

Abstract
Ellis-van Creveld (EvC) syndrome is an autosomal-recessive skeletal dysplasia, characterized by short stature and postaxial polydactyly. A series of dental abnormalities, including hypomorphic enamel formation, has been reported in patients with EvC. Despite previous studies that attempted to uncover the mechanism leading to abnormal tooth development, little is known regarding how hypomorphic enamel is formed in patients with EvC. In the current study, using Evc2/Limbin mutant mice we recently generated, we analyzed enamel formation in the mouse incisor. Consistent with symptoms in human patients, we observed that Evc2 mutant mice had smaller incisors with enamel hypoplasia. Histologic observations coupled with ameloblast marker analyses suggested that Evc2 mutant preameloblasts were capable of differentiating to secretory ameloblasts; this process, however, was apparently delayed, due to delayed odontoblast differentiation, mediated by a limited number of dental mesenchymal stem cells in Evc2 mutant mice. This concept was further supported by the observation that dental mesenchymal-specific deletion of Evc2 phenocopied the tooth abnormalities in Evc2 mutants. Overall, our findings suggest that mutations in Evc2 affect dental mesenchymal stem cell homeostasis, which further leads to hypomorphic enamel formation.
Keywords: cilium, Hedgehog signaling, ameloblast, odontoblast, Limbin, stem cells
Introduction
Ellis-van Creveld (EvC) syndrome is an autosomal-recessive chondrodysplasia (McKusick et al. 1964). Typical symptoms of EvC include shorter limbs and ribs, postaxial polydactyly, dysplastic nails, and cardiovascular defects (McKusick et al. 1964; Baujat and Le Merrer 2007). Genetic studies have linked around two-thirds of EvC-affected patients with mutations in either EVC or EVC2 (Ruiz-Perez et al. 2000, 2003). Interestingly, our previous work identified EVC2 genetic mutations (also known as LIMBIN) in Japanese brown cattle (Takeda et al. 2002). A recent study also identified EVC2 mutations in Tyrolean Grey cattle (Murgiano et al. 2014). Both cases suggest a conserved function of EVC2 across different species. In addition to the skeletal and cardiovascular symptoms, several dental abnormalities were documented in patients with EvC, including neonatal teeth, delayed eruption, hypodontia, supernumerary teeth, conical teeth, taurodontism, and hypomorphic enamel formation (Mostafa et al. 2005; Veena et al. 2011). The variable dental symptoms in these patients suggest that EVC2 plays important roles during different stages of tooth organogenesis.
Tooth development is a sequential process requiring a series of reciprocal interactions between dental epithelium and dental mesenchyme (Thesleff and Hurmerinta 1981). Despite dentition differences between mice and humans, the mouse model has been primarily used for the study of tooth development. During mouse embryonic tooth development, the tooth undergoes a series of morphological differentiations initiated from the thickening of dental epithelium to the bud stage, cap stage, and bell stage (Jussila and Thesleff 2012; Lan et al. 2014). Later in the bell stage, cells originating from the dental mesenchyme and cells originating from the dental epithelium differentiate into odontoblasts and ameloblasts, both of which will secrete dentin and enamel matrix proteins, respectively. The differentiations of odontoblasts and ameloblasts are processes that require reciprocal interactions between these 2 cell types. Initially, signaling molecules from preameloblasts induce the differentiation of mesenchymal cells into odontoblast cells. The differentiated odontoblasts will then deposit dentin matrix to further induce differentiation of preameloblasts to secretory ameloblasts (Thesleff and Hurmerinta 1981; Bei 2009).
In addition to reciprocal interactions between dental epithelium and mesenchyme, cell-autonomous factors also play important roles during ameloblast differentiation. Of many growth factors, Sonic Hedgehog (SHH)–mediated Hedgehog signaling is required for ameloblast differentiation and maturation. Shh is highly expressed in the preameloblast. Genetic ablation of Shh in the dental epithelium has resulted in limited numbers of secretory ameloblasts and limited enamel deposition (Dassule et al. 2000). Similarly, specific ablation of Smo, the Hedgehog effector protein, in dental epithelium leads to similarly defective ameloblast differentiation (Gritli-Linde et al. 2002).
EvC syndrome has been categorized as ciliopathy due to the intracellular ciliary localization of EVC and EVC2 (Baujat and Le Merrer 2007). In vitro mechanistic studies have demonstrated that EVC and EVC2 form a protein complex at the bottoms of primary cilia, a complex required for transducing Hedgehog signaling (Dorn et al. 2012; Caparrós-Martin et al. 2013). A recent report also indicated that Evc is involved in regulating symmetric responses to Hedgehog signaling during molar development (Nakatomi et al. 2013). We recently generated Evc2/Limbin mutant mice and identified abnormal tooth phenotypes therein (Zhang et al. 2015), along with other craniofacial abnormalities (Badri, Zhang, Ohyama, Venkitapathi, Alamoudi, et al. 2016; Badri, Zhang, Ohyama, Venkitapathi, Kamiya, et al. 2016). Despite these findings, the pathophysiologic mechanism leading to hypomorphic enamel formation in patients with EvC remains unknown. In the current studies, we used the mouse incisor as a model to investigate the pathophysiologic mechanism leading to hypomorphic enamel formation in the Evc2/Limbin mutant mice we generated. The results of our in vivo studies strongly suggest that these mice showed reduced numbers of dental mesenchymal stem cells and a subsequent delay in odontoblast differentiation that, secondarily, led to hypomorphic enamel formation.
