph.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Akaike A., Sasa M., Tamura Y., Ujihara H., Takaori S. Effects of protein kinase C on the muscarinic excitation of rat adrenal chromaffin cells. Jpn. J. Pharmacol. 1993;61:145–148. doi: 10.1254/jjp.61.145. [DOI] [PubMed] [Google Scholar]
- 134.Carr D.B., Day M., Cantrell A.R., Held J., Scheuer T., Catterall W.A., Surmeier D.J. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- 135.Remme C.A., Wilde A.A. Targeting sodium channels in cardiac arrhythmia. Curr. Opin. Pharmacol. 2014;15:53–60. doi: 10.1016/j.coph.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 136.Zaza A., Belardinelli L., Shryock J.C. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol. Ther. 2008;119(3):326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 137.Belardinelli L., Shryock J.C., Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92:6–14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Anger T., Madge D.J., Mulla M., Riddall D. Medicinal chemistry of neuronal voltage-gated sodium channel blockers. J. Med. Chem. 2001;44(2):115–137. doi: 10.1021/jm000155h. [DOI] [PubMed] [Google Scholar]