

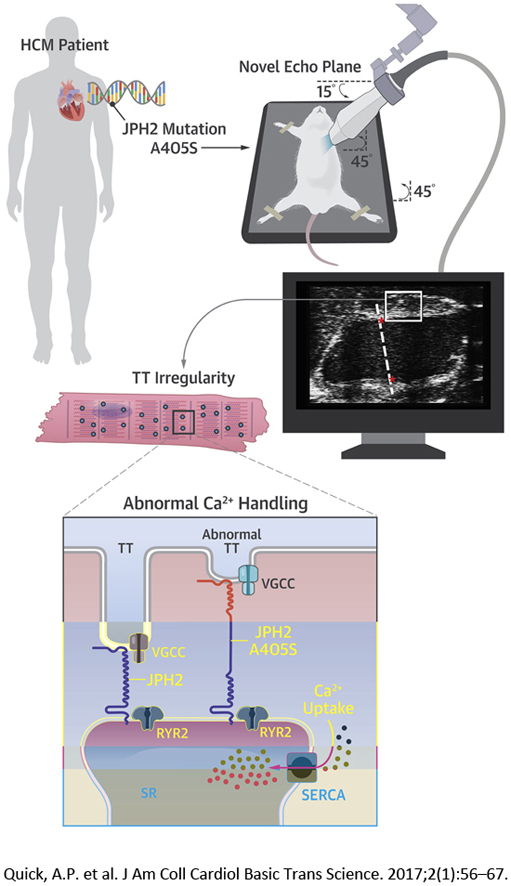
Visual Abstract
Key Words: calcium, hypertrophic cardiomyopathy, junctophilin-2, magnetic resonance imaging
Abbreviations and Acronyms: CMR, cardiac magnetic resonance imaging; HCM, hypertrophic cardiomyopathy; IQR, interquartile range; JPH2, junctophilin-2; LV, left ventricle/ventricular; PKI, pseudo-knockin; RyR2, ryanodine receptor type 2; SR, sarcoplasmic reticulum; TT, transverse tubule; WT, wild-type
Highlights
-
•
A JPH2 A405S mutation was found in a human HCM patient.
-
•
A novel echocardiographic imaging plane revealed septal hypertrophy in a mouse model bearing the human JPH2 mutation, thereby causally linking it to HCM pathogenesis.
-
•
This alpha helical JPH2 mutation resulted in decreased transverse tubule regularity and aberrant calcium handling in septal cardiomyocytes.
Summary
Junctophilin-2 (JPH2) is a structural calcium (Ca2+) handling protein, which approximates the cardiomyocyte transverse tubules (TTs) to the sarcoplasmic reticulum. This facilitates communication of the voltage-gated Ca2+ channel and the ryanodine receptor RyR2. A human patient with hypertrophic cardiomyopathy was positive for a JPH2 mutation substituting alanine-405—located within the alpha helix domain—with a serine (A405S). Using a novel mouse echocardiography plane, we found that mice bearing this JPH2 mutation developed increased subvalvular septal thickness. Cardiomyocytes from the septa of these mice displayed irregular TTs and abnormal Ca2+ handling including increased SERCA activity.
Hypertrophic cardiomyopathy (HCM) is the most common cause of sudden death in young athletes and a major cause of sudden cardiac death in the young (1). HCM is characterized by clinically unexplained left ventricular (LV) muscular thickening that can lead to diastolic dysfunction in the setting of preserved, or hyperdynamic, ejection fraction (2). HCM is also associated with varying degrees of myocyte disarray and interstitial fibrosis development, and conveys an increased risk of ventricular tachycardia and sudden cardiac arrest. Traditionally considered a genetic disease, HCM often arises from mutations in 1 or more sarcomeric genes, which encode proteins responsible for the myofilaments required for normal cardiac contraction. Despite the clear association between sarcomeric protein mutations and the HCM phenotype, controversy still exists over the molecular mechanism of HCM pathogenesis.
A growing body of evidence suggests that sarcomeric mutations may also perturb intracellular Ca2+-signaling or myofilament Ca2+-sensitivity in a prohypertrophic manner (3). Further, recent studies have linked Ca2+-regulatory and Ca2+-sensitive proteins to sarcomeric gene-negative HCM (4). This heterogeneity in both molecular substrate and genetic etiology is reflected in clinical phenotype heterogeneity. Indeed, varying morphologies of LV hypertrophy have been identified, which can cluster in families, and may be related to underlying genetic defects. Although some genotype–phenotype-based studies have linked various septal morphologies to the presence or absence of sarcomeric mutations (5), the molecular underpinnings of region-specific LV hypertrophy remain unexplored due to lack of robust experimental models.