Materials and Methods
Animals
Generations of Evc2/Limbin global mutant mice and Evc2/Limbin floxed mice were reported previously (Zhang et al. 2015). Evc2/Limbin homozygous mutant embryos and pups (Evc2ex12/ex12) were obtained through the intercrossing of Evc2ex12/+ mice. Evc2dE/dE mice are the Cre recombined allele for Evc2/Limbin mice, which do not have a lacZ expression cassette but show the same phenotype as Evc2ex12/ex12 mice (Zhang et al. 2015). Noon of the date when the vaginal plug was observed was designated as embryonic day 0.5 (E0.5). Mice homozygous for the Evc2/Limbin floxed allele (Evc2fx/fx) were bred with heterozygous Evc2/Limbin floxed mice carrying a Wnt1-Cre transgene (Evc2ex12/+;Wnt1-Cre) (Danielian et al. 1998) to generate specific deletion of Evc2/Limbin in neural crest derivatives, including dental mesenchyme. All animal experiments were performed in accordance with the policies and federal laws for the judicious use of vertebrate animals, as approved by the University Committee on Use and Care of Animals at the University of Michigan.
Histology, X-Ray, Beta-Gal Staining, Immunohistochemistry, and Alkaline Phosphatase Staining
Mouse mandibles were dissected out and fixed in 4% paraformaldehyde (PFA). Subsequently, they were embedded in paraffin, sectioned parasagittally, and stained with hematoxylin and eosin (H&E) for histologic observations. At least 4 pairs of mutants and littermate controls were used in histologic observations and immunohistochemistry analysis. X-ray images were taken by means of a Faxitron X-ray system (Faxitron). For histologic analysis of undecalcified incisors, P8 mandibles were fixed, cryoprotected, and embedded for cryosection and H&E staining. For beta-gal activity staining and immunohistochemistry, mandibles were fixed with 4% PFA and cryoprotected by 30% sucrose in phosphate-buffered saline (PBS) before being embedded for cryosection; specimens were cut into 10-µm sections. Sections were stained with 1 mg/mL X-gal at 37°C for up to 2 d for beta-gal activity. For immunohistochemistry, sections were incubated overnight at 4°C with antibody against amelogenin (C-19, 1:100; Santa Cruz Biotechnology), Ki-67 (D3B5, 1:200; Cell Signaling), Osterix (OSX, ab22552, 1:500; Abcam), and Phospho-Histone 3 (06-570, 1:500; Millipore). Mouse anti-DSP was a kind gift from Dr. Chunlin Qin (Qin et al. 2003). For immunohistochemistry, a series of cryosections was made from the buccal to the lingual side. Sections containing at least 80% of incisors were used for immunohistochemistry. Quantifications were done with an average of 4 sections used to represent 1 biological sample. The average of 4 different biological samples was then taken for the comparison of differences between controls and mutants. For alkaline phosphatase (ALP) staining, sectioned mandibles were directly stained with BM purple solution (11442074001; Roche).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
E18.5 incisor tooth germs were dissected out from mandibles. Subsequently, dental epithelium and mesenchyme were separated after Dispase (2 U/mL, 04942078001; Roche) treatment and were subjected to RNA isolation with TRIzol reagent (Life Technology) according to the manufacturer’s instructions (Thomas et al. 2000). Quantitative real-time polymerase chain reaction (PCR) was performed with Applied Biosystems ViiA7, with the following TaqMan probes: Mm00494645_m1 for Gli1, mm00436026_m1 for Ptch1, mm99999915_g1 for Gapdh, and Mm00507596-m1 for Evc2 (ex13 and ex14).
Results
Enamel Hypoplasia Is Observed in Evc2 Mutant Incisors
To understand the pathophysiologic mechanism of hypomorphic enamel formation in the EvC syndrome, we studied the enamel formation using global and tissue-specific Evc2/Limbin mutant mouse lines generated in our laboratory (Zhang et al. 2015) (Fig. 1A). The mouse incisor grows continuously and showcases all stages of tooth development from the posterior to the anterior region, which makes it a convenient model for the study of epithelial and mesenchymal interactions during enamel formation (Møinichen et al. 1996). Taking advantage of the lacZ expression cassette knocked into the Evc2 locus in the global heterozygous mutant mice (Evc2ex12/+), we identified Evc2 expression in both the dental epithelium and the dental mesenchyme (Fig. 1B). X-ray radiography of 2-mo-old heads demonstrated that Evc2ex12/ex12 mice showed a decreased length of incisor roots compared with those of wild-type and Evc2 heterozygous (Evc2ex12/+) mice (Fig. 1C). Thereafter, wild-type and Evc2 heterozygous mice were used as controls in comparison with Evc2 homozygous mutants (Evc2ex12/ex12). Molars in Evc2 mutants also showed a decreased root length, depicted in X-ray radiograms, after removal of the maxillae and mandibles from the rest of the head (Fig. 1D). In addition to the decreased root length, Evc2 mutant incisors showed less pigmentation compared with littermate controls, suggesting hypomorphic enamel formation in Evc2 mutants (Fig. 1C).
Figure 1.
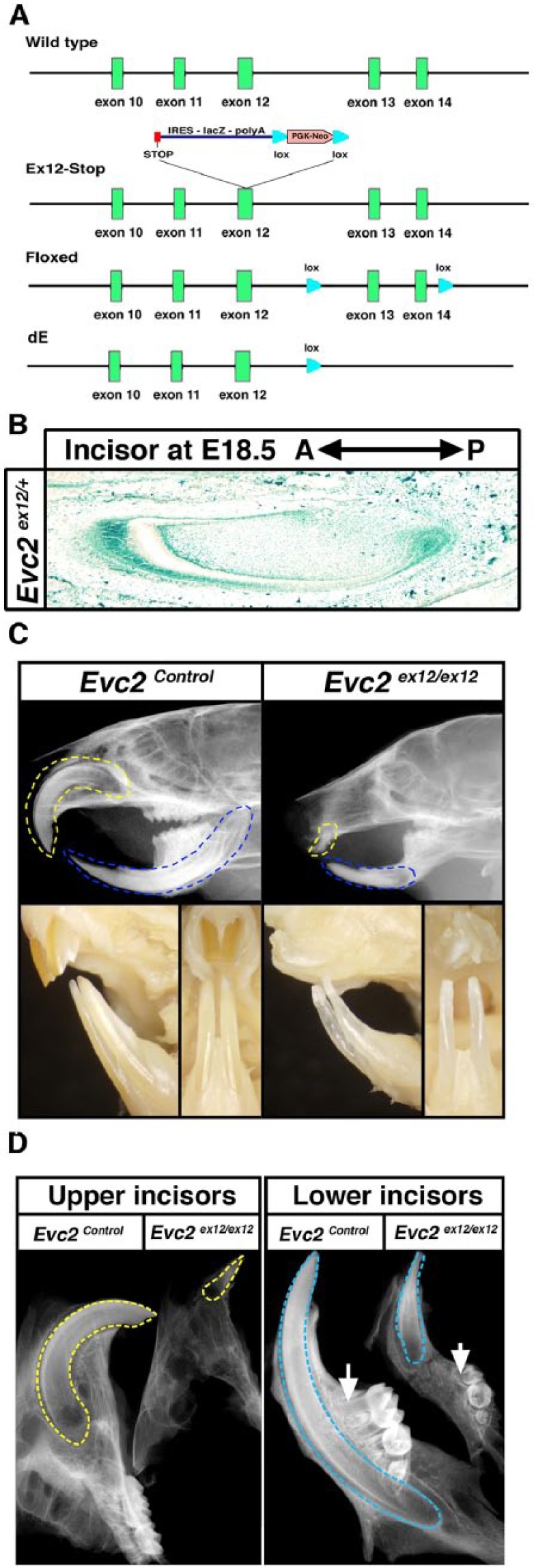
Evc2 mutant mice showed hypomorphic enamel formation. (A) Mutant alleles of Evc2/Limbin used in this study. Ex12-Stop (a global knockout allele), a stop codon, along with an IRES-LacZ cassette has been inserted into exon 12 to mimic a nonsense mutation identified in human patients. Floxed (a conditional allele) and 2 loxP sites have been introduced to flank exon 13 and exon 14. A Cre-mediated recombination results in a frame shift downstream of exon 12 of Evc2. dE, a Cre recombined allele, produces a similarly truncated EVC2 protein with the one from Ex12-Stop allele, if any. (B) LacZ staining of Evc2 ex12/+ incisor indicates that Evc2 is expressed in dental epithelium, mesenchyme, and surrounding bone tissue. (C) Lateral X-ray radiogram of 2-mo-old Evc2 mutant mice shows hypomorphic maxilla and mandible incisor formations (top). Frontal view and side view of the same mice demonstrate possible hypomorphic enamel in Evc2 mutant mice (bottom). Yellow dashed lines indicate maxilla incisors and blue dashed lines indicate mandible incisors. (D) Lateral X-ray radiogram of 2-mo-old dissected maxilla and mandible shows hypomorphic incisor and molar formation in Evc2 mutant. Yellow dashed lines indicate maxilla incisors and blue dashed lines indicate mandible incisors.
Enamel Hypoplasia in Evc2 Mutant Is Due to Delayed Ameloblast Differentiation
Well-orchestrated ameloblast proliferation, differentiation, and maturation are critical for enamel formation (Thesleff and Hurmerinta 1981). To determine if hypomorphic enamel formation in Evc2 homozygous mutant mice is due to defective ameloblast differentiation, we examined embryonic mandibular incisors at E18.5, the earliest stage at which secretory ameloblasts can be detected. Compared with controls, in which we observed gradual maturation of ameloblasts from the cervical loop to the anterior tip, we could not detect any ameloblasts with polarized nuclei in Evc2 homozygous mutant incisors (Fig. 2A). At E12.5, when tooth development was just initiated, we detected thickened epithelia in both controls and Evc2 mutants (Appendix Fig. 1A), suggesting initiation of incisor development is not affected. In contrast, starting from E15.5, we observed a smaller tooth germ with a smaller cervical loop (Appendix Fig. 1B). At postnatal day 2 (P2), we observed ameloblasts with polarized nuclei in both controls and Evc2 homozygous mutant mice (Fig. 2B). The ameloblasts with polarized nuclei were found at a more anterior region in Evc2 mutants than in controls, suggesting that maturation of ameloblasts was delayed but not absent in Evc2 homozygous mutant mice. Similarly, we also detected delayed ameloblast maturation in Evc2 mutant molars at P2 (Fig. 2C). Delayed ameloblast differentiation is supported by an observation that more cells not producing amelogenin were observed in Evc2 mutant incisors at P2 (Fig. 2D, Appendix Fig. 1C). To assess if delayed ameloblast maturation is still capable of supporting enamel formation in Evc2 mutants, we examined undecalcified incisors at postnatal day 8 (P8). Both control and Evc2 mutant incisors showed enamel formation, further supporting the hypothesis that Evc2 mutation results in delayed enamel formation in comparison with that in controls (Fig. 2E).