Initiation and coordination of myofilament contraction is a major role of Ca2+ within the cardiac myocyte and is highly regulated. Opening of the voltage-gated L-type Ca2+ channel at the sarcolemma allows for an influx of extracellular Ca2+ across the cardiac dyad. This triggers Ca2+ release from the sarcoplasmic reticulum (SR) via RYR2-encoded ryanodine receptor type 2 (RyR2) in a process known as Ca2+-induced Ca2+ release (6). Junctophilin-2 (JPH2) is a component of the junctional membrane complex, which physically and functionally couples the sarcolemma and the SR for efficient Ca2+-induced Ca2+ release and excitation–contraction coupling 7, 8. In addition to a structural role, JPH2 negatively regulates gating of RyR2 and loss of wild-type (WT) JPH2 expression is associated with cardiomyocyte hypertrophy, progressive heart failure, and deranged Ca2+ signaling through SR Ca2+ leak 9, 10.
These prior studies have suggested that JPH2 plays an important regulatory role in Ca2+ handling and can play a direct role in the pathogenesis of cardiac remodeling. This possibility is supported clinically by the identification of a small number of individuals with HCM who have mutations in JPH2, and do not host mutations in one of the canonical sarcomeric genes (11). Despite these early studies, the mechanism of JPH2-mediated hypertrophy remains unexplored, and there have been no JPH2 murine models that have recapitulated clinical HCM. Further, given recent advances genetic analysis platforms and the identification of significant variation in JPH2, and other HCM-associated genes, there is a need for experimental models that reflect genetic disease (12).
To this end, we report a novel JPH2 mutation identified in a proband with basal septal hypertrophy who was sarcomeric mutation negative. Novel advanced cardiac imaging protocols using specialized echocardiography as well as cardiac magnetic resonance imaging (CMR) of a murine model demonstrated progressive LV and septal thickening reflective of the proband’s septal morphology. Histological analysis confirmed cellular hallmarks of HCM. Studies at the cellular level revealed unperturbed excitation–contraction coupling, but a reduced level of transverse tubule (TT) organization in the JPH2 mutant transgenic mouse model. Thus, a novel JPH2 mutation was causally linked to a distinct phenotype also found in the human mutation carrier, consistent with heterogeneity in the molecular basis of HCM on the basis of specific gene defects.
Methods
Additional detailed methods are available in the Supplemental Appendix.
Genetic analysis of a large cohort of subjects with HCM
Following receipt of informed consent and enrollment of the subjects into this institutional review board approved–approved study, 778 patients were evaluated in the HCM Clinic at the Mayo Clinic (Rochester, Minnesota). Clinical evaluation of the probands comprising this cohort was conducted as previously described, and each patient met the clinical diagnostic criteria for HCM (13).
Mouse model of JPH2 variant
All animal studies were done in accordance with protocols pre-approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Pseudo-knockin (PKI) mice heterozygous for the mutant JPH2 variant were developed as described (9). Transthoracic echocardiography of anesthetized mice was performed (14). A novel echocardiography plane was developed to better image the base of the interventricular septum and the LV outflow tract. In addition, mice underwent CMR in a Bruker BioSpin 9.4-T magnet (Bruker BioSpin, Karlsruhe, Germany). ParaVision 5.1 (Bruker, Billerica, Massachusetts) was used to collect bright-blood fast low-angle shot cine images (15).
Statistical analyses
For analyses that evaluated 1 sample per subject (mouse model echocardiography and CMR, histology of interventricular septum and free wall diameter), WT and mutant JPH2 mice were compared using Mann-Whitney U test analysis or Kruskal-Wallis test when comparing more than 2 groups. In cases of repeated measures of the same subject (calcium studies, TT power, histological assessment of areas of interventricular septum and ventricular free wall, SERCA2a activation, hemodynamic studies), WT and mutant mice were compared using generalized estimating equations accounting for correlated outcomes. Analyses were performed using SPSS version 24 (IBM, Armonk, New York).
Results
Identification of the A405S mutation in JPH2 in a HCM patient
Comprehensive genetic analysis of the JPH2 gene in a large cohort of 778 HCM patients revealed an alanine to serine point mutation at residue 405 (JPH2-A405S) in single proband with HCM (Figure 1A). There was no family history of HCM or sudden death in the immediate family including all first-degree relatives. Both parents could not be reached or declined consent for genotyping. This mutation was absent in 159,358 reference alleles. Moreover, mutations in 9 canonical sarcomeric genes associated with HCM (Supplemental Appendix), as well as PRKAG2, GLA, and LAMP2 genes, were excluded in this patient. The A405S mutation is localized to the alpha helical domain of JPH2, which is thought to serve as a molecular tether and spring to maintain critical cardiac dyad distance (Figure 1B) (16). This residue and the surrounding protein domain are highly conserved across species (Figure 1C). The mouse residue corresponding to human A405 is the alanine at position 399, thus, the equivalent mouse genome mutation is A399S in JPH2.