Figure 2.
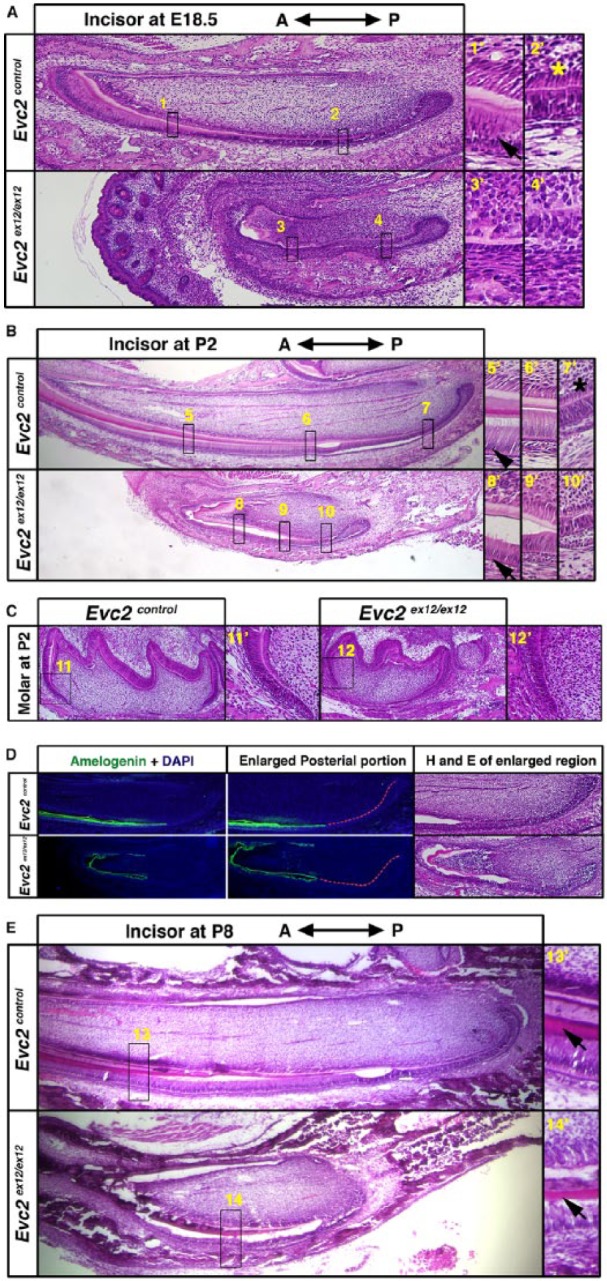
Evc2 mutant incisors showed delayed ameloblast differentiation and hypomorphic enamel formation. (A) Sagittal sections of embryonic day 18.5 (E18.5) embryonic mandible incisors. Regions in boxes in both controls and mutants were enlarged and shown on the right. Arrows indicate the ameloblasts with polarized nuclei and * indicates the odontoblasts with polarized nuclei. (B) Sagittal sections of postnatal day 2 (P2) mandible incisors. Regions in boxes in both control and mutant were enlarged and shown on the right. Arrowheads indicate ameloblasts with polarized nuclei in both controls and Evc2 mutants and * indicates the odontoblasts with polarized nuclei. (C) Sagittal sections of postnatal day 1 molar. Regions in boxes in both control and mutant were enlarged and shown. (D) Immunohistochemistry of amelogenin in mandible incisors at P2 indicates a delayed ameloblast differentiation. Posterior regions of incisors of both genotypes were enlarged and shown. Red dashed lines indicate the epithelial cells without amelogenin immunosignals. The immediate next section was processed for hematoxylin and eosin staining. (E) Sagittal sections of postnatal day 8 (P8) mandible incisors. Regions in boxes in both control and mutant were enlarged and shown on the right. Arrows indicate enamel formation in both control and Evc2 mutants.