Figure 1.

JPH2 Mutation A405S Occurred De Novo and Affects a Conserved Residue
(A) Pedigree of HCM proband with JPH2 mutation A405S. (B) Cartoon showing that the JPH2 mutation A405S is localized within the alpha helix domain. (C) Alignment showing conservation of JPH2 sequences across species, the mutant residue is marked by the red box. HCM = hypertrophic cardiomyopathy; JPH2 = junctophilin-2; LVOTO = left ventricular outflow tract obstruction; MORN = membrane occupation and recognition nexus; PM = plasma membrane; RyR2 = ryanodine receptor type-2; SR = sarcoplasmic reticulum; TMD = transmembrane domain; VGCC = voltage-gated calcium channel.
Clinical imaging studies reveal distinct HCM phenotype in proband
The proband carrying the JPH2 mutation is a Caucasian male diagnosed with HCM at 16 years of age after presenting with shortness of breath, chest pain, and dizziness. His electrocardiogram revealed sinus rhythm with left anterior fascicular block with ST-segment and T-wave abnormalities and a QTc of ∼510 ms (Supplemental Figure 1). Transthoracic echocardiography demonstrated basal septal hypertrophy with an interventricular septal thickness of 23 mm (Figures 2A and 2B). Dynamic left ventricular outflow tract obstruction with systolic anterior movement of the mitral valve was noted at diagnosis which progressed to a resting gradient of 41 mm Hg with Valsalva, which increased to 88 mm Hg with amyl nitrate (Figures 2C and 2D).
Figure 2.

Patients With JPH2 Mutation A405S Develop HCM With LVOTO
(A and B) Echocardiography of a patient with HCM JPH2 mutation A405S showing asymmetrical septal hypertrophy denoted by a red arrow, and (C) LVOTO evidenced by (D) color Doppler flow mapping. (E to H) Bright-blood CMR depicting left ventricular basal septal hypertrophy (red arrow) in diastole (E) and (F) systole, with corresponding transverse CMR in diastole (G) and systole (H). Scale bars = 2.0 cm. CMR = cardiac magnetic resonance imaging; other abbreviations as in Figure 1.
Bright-blood CMR demonstrated asymmetric LV hypertrophy, which was most pronounced in the basal septum with dynamic subaortic obstruction (Figures 2E and 2F). Functional CMR with gadolinium demonstrated delayed myocardial enhancement indicative of fibrosis (Figures 2G and 2H). Due to the pronounced septal hypertrophy, systolic anterior movement of the mitral valve leaflet, and symptoms of severe exertional dyspnea, chest pain, and dizziness, the proband ultimately underwent surgical myectomy for symptomatic management.
Development of a mouse model of the HCM-associated JPH2 mutation
To assess whether the JPH2 mutation A405S in the patient with HCM is causally linked to this cardiac disorder, we generated a mouse model of the corresponding mutation, A399S, in mice. First, transgenic mice were generated expressing transgenic A399S mutant or WT JPH2 in the heart only, which increased the total cardiac JPH2 levels (Supplemental Figure 2). Therefore, the transgenic mice were crossed with cardiac-specific JPH2 knockdown mice to generate PKI mice that express stoichiometric JPH2 levels similar to nontransgenic (control) mice (Supplemental Appendix, and Beavers et al. [9]). Western blot analysis revealed that cardiac JPH2 levels were similar in WT-PKI (WT from hereon in paper) and A399S-PKI (A399S) mice (Supplemental Figure 2).
To determine whether A399S mice developed signs of HCM, echocardiographic imaging was performed using standard short-axis B-mode measurements at the level of the LV papillary muscles 14, 17. There was no significant change in the LV outer diastolic diameter in A399S mice (median 5.93 mm, interquartile range [IQR]: 5.70 to 6.14 mm, n = 10) compared with WT mice (median 5.83 mm, IQR: 5.79 to 5.94 mm, n = 10; p = 0.39) (Figures 3A and 3B). There were also no significant differences in LV posterior wall thickness in systole and diastole, as well as other structural indices (Supplemental Table 1). Moreover, there were no significant differences in cardiac contractility using M-mode measurements. The ejection fraction was not significantly reduced in A399S (median 52%, IQR: 48% to 56%, n = 10) compared with WT (median 55%, IQR: 54% to 56%; p = 0.165).