Odontoblast Differentiation Is Delayed in Evc2 Mutants
Mutual interactions between ameloblasts and odontoblasts are required for the maturation of both cell types. Particularly, the maturation of ameloblasts depends on dentin secreted by odontoblasts (Linde and Granstrom 1978; Thesleff and Hurmerinta 1981). Along with delayed ameloblast maturation in Evc2 homozygous mutant mice, we detected delayed odontoblast maturation as well, evidenced by the presence of more preodontoblasts with nonpolarized nuclei in the mesenchyme of Evc2 homozygous mutant mice at P2 (Fig. 2B). ALP and OSX (aka SP-7) are marker proteins highly produced in differentiated odontoblasts (Linde and Granstrom 1978; Chen et al. 2009). ALP staining results indicated more non–ALP-producing cells in the dental mesenchyme of Evc2 homozygous mutant mice (Fig. 3A). Similarly, more non–OSX-producing cells were detected in the dental mesenchyme of Evc2 homozygous mutants (Fig. 3B, C). In summary, along with delayed ameloblast differentiation, we also detected delayed odontoblast differentiation in Evc2 homozygous mutant incisors.
Figure 3.

Evc2 mutant incisors showed delayed odontoblast differentiation. (A) Mandible incisors from postnatal day 2 (P2) were sectioned and processed for alkaline phosphatase (ALP) staining. Black dashed lines indicate a boundary between dental epithelium and mesenchyme. Yellow dashed lines indicate ALP-stained cells in the odontoblast layer. (B, C) Mandible incisors from P2 were sectioned and processed for immunohistochemistry for Osterix (OSX). White dashed lines separate dental epithelium and mesenchyme. Red dashed lines indicate OSX-expressing dental mesenchymal cells. Yellow dashed lines indicate OSX-stained cells in the odontoblast layer. The number of OSX-expressing dental mesenchymal cells was quantified and is shown in B (n = 4, P < 0.01).
Delayed Odontoblast Differentiation in Evc2 Mutant Is Due to Limited Numbers of Dental Mesenchymal Stem Cells
In mouse incisors, Gli1 has been reported as a marker for both epithelial and mesenchymal stem cells, which give rise to both ameloblasts and odontoblasts during incisor growth (Seidel et al. 2010; Zhao et al. 2014). To examine if Evc2 mutation has an impact on maintaining stem cells in both dental epithelium and mesenchyme, we introduced Gli1-lacZ into the Evc2 mutant background by breeding them with mice carrying the Cre recombined allele for Evc2/Limbin (Evc2dE/+). Evc2dE/dE mice, which do not have a lacZ expression cassette, showed the same phenotype as Evc2ex12/ex12 mice (Zhang et al. 2015) (Fig. 1A). The resulting mice (Evc2dE/dE; Gli1-lacZ) showed a significantly decreased number of lacZ-positive cells in both dental epithelium and mesenchyme (Fig. 4A, B). The mutant incisors appeared to show less intense lacZ staining compared with controls (Fig. 4A), suggesting decreased Hedgehog signaling in the mutant incisors. Previous reports indicated that dental epithelial stem cells are located in the cervical loop, while dental mesenchymal stem cells are located in the mesenchyme next to the cervical loops (Seidel et al. 2010; Zhao et al. 2014). Both groups of stem cells give rise to transient amplifying (TA) cells in the dental epithelium and the mesenchyme, respectively. The TA cells in both the epithelium and the mesenchyme will undergo further differentiation and eventually become postmitotic cells responsible for secreting enamel and dentin. Since Ki-67 is present in the cells within active cell cycles but is absent in the cells out of cell cycles, we used Ki-67 to label the TA cells and to differentiate them from the postmitotic population. In Evc2 mutant incisors, we observed significantly increased Ki-67–positive cells in the odontoblast layer compared with controls, suggesting that the loss of Evc2 keeps cells in the mitotic state, resulting in delayed odontoblast differentiation (Fig. 4C, red dashed lines, 4D). Conversely, no significant difference was observed in epithelial TA cells in the ameloblast layer in Evc2 mutant incisors (Fig. 4C, orange dashed lines, 4D). To examine if the small incisors in Evc2 mutants are due to differential cell proliferation or cell death, we further examined cell proliferation using antibody against phospho-histone H3 (P-H3) and cell death using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. In Evc2 mutant incisors at E18.5, we did not detect obvious differences in the numbers of P-H3- and TUNEL-positive cells compared with those in controls (Fig. 4E–G). Given the previous reports that Hedgehog signaling positively regulates odontoblast differentiation (Zhao et al. 2014), the compromised Hedgehog signaling detected in Evc2 mutant incisors is the likely reason for the delayed odontoblast differentiation observed. Since no differential cell proliferation and apoptosis were detected in Evc2 mutant incisors, we concluded that the decreased numbers of stem cells in the dental mesenchyme led to subsequent shorter incisors.
Figure 4.

Evc2 mutant incisors showed decreased dental epithelial and mesenchymal stem cells. (A) A Gli1-LacZ reporter was introduced to label dental epithelial and mesenchymal stem cells in the incisor. E18.5 mandible incisors of indicated genotypes were processed for LacZ staining. (B) Quantification of Gli1-LacZ–positive cells in A. n = 3, **P< 0.01. (C) Immunohistochemistry of Ki-67 was applied to mark the transient amplifying (TA) cells in controls and Evc2 mutants. Red arrowheads in the left panels indicate the 2 ends of the zone of Ki-67–positive cells in the odontoblast layers. White dashed lines in the right panels indicate a boundary between dental epithelium and mesenchyme. Red dashed lines indicate areas of Ki-67–positive cells in preodontoblasts (areas between 2 red arrowheads shown in the left panels). Yellow dashed lines indicate Ki-67–positive cells in preameloblasts. (D) Quantification of Ki-67–positive cells in B. n = 4, *P < 0.05; #not significant. (E) Immunohistochemistry of phospho-histone3 (P-H3) depicts proliferating cells in control and Evc2 mutant incisors at postnatal day 2 (P2). White dashed line indicates a boundary between dental epithelium and dental mesenchyme. (F) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining indicates apoptotic cells in control and Evc2 mutant incisor at P2. (G) Quantification of P-H3–positive cells in C. n = 4, #not significant.
Mesenchymal-Specific Deletion of Evc2 Phenocopies Hypomorphic Enamel in Evc2 Global Mutant
Our analysis demonstrated decreased Hedgehog signaling in dental epithelium in Evc2 mutant incisors (Fig. 4A), which may lead to hypomorphic enamel formation through affecting ameloblast differentiation. Our analysis also demonstrated evidence of delayed odontoblast differentiation in association with a limited number of dental mesenchymal stem cells, which may also affect ameloblast differentiation and lead to subsequent hypomorphic enamel formation. To determine which alteration(s) critically contribute to hypomorphic enamel formation in Evc2 mutants, we took advantage of the conditional Evc2 allele with Wnt1-Cre to specifically delete Evc2 in the dental mesenchyme (Chai et al. 2000). At P28, similar to what was observed in Evc2ex12/ex12 mice, incisors from the Evc2fx/fx;Wnt1-Cre mice (conditional mutants) showed less pigmentation (Fig. 5A). Mandibular radiography clearly demonstrated a decreased length of the incisor root (Fig. 5B). Micro–computed tomography (CT) of molars clearly indicated hypomorphic enamel in Evc2 conditional mutants. Histologic assessment of conditional mutants at the newborn stage consistently showed delayed ameloblast and odontoblast differentiation (Fig. 5C). We isolated an embryonic incisor at postnatal day 0 (NB) and physically separated dental epithelium (negative for Cre activity) from mesenchyme (positive for Cre activity). As expected, levels of Evc2 expression clearly indicated that is Wnt1-Cre–mediated deletion is restricted in the dental mesenchyme (Fig. 5D). We also confirmed a reduction of Gli1 expression only in the dental mesenchyme but not in the epithelium (Fig. 5D), suggesting that Hedgehog signaling activity is compromised in a mesenchyme-specific manner. Conversely, specific deletion of Evc2 in dental epithelium mediated by K14-Cre did not lead to overt abnormalities in incisors (data not shown). These results strongly suggest that in Evc2 mutant incisors, delayed odontoblast differentiation in dental mesenchyme secondarily leads to hypomorphic enamel formation.
Figure 5.

Mesenchyme-specific deletion of Evc2 phenocopied hypomorphic tooth formation found in Evc2 global knockout incisors. (A) Frontal view of maxilla and mandible incisor of Evc2 conditional knockout mice at postnatal day 28 (P28). (B) X-ray radiogram of Evc2 conditional mutants showed hypomorphic tooth formation. Mandibles of Evc2 conditional mutants and littermate controls at P28 were dissected and sagittal X-ray radiography was performed. Yellow dashed lines indicate incisors. (C) Micro–computed tomography (CT) of Evc2 conditional mutants and littermate controls at P28 indicates hypomorphic enamel in molars. (D) Mandible incisors of mesenchyme-specific Evc2 mutants and littermate controls were sagittally sectioned and processed for hematoxylin and eosin staining. Regions in boxes in both control and mutant were enlarged and shown on the right. (E) Quantitative reverse transcribed polymerase chain reaction from incisor epithelium and mesenchyme indicated that Wnt1-Cre–mediated deletion of Evc2 is specific for dental mesenchyme. EPI and MES stand for epithelial and mesenchymal tissues, respectively. n = 3, **P < 0.001. (F) A model to explain how mesenchymal function of Evc2 secondarily affects enamel formation. EP-TA, epithelial transient amplifying cells; ESC, dental epithelial stem cells; MC-TA, mesenchymal transient amplifying cells; MSC, dental mesenchymal stem cells. See text for details.
Discussion
A wide spectrum of dental abnormalities can be found in patients with EvC. In the current study, we used both global and conditional mutant mouse lines for Evc2/Limbin as models to investigate the pathophysiologic mechanism leading to hypomorphic enamel formation. Our in vivo studies on the Evc2 ex12/ex12 mutant mice and mesenchymal-specific knockout of Evc2 suggested that loss of Evc2 in the dental mesenchyme resulted in decreased numbers of dental mesenchymal stem cells, leading to delayed odontoblast differentiation, which secondarily leads to hypomorphic enamel formation through delayed ameloblast differentiation. Overall, our work elucidated, for the first time, the pathophysiologic mechanism leading to hypomorphic enamel formation in patients with EvC and suggested a mechanism whereby Evc2 mutations lead to hypomorphic tooth development through compromised dental mesenchymal stem cells (Fig. 5E).