Figure 3.

Standard Echocardiographic Analysis of A399S Mice
(A) Representative echocardiograms of traditional mouse SAX mid-level B-modes; asterisks mark papillary muscles with corresponding standard M-mode tracings. Quantifications revealed no differences in (B) left ventricular outer diastolic diameter (LVOD;d) or (C) ejection fraction (EF) between A399S and wild-type (WT) mice. Circles in box plots represent outliers. IVS = intraventricular septum; LV = left ventricle; N = number of animals; SAX = short axis.
Novel echocardiographic imaging plane reveals asymmetrical septal hypertrophy in A399S mice
Given the inadequate basal septum visualization by traditional M-mode measurements and the prominent basal septal hypertrophy in the proband carrying the JPH2 mutation, a novel echocardiographic imaging plane was developed to more thoroughly interrogate the LV outflow tract and basal interventricular septal region in the mouse (Figure 4). The imaging plane is analogous to the human parasternal long-axis view and clearly visualizes the basal portion of the interventricular septum and the LV outflow tract. Application of this imaging plane demonstrated a decrease in end-diastolic LV chamber area (Figures 4A and 4B). Additional analysis revealed that this is mainly attributable to an increased end-diastolic basal interventricular septal thickness, which was a median 1.22 mm (IQR: 1.16 to 1.22 mm, n = 7) in A399S mice compared with median 0.89 mm (IQR: 0.87 to 0.98 mm, n = 10) in controls (p < 0.001) (Figure 4C). This ∼37% increase in local septal thickness is a clear pathognomonic feature of HCM in patients.
Figure 4.

Novel Echocardiographic Imaging Plane Reveals Asymmetrical Interventricular Septal Hypertrophy in A399S Mice
(A) Representative modified long-axis (mLAX) echocardiograms were used to visualize the LV and IVS; red stars mark papillary regions. White boxes indicate region of zoom shown in panels below. Dotted lines delineate the traditional mouse echocardiography plane, for comparison. (B) Quantification showing significantly reduced LV chamber area in diastole (endoArea;d) in A399S mice compared with WT and (C) increased maximal interventricular septum diameter in diastole (IVSD;dmax) in A399S mice compared with WT. Circle in box plot represents outlier. *p < 0.05, ***p < 0.001. Ao = aorta; other abbreviations as in Figure 3.
CMR confirms subvalvular septal hypertrophy in A399S mice
To validate the novel mouse echocardiographic imaging plane, CMR was conducted in an independent cohort of A399S and WT control mice. The long-axis 4-chamber view revealed evidence for LV hypertrophy (Figure 5A). Quantification of the LV end-diastolic mass demonstrated enlarged LV end-diastolic mass of median 0.091 g (IQR: 0.085 to 0.099 g, n = 10) in A399S mice compared with 0.073 g (IQR: 0.069 to 0.076 g, n = 9) in WT controls (p = 0.013) (Figure 5B). Quantification revealed significantly higher septal diastolic diameters in A399S (median 1.09 mm, IQR: 1.02 to 1.21 mm, n = 11) compared with control mice (median 0.81 mm, IQR: 0.71 to 0.95 mm, n = 9; p < 0.001) (Figure 5C). This increase in septal size was not due to increased animal size because body weights were unchanged between A399S (median 29.7 g, IQR: 27.7 to 32.7 g) and WT mice (median 30.8 g, IQR: 30.6 to 32.6 g) (Supplemental Table 2).
Figure 5.

Confirmation of IVS Hypertrophy in A399S-PKI Mice Using CMR
(A) Representative long-axis view (LAX) magnetic resonance images demonstrating IVS hypertrophy. Quantifications of (B) left ventricular end-diastolic mass (LV Mass) and (C) maximal IVSD;dmax. Circles in box plot represent outliers. *p < 0.05, ***p < 0.001. FW = free wall; PKI = pseudo-knockin; RV = right ventricle; other abbreviations as in Figures 2, 3, and 4.
JPH2-A399S mice demonstrate diastolic dysfunction and strain
A hallmark of hypertrophic cardiomyopathy is diastolic dysfunction in the setting of intact systolic function. Although echocardiography can be used to indirectly measure diastolic function in humans, these parameters have not been translated into murine studies. Therefore, we used Langendorff perfusion to obtain hemodynamic pressure–volume relationships of hearts from A399S and WT control mice. There was no overall difference in negative dP/dt in A399S (mean −938 ± 166 mm Hg/s, n = 60 samples in 3 mice) compared with WT controls (mean −1,056 ± 397 mm Hg/s, n = 70 samples in 3 mice; p = 0.78) (Supplemental Figure 3), although 1 WT mouse had very decreased negative dP/dt (mean for 20 samples −2,022 ± 191 mm Hg/s). There was a large, but not statistically significant, increase in LV end-diastolic pressure in A399S mice (mean 21.0 ± 6.2 mm Hg, n = 45 samples in 3 mice) compared with WT mice (mean 5.7 ± 1.2 mm Hg, n = 45 samples in 3 mice; p = 0.016).
Cellular remodeling consistent with HCM in A399S mice
Echocardiography and CMR demonstrated morphological aberrations at the macroscopic level in A399S mice, consistent with HCM. We also performed histological analysis to correlate changes at the cellular level with macroscopic disease manifestations. Hematoxylin and eosin staining of heart sections confirmed the increase in interventricular septal diameter seen using imaging modalities (Figure 6A). The interventricular septal and free wall diameter measurements confirmed a significant increase in septal size relative to the free wall (median 1.45 mm, IQR: 1.44 to 1.75 mm, n = 3) compared with WT controls (median 0.96 mm, IQR: 0.72 to 1.00 mm, n = 3; p = 0.05) (Figures 6B and 6C). We assessed cell size in order to better understand the underlying septal thickening in A399S mice. Sarcolemma staining with wheat germ agglutinin revealed an increased average cross-sectional area of the septal myocytes from A399S mice (475 ± 80 μm2, n = 9 total samples from 3 mice) compared with myocytes from WT mice (272 ± 48 μm2, n = 9 total samples from 3 mice; p < 0.001). Whereas, average myocyte area from the LV free wall of A399S hearts remained unchanged (279 ± 41 μm2) compared with WT free wall myocyte area (293 ± 17 μm2; p = 0.509) (Figures 6D and 6F). These findings suggest that thickening of the interventricular septum is caused by localized hypertrophy of cardiomyocytes. Finally, analysis of magnified images of the interventricular septum (Figure 6G) revealed a greatly increased degree of myocardial fiber disarray and interstitial fibrosis (mean 8.9%, 95% confidence interval: 8.6% to 9.3%) in A399S hearts compared with WT hearts (mean 2.3%, 95% confidence interval 0.9% to 3.7%; p < 0.001) (Figure 6H). This pronounced level of myofiber disarray and fibrosis is often seen in the context of HCM.
Figure 6.

Cardiomyocyte Hypertrophy and Disarray in IVS of A399S-PKI Mice
(A) Long-axis hematoxylin and eosin–stained paraffin sections. (B) Box plots showing median IVS diameter and (C) LV free wall diameter (FWD). (D) Representative images of wheat germ agglutinin–stained cardiomyocyte cross sections at 40× magnification from the IVS and LV FW with (E and F) quantification in bar graphs. (G) Representative 40× magnifications of Masson’s Trichrome–stained paraffin sections of cardiomyocytes showing disarray and interstitial fibrosis with (H) quantification in bar graph. For B and C, n = number animals. For E, F, and H, n = number images, and number in parentheses = animals. ***p < 0.001. IVSD = interventricular septum diameter; other abbreviations as in Figures 3 and 5.
Calcium handling and TT changes in A399S-PKI mice
To gain more insight into the possible mechanisms by which a JPH2 mutation causes HCM, we studied intracellular Ca2+ handling in ventricular myocytes isolated from WT and A399S mice. Isolated myocytes were paced at 1 Hz to obtain steady-state Ca2+ cycling (Figure 7A). The amplitude of the Ca2+ transients in myocytes from A399S mice (mean 1.95 ± 0.23, 24 total samples from 3 mice) was similar to those from WT mice (mean 1.75 ± 0.12, 26 total samples from 3 mice; p = 0.455) (Figure 7B). The SR Ca2+ content assessed using the caffeine dump protocol revealed no significant differences between myocytes from A399S mice (3.68 ± 0.41, n = 26 total samples from 3 mice) and WT mice (3.84 ± 0.97; n = 24 total samples from 3 mice; p = 0.88). On the other hand, there was a significantly higher SERCA2a activity consistent with faster Ca2+ reuptake into the SR in A399S myocytes, with a relative increase of SERCA2a activity in A399S cells compared with WT cells of mean 28 ± 10% (p = 0.006) (Figure 7C).
Figure 7.

Unaltered SR Calcium Handling, But Reduced TT Power, in A399S-PKI Mice
(A) Representative fluorescent tracings during 1-Hz pacing showing Ca2+ transient amplitudes and summary results (B). The SERCA2a activity, defined as the inverse of the decay time constant of the caffeine-induced SR Ca2+ dump, was increased in A399S-PKI mice (C). Representative confocal line-scan images of individual Ca2+ sparks (D) and summary results (E). Di-8-ANEPPS staining of ventricular myocytes isolated from WT and A399S mice showing a reduction in T-tubule (TT) power (F) and summary results (G). N = cells, and number in parentheses = animals. **p < 0.01. Abbreviations as in Figures 1, 3, and 5.
Next, the incidence of spontaneous SR Ca2+ release events known as Ca2+ sparks was assessed following the 1-Hz pacing train (Figure 7D). Quantification of the frequency of Ca2+ sparks revealed no significant differences comparing A399S mice (6.73 ± 0.84 sparks/100 μm/s, n = 20 total samples in 3 mice) and WT (6.13 ± 0.46 sparks/100 μm/s, n = 22 total samples in 3 mice; p = 0.54) mice (Figure 7E). Finally, we aimed to determine whether the A399S mutation in JPH2 altered the organization of TTs in isolated cardiomyocytes (Figure 7F). Compared with myocytes from WT mice, the TT power—a measure of TT regularity—was significantly reduced in cells from A399S mice (mean 15 ± 9% reduction; p = 0.097) (Figure 7G). These findings suggest that the genetic JPH2 variant might affect TT organization, which in turn might promote prohypertrophic growth in the ventricular septum.
Discussion
Genetic basis of hypertrophic cardiomyopathy
HCM is a common genetic form of cardiomyopathy characterized by asymmetric cardiac enlargement with a reduction in ventricular chamber dimensions, hypertrophy of the interventricular septum, and diastolic dysfunction. Although more than 18 genes have been linked to inherited HCM, the most common defects are mutations in sarcomeric genes (18). Non-sarcomeric genes have also been linked to genetic HCM, including some genes involved in intracellular Ca2+ handling such as calreticulin 3 (CALR3), phospholamban (PLN), and also junctophilin-2 (JPH2) 9, 11.
Because JPH2 mutations are a rare cause of HCM, and benign polymorphism has been reported in healthy individuals, we sought to establish whether the JPH2 variant A405S identified in the proband was causally linked to the disease phenotype. Transgenic mouse models have been used extensively to establish gene–disease causality and to gain deeper insights into the mechanistic pathways underlying cardiomyopathies (19). Therefore, we generated a novel HCM model caused by a JPH2 variant. Our studies revealed that A399S mice exhibited various features typically associated with HCM, including a hypertrophic interventricular septum, an increased LV mass, asymmetric LV hypertrophy with a reduced diastolic filling, and myofiber disarray. Because these findings in the mouse model mimicked clinical findings in the proband, a causal relationship between the JPH2 variant and HCM was established.
Highly sensitive cardiac imaging to uncover cardiomyopathy in a mouse model
Clinically, HCM is diagnosed on the basis of echocardiography parameters of LV hypertrophy in the absence of a clinically identifiable cause. However, due to the small size and rapid breathing of rodents, the detection of subtle asymmetric remodeling in mouse models is very challenging. Further, given the variable nature of the septal hypertrophy, traditional mouse echocardiography consisting of papillary-level transverse B-modes and corresponding M-modes may underappreciate certain configurations. Traditional long-axis views of the mouse heart provide poor visualization of the intraventricular septum, because the LV outflow tract is not well seen. Due to the ease of use of ultrasound and the relatively low cost, we found that devising an echocardiographic plane similar to the parasternal long axis in humans would prove beneficial in septal visualization in mice and thus improve identification of HCM. Indeed, this echocardiographic view of the mouse heart allowed us to see the full width of the septum and the LV outflow tract, and detect regional septal hypertrophy in the A399S mice (Figure 4). This human-like observation is quite unique among mouse models of HCM. It would be interesting to assess whether our novel imaging approach may also uncover local septal defects in other mouse models of HCM in which traditional echo protocols failed to uncover pathological findings (20).
CMR is the gold standard clinical test for cardiomyopathic disease that may appear normal on echocardiography, and can be used to establish a diagnosis in these false-negative cases (21). Further, CMR offers the possibility of more clearly defining cardiac anatomy and evaluating functional changes in myocardial contractility, stiffness, and strain, among other parameters, that are superior to traditional echocardiography. Here, we demonstrated that this imaging modality correlates well with echocardiography in the evaluation of the A399S mouse (Figure 5). Like the human patient, the A399S mice maintained normal systolic function but developed signs of diastolic dysfunction (22). These features of the myocardium are associated with clinical diastolic dysfunction and sudden cardiac death, especially among young HCM patients 23, 24.
New insights into HCM pathogenesis obtained from our JPH2 mutant mouse model
Because the first HCM-associated genes were in genes encoding sarcomeric proteins, a final common pathway was proposed in which sarcomere disruption was the key pathogenic mechanism (25). Sarcomeric gene mutations disrupt the fine balance between intracellular Ca2+ cycling and mechanical force generation by the myofilaments. For example, many HCM-linked mutations in sarcomeric genes sensitize the myofilaments to greater Ca2+ adherence, thereby delaying SR Ca2+ reuptake and prolonging mechanical relaxation (26). In a recent study, Davis et al. (26) proposed a new tension-based model to predict which sarcomeric mutations cause HCM as compared with dilated cardiomyopathy. This model predicts that with HCM mutations, increased myofilament Ca2+ sensitization leads to a lower cytosolic Ca2+ requirement. Thus, Ca2+ leak typically seen in dilated cardiomyopathy may not necessarily occur in HCM pathogenesis.
The mechanism underlying hypertrophic remodeling associated with JPH2 mutations has remained elusive in part due to the lack of available animal models. We found that the A399S mutation in JPH2 affects the organization of the TT network, whereas there were no overt changes in systolic and diastolic SR Ca2+ handling. These results obtained in myocytes isolated from A399S mutant mice are corroborated by the previously mentioned Davis et al. (26) HCM model and correlated with the echocardiographic findings that ejection fractions were not altered in the mutant mice. However, contrary to the HCM model presented in the preceding text, SERCA2a activity was surprisingly enhanced in our mutants. This may be due to the specific nonsarcomeric nature of the A399S JPH2 mutation in contrast to the sarcomeric mutation model presented by Davis et al. (26). Because JPH2 is critical for TT maintenance, it is not surprising that a key cellular feature of the A399S mice is reduced TT regularity. This change in TTs may precipitate downstream events leading to HCM pathogenesis. The reduction in TT density may over time affect SR Ca2+ handling and affect excitation-contraction coupling, because prior studies have correlated a loss of TTs with the development of heart failure (27). Additional studies are required to determine whether and how JPH2 mutations linked to human HCM promote hypertrophic remodeling.
Conclusions
Our translational studies revealed that a novel mutation in JPH2— identified in a patient with HCM—also caused clear, unique features of HCM in the equivalent mutant mouse model, thereby validating a causal relationship between the JPH2 variants and HCM. The affected gene (JPH2) is not one of the canonical sarcomeric genes, and therefore underscores the heterogeneity in the molecular basis of this disease. Structural evaluation of cardiac function in the mutant mice required the development of novel imaging planes and techniques for mouse models of heritable cardiomyopathies will facilitate future rodent studies of the pathogenicity of novel gene mutants linked to human disease. These imaging approaches enabled us to uncover basal septal hypertrophy and diastolic dysfunction, signs of HCM that would have been likely missed on traditional echocardiographic approached used in mice. Future studies are warranted to determine what aspects of abnormal Ca2+ handling are amenable to therapeutic intervention to normalize cardiac function in animals or patients with JPH2-linked HCM.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: HCM is one of the most common heritable cardiovascular diseases and a common cause of sudden cardiac death in young individuals. The disease is often identified by imaging studies that show thickened walls of the heart muscle, usually most prominently in the ventricular septum.
TRANSLATIONAL OUTLOOK 1: The validation of a causal relationship between a mutation in the junctophilin-2 gene—a gene other than the typical sarcomeric genes linked to hypertrophic cardiomyopathy—underscores heterogeneity in the molecular basis of this disease.
TRANSLATIONAL OUTLOOK 2: The development of novel imaging planes and techniques for mouse models of heritable cardiomyopathies will facilitate future rodent studies of the pathogenicity of novel gene mutants linked to human disease.
Acknowledgments
The Genetically Engineered Mouse (GEM) Core at Baylor College of Medicine developed the mice used for breeding the PKI mice in this study. Mouse echocardiography was performed at the Mouse Phenotyping Core of Baylor College of Medicine, and mouse cardiac magnetic resonance imaging (CMR) was completed at the Small Animal Imaging Facility at Texas Children’s Feigin Center. CMR analysis was completed at Texas Children’s 3D analysis lab.
Footnotes
Supported by grants from the National Institute of Health and American Heart Association. Ms. Quick was funded by NIH training grant T32-HL007676, and American Heart Association 14PRE20490083. Dr. Landstrom is supported by a grant from the Pediatric and Congenital Electrophysiology Society. Dr. Wehrens is supported National Institutes of Health grants R01-HL089598, R01-HL091947, R01-HL117641, and R41-HL129570, and American Heart Association grant 13EIA14560061. Dr. Ackerman is a consultant for Biotronik, Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia, St. Jude Medical, and Transgenomic; and receives royalties from Transgenomic. Intellectual property derived from Dr. Ackerman’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals, now Transgenomic). Dr. Wehrens is a founding partner of Elex Biotech, a start-up company that developed drug molecules that target ryanodine receptors for the treatment of cardiac arrhythmia disorders. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Go A.S., Mozaffarian D., Roger V.L. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maron B.J. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Baudenbacher F., Schober T., Pinto J.R. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landstrom A.P., Ackerman M.J. Beyond the cardiac myofilament: hypertrophic cardiomyopathy- associated mutations in genes that encode calcium-handling proteins. Curr Mol Med. 2012;12:507–518. doi: 10.2174/156652412800620020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binder J., Ommen S.R., Gersh B.J. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc. 2006;81:459–467. doi: 10.4065/81.4.459. [DOI] [PubMed] [Google Scholar]
- 6.Landstrom A.P., Beavers D.L., Wehrens X.H. The junctophilin family of proteins: from bench to bedside. Trends Mol Med. 2014;20:353–362. doi: 10.1016/j.molmed.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 8.Beavers D.L., Landstrom A.P., Chiang D.Y., Wehrens X.H. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res. 2014;103:198–205. doi: 10.1093/cvr/cvu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beavers D.L., Wang W., Ather S. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010–2019. doi: 10.1016/j.jacc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landstrom A.P., Kellen C.A., Dixit S.S. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail. 2011;4:214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landstrom A.P., Weisleder N., Batalden K.B. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapplinger J.D., Landstrom A.P., Bos J.M., Salisbury B.A., Callis T.E., Ackerman M.J. Distinguishing hypertrophic cardiomyopathy-associated mutations from background genetic noise. J Cardiovasc Transl Res. 2014;7:347–361. doi: 10.1007/s12265-014-9542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landstrom A.P., Parvatiyar M.S., Pinto J.R. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J Mol Cell Cardiol. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Respress J.L., Wehrens X.H. Transthoracic echocardiography in mice. J Vis Exp. 2010:1738. doi: 10.3791/1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heijman E., de Graaf W., Niessen P. Comparison between prospective and retrospective triggering for mouse cardiac MRI. NMR Biomed. 2007;20:439–447. doi: 10.1002/nbm.1110. [DOI] [PubMed] [Google Scholar]
- 16.Garbino A., van Oort R.J., Dixit S.S., Landstrom A.P., Ackerman M.J., Wehrens X.H. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Oort R.J., Respress J.L., Li N. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension. 2010;55:932–938. doi: 10.1161/HYPERTENSIONAHA.109.146449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidman C.E., Seidman J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–750. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCauley M.D., Wehrens X.H. Animal models of arrhythmogenic cardiomyopathy. Dis Model Mech. 2009;2:563–570. doi: 10.1242/dmm.002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bang M.L. Animal models of congenital cardiomyopathies associated with mutations in Z-line proteins. J Cell Physiol. 2017;232:38–52. doi: 10.1002/jcp.25424. [DOI] [PubMed] [Google Scholar]
- 21.Moon J.C., Fisher N.G., McKenna W.J., Pennell D.J. Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart. 2004;90:645–649. doi: 10.1136/hrt.2003.014969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gwathmey J.K., Warren S.E., Briggs G.M. Diastolic dysfunction in hypertrophic cardiomyopathy. Effect on active force generation during systole. J Clin Invest. 1991;87:1023–1031. doi: 10.1172/JCI115061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menon S.C., Eidem B.W., Dearani J.A., Ommen S.R., Ackerman M.J., Miller D. Diastolic dysfunction and its histopathological correlation in obstructive hypertrophic cardiomyopathy in children and adolescents. J Am Soc Echocardiogr. 2009;22:1327–1334. doi: 10.1016/j.echo.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 24.Varnava A.M., Elliott P.M., Mahon N., Davies M.J., McKenna W.J. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. 2001;88:275–279. doi: 10.1016/s0002-9149(01)01640-x. [DOI] [PubMed] [Google Scholar]
- 25.Towbin J.A., Bowles N.E. The failing heart. Nature. 2002;415:227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 26.Davis J., Davis L.C., Correll R.N. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell. 2016;165:1147–1159. doi: 10.1016/j.cell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Oort R.J., Garbino A., Wang W. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.