In vitro mechanistic studies have suggested that EVC and EVC2 can form a protein complex, which is required for their mutual localization at the bottoms of the primary cilia (Dorn et al. 2012; Caparrós-Martin et al. 2013). This is consistent with genetic observations that a homozygous mutation of either of them leads to similar mouse phenotypes and that genetic deletion of both of them does not increase the severity of the phenotypes (Caparrós-Martin et al. 2013). In our Evc2ex12/ex12 mutant mice, we observed pale mandibular incisors and decreased root size (Fig. 1C), which were also observed in Evc mutant mice (Nakatomi et al. 2013). These observations are consistent with what was demonstrated in mechanistic studies that EVC and EVC2 are reciprocally required to form a protein complex at the bottoms of primary cilia.
Results from previous studies suggested that the EVC/EVC2 protein complex is required for transducing Hedgehog signaling (Dorn et al. 2012; Nakatomi et al. 2013). Mutation in Evc or Evc2 makes Hedgehog responding cells bear a compromised but not abolished response to the Hedgehog ligand (Zhang et al. 2015). Hedgehog signaling mediated by SHH is required for ameloblast differentiation and maturation and for maintenance of dental epithelial stem cells (Dassule et al. 2000; Gritli-Linde et al. 2002; Seidel et al. 2010). In the Evc2 mutant mice, we also detected decreased numbers of dental epithelial stem cells. A logical speculation is that decreased Hedgehog signaling in the dental epithelium leads to delayed ameloblast differentiation and hypomorphic enamel formation and that specific deletion of Evc2 in dental epithelium would phenocopy the tooth abnormalities in Evc2 mutant mice. In contrast, results from our studies demonstrated that a dental epithelial-specific deletion of Evc2 does not lead to overt tooth abnormalities. This is likely due to the fact that the remaining Hedgehog signaling in the Evc2 mutant dental epithelium is still sufficient to maintain dental epithelial stem cells and to drive ameloblast differentiation.
In Evc2 mutant mice, associated with delayed ameloblast differentiation, we also detected delayed odontoblast differentiation. Since ameloblast differentiation depends on the dentin secreted by odontoblasts (Thesleff and Hurmerinta 1981), the delayed ameloblast differentiation in Evc2 mutant mice likely results from delayed odontoblast differentiation. This speculation is also supported by our observation that Evc2 fx/fx;Wnt1-Cre mice phenocopy the tooth abnormalities in Evc2 mutant mice (Fig. 5A, C, D).
Evc2 mutants apparently feature short incisors compared with those of controls, which can be detected at as early as E15.5. It is possible that at cup stage or earlier, compromised Hedgehog signaling in the tooth mesenchyme leads to this small incisor germ. However, currently we are not able to rule out the possibility that Evc2 loss of function leads to abnormal Shh expression at early stages. During minimization stages, our analysis indicated no apparent differences in cell proliferation and cell death in Evc2 mutant incisors. The enlarged TA zones observed in dental mesenchyme in Evc2 global mutant mice represented delayed transition into the postmitotic stages of preodontoblasts. A recent report indicated that Hedgehog ligands secreted from neural bundles support the maintenance of dental mesenchymal stem cells (Zhao et al. 2014), which give rise to odontoblasts in adult mouse incisors. Similarly, in our studies, we detected fewer Gli1-lacZ–labeled cells in the dental mesenchyme. Limited availability of the dental mesenchymal stem cells may account for delayed odontoblast differentiation and thus the shorter incisor length, since no apparent differential cell proliferation and apoptosis have been detected in Evc2 mutant incisors.
In this study, we used mouse incisors as a model to investigate the pathophysiologic mechanism leading to hypomorphic enamel formation in EvC patients. Our in vivo data from genetic studies are summarized in Figure 5E. Compared with controls, Evc2 mutations lead to a limited number of dental mesenchymal stem cells, in association with delayed odontoblast differentiation. The underlying mechanism relating these 2 phenotypes may be an interesting topic for future study. Subsequently, delayed odontoblast differentiation leads to reduced deposition of dentin (Fig. 5E, green arrow), which may result in delayed ameloblast differentiation (Fig. 5E, yellow arrow) and subsequent hypomorphic enamel formation (Fig. 5E, pink arrow).
Author Contributions
H. Zhang, Y. Mochida, contributed to conception, design, and data analysis, drafted and critically revised the manuscript; H. Takeda, T. Tsuji, T. Kunieda, contributed to data analysis, critically revised the manuscript; N. Kamiya, Y. Mishina, contributed to conception, design, and data analysis, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material

Acknowledgments
We gratefully acknowledge Drs. Ray Manas, Scott Greg, Sudha Rajderkar, Ke’Ale Louie, and Crystal Collier for their contributions during earlier stages of this work; Drs. Satoru Hayano, Ce Shi, and Jingwen Yang for advice on research strategy; Drs. Satoru Hayano, Ce Shi, and Wanida Ono for critical reading of this manuscript; and Dr. Chunlin Qin for generous help on DSPP antibody.
Footnotes
A supplemental appendix to this article is available online.
This study is supported by the National Institutes of Health (R01DE019527 to Y. Mochida and R01DE020843 to Y. Mishina).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Badri MK, Zhang H, Ohyama Y, Venkitapathi S, Alamoudi A, Kamiya N, Takeda H, Ray M, Scott G, Tsuji T, et al. 2016. Expression of Evc2 in craniofacial tissues and craniofacial bone defects in Evc2 knockout mouse. Arch Oral Biol. 68:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badri MK, Zhang H, Ohyama Y, Venkitapathi S, Kamiya N, Takeda H, Ray M, Scott G, Tsuji T, Kunieda T, et al. 2016. Ellis van Creveld2 is required for postnatal craniofacial bone development. Anat Rec (Hoboken). 299(8):1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baujat G, Le Merrer M. 2007. Ellis-van Creveld syndrome. Orphanet J Rare Dis. 2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei M. 2009. Molecular genetics of ameloblast cell lineage. J Exp Zool B Mol Dev Evol. 312B(5): 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparrós-Martin JA, Valencia M, Reytor E, Pacheco M, Fernandez M, Perez-Aytes A, Gean E, Lapunzina P, Peters H, Goodship JA, et al. 2013. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum Mol Genet. 22(1):124–139. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. 2000. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 127(8):1671–1679. [DOI] [PubMed] [Google Scholar]
- Chen S, Gluhak-Heinrich J, Wang YH, Wu YM, Chuang HH, Chen L, Yuan GH, Dong J, Gay I, MacDougall M. 2009. Runx2, osx, and dspp in tooth development. J Dent Res. 88(10):904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. 1998. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 8(24):1323–1326. [DOI] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. 2000. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 127(22):4775–4785. [DOI] [PubMed] [Google Scholar]
- Dorn KV, Hughes CE, Rohatgi R. 2012. A smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. 23(4):823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A, Bei M, Maas R, Zhang XM, Linde A, McMahon AP. 2002. Shh signaling within the dental epithelium is necessary for cell proliferation, growth and polarization. Development. 129(23):5323–5337. [DOI] [PubMed] [Google Scholar]
- Jussila M, Thesleff I. 2012. Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb Perspect Biol. 4(4):a008425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Jia S, Jiang R. 2014. Molecular patterning of the mammalian dentition. Semin Cell Dev Biol. 25–26:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde A, Granstrom G. 1978. Odontoblast alkaline phosphatases and Ca2+ transport. J Biol Buccale. 6(4):293–308. [PubMed] [Google Scholar]
- McKusick VA, Egeland JA, Eldridge R, Krusen DE. 1964. Dwarfism in the Amish I. The Ellis-van Creveld syndrome. Bull Johns Hopkins Hosp. 115:306–336. [PubMed] [Google Scholar]
- Møinichen CB, Lyngstadaas SP, Risnes S. 1996. Morphological characteristics of mouse incisor enamel. J Anat. 189(Pt 2): 325–333. [PMC free article] [PubMed] [Google Scholar]
- Mostafa MI, Temtamy SA, el-Gammal MA, Mazen IM. 2005. Unusual pattern of inheritance and orodental changes in the Ellis-van Creveld syndrome. Genet Couns. 16(1):75–83. [PubMed] [Google Scholar]
- Murgiano L, Jagannathan V, Benazzi C, Bolcato M, Brunetti B, Muscatello LV, Dittmer K, Piffer C, Gentile A, Drögemüller C. 2014. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean Grey Cattle. PLoS One. 9(4):e94861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatomi M, Hovorakova M, Gritli-Linde A, Blair HJ, MacArthur K, Peterka M, Lesot H, Peterkova R, Ruiz-Perez VL, Goodship JA, et al. 2013. Evc regulates a symmetrical response to Shh signaling in molar development. J Dent Res. 92(3):222–228. [DOI] [PubMed] [Google Scholar]
- Qin C, Brunn JC, Baba O, Wygant JN, McIntyre BW, Butler WT. 2003. Dentin sialoprotein isoforms: detection and characterization of a high molecular weight dentin sialoprotein. Eur J Oral Sci. 111(3):235–242. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, et al. 2000. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 24(3):283–286. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ, et al. 2003. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 72(3):728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Ahn CP, Lyons D, Nee A, Ting K, Brownell I, Cao T, Carano RA, Curran T, Schober M, et al. 2010. Hedgehog signaling regulates the generation of ameloblast progenitors in the continuously growing mouse incisor. Development. 137(22):3753–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Takami M, Oguni T, Tsuji T, Yoneda K, Sato H, Ihara N, Itoh T, Kata SR, Mishina Y, et al. 2002. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc Natl Acad Sci U S A. 99(16):10549–10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thesleff I, Hurmerinta K. 1981. Tissue interactions in tooth development. Differentiation. 18(2):75–88. [DOI] [PubMed] [Google Scholar]
- Thomas BL, Liu JK, Rubenstein JL, Sharpe PT. 2000. Independent regulation of Dlx2 expression in the epithelium and mesenchyme of the first branchial arch. Development. 127(2):217–224. [DOI] [PubMed] [Google Scholar]
- Veena KM, Jagadishchandra H, Rao PK, Chatra L. 2011. Ellis-van Creveld syndrome in an Indian child: a case report. Imaging Sci Dent. 41(4):167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Takeda H, Tsuji T, Kamiya N, Rajderkar S, Louie KA, Collier C, Scott G, Ray M, Mochida Y, et al. 2015. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis. 53(9):612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Feng J, Seidel K, Shi S, Klein O, Sharpe P, Chai Y. 2014. Secretion of shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell. 14(2):160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials