

Real-time NMR, EM, and native MS studies revealed intermediates and parallel pathways in the assembly of a dodecameric peptidase.
Keywords: Structural Biology, Nuclear magnetic resonance, Electron microscopy, mass spectrometry, Real-time structural study, self-assembly, quaternary structure
Abstract
The spontaneous formation of biological higher-order structures from smaller building blocks, called self-assembly, is a fundamental attribute of life. Although the protein self-assembly is a time-dependent process that occurs at the molecular level, its current understanding originates either from static structures of trapped intermediates or from modeling. Nuclear magnetic resonance (NMR) spectroscopy has the unique ability to monitor structural changes in real time; however, its size limitation and time-resolution constraints remain a challenge when studying the self-assembly of large biological particles. We report the application of methyl-specific isotopic labeling combined with relaxation-optimized NMR spectroscopy to overcome both size- and time-scale limitations. We report for the first time the self-assembly process of a half-megadalton protein complex that was monitored at the structural level, including the characterization of intermediate states, using a mutagenesis-free strategy. NMR was used to obtain individual kinetics data on the different transient intermediates and the formation of final native particle. In addition, complementary time-resolved electron microscopy and native mass spectrometry were used to characterize the low-resolution structures of oligomerization intermediates.
INTRODUCTION
Self-assembly is an essential process whereby nature creates biomolecular machines, such as large proteases, chaperonins, or unfoldases. Molecular self-assembly reflects information encoded in protein three-dimensional (3D) structure as a combination of noncovalent intermolecular interactions (1). A repetitive protein-protein association process, which may involve conformational rearrangement of building blocks, is required to assemble functional machinery (2). Tracking the conformational changes and oligomerization states under off-equilibrium conditions is challenging because of the added time dimension and the low abundance of the different intermediate states. Only a few experimental tools have been developed for time-resolved structural studies. Nuclear magnetic resonance (NMR) spectroscopy can probe conformational changes in real time, but its use in self-assembly studies has been limited by the size of the particles (3–5). Electron microscopy (EM) is now able to resolve the 3D structure of large biomolecules at quasi-atomic resolution (6, 7), but time resolution is still only mainly possible in silico and if the different states that are coexisting in solution are present in a discreet and limited number (8). Native mass spectrometry (MS) can determine the mass of intact protein complexes and their precise stoichiometry (9) but provides no direct atomic information.
This work combines these techniques to investigate the oligomerization process of a half-megadalton homododecameric tetrahedral (TET) aminopeptidase. TET-like aminopeptidases participate to maintain cellular homeostasis by hydrolyzing small peptides into amino acids in Archaea and bacteria (10). The functional TET2 quaternary structure is composed of six dimers forming the edges of a hollow slightly truncated tetrahedron (Fig. 1A and fig. S1A) (11). Considering the large size of a TET2 particle, the NMR study of its assembly in solution is challenging. This is due to the fast relaxation of NMR signals, typical of such slow overall-tumbling system. To get insight into the oligomerization mechanism of this 0.5-MDa biological machine, we performed an NMR study at high temperature, taking advantage of the high stability of TET2 from a hyperthermophilic Archaea (Pyrococcus horikoshii). Whereas previous studies of high–molecular weight assemblies have used mutagenesis to destabilize interfaces to divide the large machineries into smaller building blocks (12, 13) amenable for biochemical and biophysical studies under steady-state conditions, in our studies we prepared wild-type TET2 as a monomeric unfolded chain and followed its folding and self-assembling processes in a time-resolved manner using a combination of structural biology approaches (Fig. 1B). Native MS and fast 2D NMR were combined with optimized isotopic labeling schemes to characterize the short-lived initial state of the off-equilibrium folding pathway. Time-resolved EM was set up to identify the shape of larger oligomeric intermediates appearing during the self-assembly pathway. In addition, the real-time relaxation-optimized NMR experiments were used to extract kinetic parameters characterizing the disappearance of unfolded monomer, evolution of transient oligomeric species, and progressive appearance of native dodecameric assemblies (fig. S1B). These three complementary structural techniques allowed us to elucidate the self-assembly pathways of this half-megadalton protein particle.
Fig. 1. Real-time kinetic NMR and TET2 self-assembly progress.
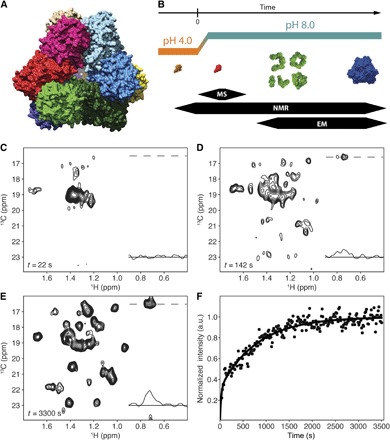
(A) The surface representation of the TET2 structure (11) viewed from the facet of the tetrahedron. (B) Schematic representation of TET2 self-assembly reaction observed by time-resolved EM, NMR, and MS. (C) The first Alaβ TET2 NMR spectrum after self-assembly initiation showing the broad peak of the flexible monomer (processed free induction decays used to generate spectrum were acquired between 5 and 37 s after pH jump). ppm, parts per million. (D) The TET2 Alaβ spectrum (data acquired between t = 127 and 157 s) of the intermediate self-assembly states comprises the peaks of the flexible monomer, assembly intermediates, and dodecamer. (E) Spectrum of the self-assembled Alaβ TET2 (acquired 55 min. after the initiation of oligomerization). Traces in the NMR spectra show 1H 1D projections at position of 13C resonance of dodecamer A194. (F) Buildup of the nonoverlapping peak of A194 in assembly intermediates and dodecamer. Circles represent the evolution of intensity of A194 signal, and data were fitted as a double exponential (solid line in fig. S3). a.u., arbitrary units.
RESULTS
The self-assembly of TET2 was initiated by a pH jump and observed in real time
The native dodecameric machinery was first disassembled as a monomer by zinc addition in acidic conditions. The acid-stabilized TET2 state was characterized by NMR spectra with well-dispersed and narrow signals but was too small to be observed by EM, suggesting the presence of folded monomers (fig. S1C). The existence of the monomeric form was confirmed by size exclusion chromatography coupled to multiangle light scattering analysis (fig. S1). To investigate the folding and oligomerization mechanism of a half-megadalton protein particle with time and site resolution, we used methyl-specific labeling in a perdeuterated background (14–16) with SOFAST (band-selective optimized flip-angle short transient)–methyl–TROSY (transverse relaxation-optimized spectroscopy) experiments (17, 18) for real-time NMR investigation (12, 19, 20). Because the Alaβ chemical shifts are well-known reporters of secondary structures (21), we selected the methyl groups as optimal probes (22) to follow the structural changes during the self-assembly process. Reassembly of TET2 was initiated by a rapid pH jump, using a stopped-flow device inside the NMR magnet (23). To follow the oligomerization process, a series of Alaβ 2D 1H,13C spectra were collected with time resolution of 15 s (fig. S1B) using a 0.2 mM sample of U-[2H],[13CH3]β-Ala–labeled TET2. The initial data collected immediately after the jump to neutral pH revealed spectral characteristics of a poorly structured protein (Fig. 1C), indicating that the crossing of the isoelectric point destabilized the monomer. This first intermediate state disappeared rapidly (fig. S2) with the concomitant buildup of well-dispersed NMR signals corresponding to folded oligomeric intermediates and native dodecameric TET2 peptidase (Fig. 1, D to F). NMR data were complemented and corroborated by “isotopic hybridization” MS analyses to identify the oligomerization state of the short lifetime initial intermediate (Fig. 2). Negative staining EM snapshots were also captured every 30 s to identify the shapes and oligomeric states of the TET2 protein during the self-assembling process.
Fig. 2. Oligomeric state characterization of the flexible intermediate by native MS.
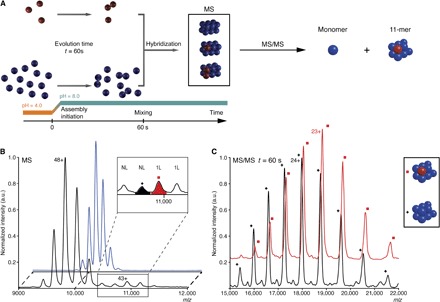
(A) Schematics of the isotopic chase experiment. Red and blue colors represent U-[15N,13C,2H]–labeled and unlabeled proteins, respectively. The self-assembly of the labeled and unlabeled samples was initiated in parallel in separate experiments by a synchronized pH jump. After 60 s, the labeled sample was mixed with an 11-fold excess of unlabeled sample, which assembled into isotopically hybrid dodecamers. (B) MS spectra of native unlabeled (blue) and “isotopically chased” hybrid (black) TET2 assembled as described above. The inset shows additional peaks appearing in hybrid dodecamers, which were subjected to the further tandem MS analyses. The major species were annotated above each peak: 1L and NL correspond to TET2 dodecamer containing one labeled monomer or fully unlabeled assembly, respectively. (C) Tandem mass spectra generated from the ions displayed on (B) at m/z 10,650 (black spectrum) and 10,980 (red spectrum). After dissociation in the gas phase, the 10,650 ions generated fully unlabeled 11-mers (11-mer) (black +, peak annotated 24+ is at m/z 17,890). The 10,980 ions dissociated into 11-mer, containing a labeled subunit and 10 unlabeled proteins (red square, 23+ peak is at m/z 18,832).
A flexible monomer is the initial intermediate in the TET2 self-assembly
The first NMR spectrum acquired after the initiation of the assembly process contained a major peak with broad linewidth located in the central region (Fig. 1C), which decayed during the self-assembly process. This spectrum did not resemble the spectrum of the low pH–stabilized monomer (fig. S1) and significantly differed from the dodecamer one (Fig. 1E). The broad peak in the center of the spectrum is characteristic of an unstructured protein (24), lacking stable secondary structures (named here as “flexible intermediate”). The peak intensity time course is typical of an intermediate state, showing zero intensity before the pH jump in the starting spectrum, a maximum in the first spectrum after self-assembly initiation, and a decay over time (fig. S2). However, the broad peak and dodecamer peaks are overlapped and influenced the analysis of one another as they simultaneously decayed and increased, respectively. Therefore, the scaled spectrum of the final dodecamer (fig. S3) was subtracted from each spectrum of the series to extract the decay of the flexible intermediate (fig. S2). The flexible intermediate could be either a monomer or a poorly structured oligomer. To uncover the oligomeric state of this flexible intermediate, we used EM and native MS. The EM projections of the sample stained immediately after the initiation of the assembly revealed heterogeneous particles without well-defined shapes (fig. S4, A and B). However, the small size of the visible particles, at the limit of EM resolution, was compatible with either a monomer or a dimer. To differentiate between these two cases, we coupled an “isotopic chase experiment” with native MS. In this approach, the oligomeric intermediate was characterized by mixing labeled TET2 with an excess of unlabeled proteins 60 s after the beginning of the self-assembly. This reaction generated hybrid dodecamers that contained labeled and unlabeled subunits (Fig. 2A). If the flexible intermediate was monomeric, the labeled subunit would be mainly embedded in the hybrid dodecamer, containing only one heavy subunit. On the contrary, a dimeric intermediate would generate final assemblies, which would accommodate only labeled dimers. Hybrid dodecamers, identified at a mass-to-charge ratio (m/z) of 10,980 and 11,240, contained only one labeled subunit, revealing that the flexible intermediate was a monomer (Fig. 2B). The 10,980 peak was further analyzed by tandem MS (Fig. 2C), confirming the presence of one labeled subunit (25).
TET2 self-assembly proceeds through an ensemble of oligomeric intermediates
The NMR spectrum series, after the subtraction of dodecamer evolution, contained an additional set of peaks overlapped with those of the flexible intermediate (Fig. 3A and fig. S2). In contrast to the signal of the first intermediate, the spectra showed well-dispersed signals (Fig. 3A), indicating the presence of one or several folded species. The time evolution of these peaks was characterized by an initial buildup followed by a decay (Fig. 4), revealing the presence of several intermediates that are also visible in the EM snapshots (Fig. 3B). The additional NMR signals were located next to the peaks corresponding to the dodecamer. This indicates that the units in these intermediates have similar fold to those in the dodecameric assembly but exhibit an increased structural heterogeneity, which is characteristic of different oligomeric states. The EM snapshots obtained from the samples stained at a time when the concentration of the folded intermediate was at its maximum (that is, 120 s after the initiation of the reassembly reaction) showed well-structured particles with high heterogeneity in terms of size and shape (Fig. 3B and fig. S4, C and D). This confirmed that the increased linewidths in the NMR spectra originate from a superposition of different assembly intermediates with various oligomeric states. Distinct particle projections could be recognized, such as triangle, V-shaped, horseshoe, and square-like forms. These projections were strikingly similar to the back projections of various oligomeric states and topologies generated from the TET2 structure (Fig. 3B). Using these projections, we assigned the oligomeric states of the EM particles as tetramer (V-shaped), hexamer (triangle and horseshoe), and octamer (square). Particles with a global shape of tetrahedral aminopeptidase but with some missing density were also detected and interpreted as decamer (fig. S5). Furthermore, the EM-determined intermediates contained an even number of monomers (Fig. 3B) with topologies of a broken tetrahedron with preserved edges, indicating that the initial flexible intermediate rapidly stabilizes and forms dimers, which are the building blocks of TET2 self-assembly.
Fig. 3. TET2 self-assembly intermediate states.
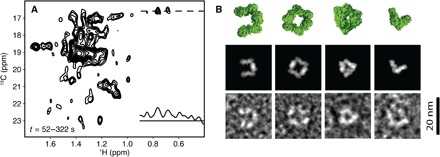
(A) Sum of the dodecamer-subtracted spectra between 52 and 322 s, including a 1H 1D slice extracted at the position of the cross peak of A194 (see Fig. 1). (B) EM projections of oligomeric intermediates captured at 2 min (bottom) and back projections (middle) of corresponding oligomeric structures (top). From left to right: horseshoe-like hexamer, square octamer, triangle hexamer, and V-shaped tetramer.
Fig. 4. Kinetic model of TET2 self-assembly.
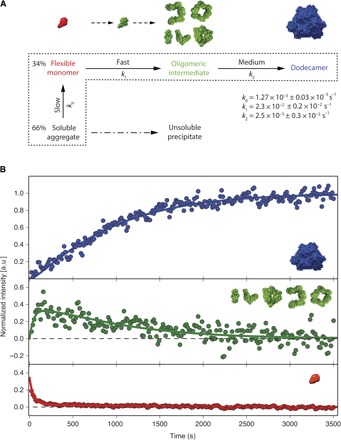
(A) Kinetic model of the TET2 self-assembly initiated by a pH jump. The jump from pH 4 to 8 induced the conversion of the acid-stabilized monomer to the flexible monomeric intermediate and soluble aggregate. Dashed area surrounds the states included in the fit. Dash-dotted arrow indicates precipitation of the soluble aggregate. The structures of the corresponding states are visualized above the kinetic model. The monomer with stable tertiary structure (green) represents the necessary step in the transition of flexible monomer (red) to ensemble of oligomeric states (green). (B) Fit of the progress curves into time-dependent peak intensities of corresponding states. The solid green line represents the progress curve of oligomeric intermediates (green). Flexible intermediate evolution is shown in red, whereas the stable dodecamer buildup is represented in blue.
Evolution of the flexible monomer intermediate to the native dodecameric particle was derived from NMR kinetic curves
The minimal kinetic model (Fig. 4A), which satisfies all data together after the pH jump, comprises (i) an initial superposition of the flexible intermediate and the NMR invisible soluble aggregate, (ii) the ensemble of oligomeric intermediates with an even number of subunits, and (iii) the final dodecamer. The jump from pH 4 to 8 induced the conversion of the acid-stabilized monomer to the flexible monomeric intermediate and soluble aggregate (fig. S4, G and H) at a rate beyond the time resolution of the fast-acquisition NMR. Self-assembly of the TET2 dodecamer from the flexible monomer and soluble aggregate followed a sigmoidal evolution (Fig. 4B) and comprised events with fast, medium, and slow kinetics. The fast event corresponds to the transition of the flexible monomer to properly folded assembly intermediates of various oligomeric states. This complex event comprises several individual processes, such as stabilization of monomeric tertiary structure, dimerization, and subsequent oligomerization of dimer building blocks (Fig. 4A). These processes cannot be resolved individually and are described by an apparent rate constant k1 = 2.3 × 10−2 ± 0.2 × 10−2 s−1. In this complex event, the transition of the flexible monomer to a properly folded monomer should be the rate-limiting step, which corresponds to the decrease in flexible monomer population (Fig. 4B, red curve). Once the monomers are properly folded at neutral pH, they exhibit a large number of charged residues positioned at the dimerization interface (fig. S5A), leading to the fast formation of stable dimer building blocks. The medium event (k2 = 2.5 × 10−3 ± 0.3 × 10−3 s−1) represents the progressive association of oligomeric intermediates, with even number of monomers, to the final dodecamer. The slow event (k0 = 1.27 × 10−3 ± 0.03 × 10−3 s−1) describes the partial replenishment of the flexible monomer pool from the soluble aggregate. This soluble aggregate belongs to a misassembly pathway that leads to the formation of macroscopic precipitate observed at the end of the self-assembly experiment.
DISCUSSION
The self-assembly can be viewed as a sequence of steps determined by the interface size
In this model (26, 27), the hierarchy of interface sizes and their interaction strengths define the order during the self-assembly. The TET2 dimerization interface comprises 51 amino acids (1855 Å2 solvent-accessible area per subunit), with 10 salt bridges and 14 hydrogen bond interactions (fig. S6, A and B). In contrast, there are two trimerization interfaces per monomer, each one comprising, on average, 31 amino acids (1102 Å2 solvent-accessible area) stabilized by only one salt bridge and two hydrogen bonds (fig. S6C). Therefore, the comparison of TET2 dimerization and trimerization interface suggests a fast formation of dimer, followed by the random association of dimers to the final dodecamer. This is corroborated by the fact that the initial build-up rate of oligomeric intermediates with an even number of subunits corresponds to the initial decay rate of the flexible monomer (Fig. 4), implying that the folding of the flexible monomer into a structured monomer is the rate-limiting step of the fast event. The folded monomer, with a native-like tertiary structure, exhibits a large number of accessible residues at the dimerization surface, and rapidly dimerizes, at a rate too fast to give rise to a detectable population of native-like structured monomer. This is followed by the association of stable dimeric building blocks to assembly intermediates using a smaller trimerization interface. This last process is slower and can be detected by NMR.
Previous studies of TET2 self-assembly have made use of mutagenesis to destabilize the trimerization interface (13) and to hamper oligomerization. From the studies of these mutants, the authors derived an assembly pathway model in which two identical hexamers assemble to form the final dodecameric particle (10, 13). In our work, we observed an ensemble of assembly intermediates comprising different oligomerization states and topologies, with the presence of intermediates with the same oligomerization state but different topologies, implying that they belong to different assembly pathways (for example, hexamers with triangle and horseshoe forms) (Fig. 3B). Although the previous study observed triangle and horseshoe hexamers and misassembled octamers (13), the authors assumed that the triangle hexamers and octameric intermediates were not productive assembly intermediates. This former study further suggests a desoligomerization of the nonproductive triangle and octameric intermediates to obtain assembly-capable horseshoe hexamers. In our study, besides the two types of hexamers, we also observed V-shaped tetramer, square octamer (Fig. 3), and decamer (fig. S5) compatible with dodecameric topology, pointing toward the simultaneous occurrence of multiple assembly pathways. These results are corroborated with an analysis of TET2/TET3 heterododecamer using small-angle neutron scattering, where both the productive assembly octamer and horseshoe hexamers were detected (28). The apparent discrepancy between the studies using the native and the mutant TET2 sequences suggests that the mutation introduced at the trimerization interface may have substantially altered self-organization. The five mutations were introduced at the trimerization interface (fig. S6D) (13), suppressing the unique salt bridge, one of the two hydrogen bonds, and hydrophobic interactions, which are required to stabilize the TET2 native quaternary structure (11).
The occurrence of multiple assembly pathways indicates that the assembly proceeds through parallel sequential addition or completely stochastic assembly (29, 30). The lack of a single kinetic pathway in the stochastic mechanism is distinct from the self-organization into dihedral oligomers, which progresses by serial second-order associations of two identical well-defined intermediates (26, 31). In the dihedral complexes, the hierarchy of interfaces dictates the order of sequential associations of two identical intermediates. In contrast, the stochastic assembly of TET2 includes the initial formation of dimeric building blocks through the association of larger interfaces, which is followed by the slower random association of dimer building blocks through the equally sized smaller trimerization interfaces to the final structure. Our model of stochastic assembly includes the pathway in which two identical hexamers assemble into a final dodecamer (13) as one of the multiple pathways occurring simultaneously.
The paradigm of self-assembly has been recently transferred from nature to nanotechnology as a bottom-up approach. In addition, development in protein and DNA technologies led to de novo design of self-assembled particles and materials, demonstrating significant progress in bionanotechnology (32, 33). Our study illustrates that the key processes of self-assembly to higher-order structures can be described and understood through direct observation at the structural level. These results highlight the potential of integrating time-resolved structural approaches, such as NMR, EM, and MS to increase our understanding of protein self-assembly and its use to develop novel therapeutic approaches and rational design of bioinspired systems.
MATERIALS AND METHODS
Protein expression and purification
Expression of U-[2H], U-[15N], U-[12C], U-[13C1H3]-Alaβ–labeled PhTET2 was performed in the Escherichia coli BL21(DE3) RIL strain. Detailed protocol is described in the study by Amero et al. (18). E. coli BL21(DE3) RIL were transformed with pET41c-PhTET2 and progressively adapted to M9/D2O medium (three stages in 24 hours). Constituents of the basal M9 medium (Sigma-Aldrich) were anhydrous. All of the 15NH4Cl, [1,2,3,4,5,6,6-2H7, U-12C]glucose, kanamycine, isopropyl-β-d-thiogalactopyranoside (IPTG), and M9 complements were resuspended in D2O (99.85%) and lyophilized twice. The final culture was grown at 37°C in M9 medium prepared with 99.85% D2O (Euriso-top). When the optical density (600 nm) reached 0.6, a D2O solution containing 2-[2H], 3-[13C] l-alanine and other perdeuterated precursors was added (22). After 1 hour, protein expression was induced by the addition of IPTG to a final concentration of 0.5 mM and then cells were grown at 37°C for 4 hours before harvesting.
Cell pellet was resuspended in lysis buffer [50 mM tris, 150 mM NaCl, 0.1% Triton X-100, lysozyme (0.25 mg/ml), deoxyribonuclease (0.05 mg/ml), 20 mM MgSO4, and ribonuclease (0.2 mg/ml) (pH 8)]. Cells were disrupted by decompression carried out by the Microfluidizer using three passes at 15,000 psi (pounds per square inch). The crude extract was heated at 85°C for 15 min and then centrifuged at 17,500 relative centrifugal force (rcf) for 1 hour at 4°C. The supernatant was dialyzed overnight at room temperature against 20 mM tris and 100 mM NaCl (pH 7.5). The dialyzed extract was centrifuged at 17,500 rcf for 10 min. at 4°C, and the supernatant was loaded on Resource Q column (GE Healthcare). The PhTET2 was eluted with linear gradient [0 to 1 M NaCl in 20 mM tris (pH 8) over 10 column volumes]. The fractions containing protein with similar mass (39 kDa), according to SDS–polyacrylamide gel electrophoresis (12.5% polyacrylamide), were pooled and concentrated using an Amicon cell (Millipore) with a molecular mass cutoff of 30 kDa. The protein solution was then loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with 20 mM tris (pH 8) and 100 mM NaCl.
Disassembly and reassembly of TET2
Classical procedures consisting of solubilizing a protein complex by adding a high concentration of chaotropic agents (for example, urea) followed by initiation of refolding by flash dilution are not suitable for real-time NMR studies. This is because dilution of the NMR sample by a factor F will require increasing the acquisition time by a factor F2 to maintain the NMR signal-to-noise ratio constant (the main limiting factor for real-time NMR studies). To preserve time resolution of NMR experiments, the initiation of the refolding using in situ pH jump with minimal dilution of NMR sample is preferred (3, 23). To optimize desoligomerization and reoligomerization protocols of the TET2 assembly, we systematically screened the pH conditions. We noticed that basic buffer (pH > 10) induces a partial disassembly of the dodecamer to dimers, whereas acidic conditions (pH < 4) led to the production of soluble monomeric TET2 protein. Previously, it has been reported in the study by Borissenko and Groll (11) that the addition of an excess of Zn2+ also induces the TET2 disassembly. We thus decided to combine both factors (that is, pH and Zn2+ concentration) to control the disassembly/reassembly process of TET2. In acidic conditions, the addition of large excess of Zn2+ led to the total TET disassembly to monomers.
First, the TET2 sample [0.2 mM in 20 mM tris and 20 mM NaCl (pH 7.8) in D2O] was disassembled into monomers by adding a 3 M ZnSO4 solution in a volume ratio of 1:10 (ZnSO4/protein). Resolubilization of the precipitated zinc hydroxide, formed by excess of ZnSO4, was carried out by chelating Zn2+ ions with 70 mM EDTA in 20 mM tris and 20 mM NaCl in D2O in a volume ratio of 1:13 (protein/EDTA). Monomeric TET2 was concentrated and buffer-exchanged to 20 mM tris-d11, 20 mM NaCl, and 500 μM ZnCl2 (pH 4) in D2O to stabilize the monomeric state. The self-assembly was initiated by stopped-flow pH jump within the magnet (23) by rapid injection of a 75-μl concentrated buffer containing 283 mM tris-d11 (pH 8) in D2O. After this rapid injection, the final buffer of TET2 was composed of 66 mM tris-d11, 0.4 mM ZnCl2, and 16 mM NaCl in D2O, corresponding to optimal conditions to initiate self-assembly and to stabilize the final dodecameric assembly.
NMR data acquisition and processing
All 1H NMR experiments were performed on an Inova spectrometer (Agilent) operating at 800-MHz frequency and equipped with a cryogenic probe. All experiments were acquired at 50°C. SOFAST-methyl-TROSY (18) was acquired with PC9 shape pulse with flip angle of 30° and length δ of 4.26 ms. The interscan delay (d1) was set to 290 ms. Each spectrum in kinetic dimension was acquired with spectral width of 1600 Hz and 20 complex points in 13C dimension. The total acquisition time of one 2D spectrum was 14.4 s. Each experiment was performed with only one scan per increment in t1, ensuring highest repetition rates of experiments, and the sign of the 13C excitation pulse phase and receiver phase in SOFAST–methyl–HMQC (heteronuclear multiple-quantum coherence) was alternated between subsequent experiments (18).
Spectra were processed and analysed using NMRPipe (34) and nmrglue (35) module of the Python programming language (36). Two succeeding spectra (1 and 2, 2 and 3, 3 and 4,…, n − 1 and n) along the kinetic time were added before processing of 2D kinetic planes. Such data treatment removes spectral artifacts, such as axial peaks or t1 noise, at the water frequency and improves the baseline, allowing a more accurate and precise measurement of spectral parameters, such as peak positions and intensities (18).
NMR data fitting
All numerical modeling and fitting were performed using the NumPy and SciPy (37) Python modules. Signals of Ala194, Ala281, Ala145, and Ala314 in the spectra of the dodecamer (38) were integrated (fig. S2A, signals delimited by cyan boxes) and were used as reporters for the evolution of the population of final dodecamer and oligomeric intermediates (Fig. 1F). The normalized buildup of dodecamer and dodecamer-like species was numerically fitted with double exponential (fig. S3) and used for subtraction of dodecamer/dodecamer-like contribution from the kinetic spectral series (fig. S2). To minimize noise contribution introduced by these combinations of spectra on each 2D kinetics plane, we performed the subtraction using a 2D SOFAST-methyl-HMQC (fig. S2B) of the final dodecamer recorded at the end of the self-assembly experiment with high signal-to-noise ratio (acquisition time, 1 hour). As a result, subtracted processed 2D spectral series (fig. S2C) were used to independently extract the contribution of flexible monomeric intermediate by integrating peaks in the red box and the oligomeric intermediate contribution by integrating signal in green boxes. The normalized buildup of dodecamer/dodecamer-like species (black points in Fig. 1F), oligomeric intermediates (green points in Fig. 4B), and flexible monomer intermediate (red points in Fig. 4B) were used as input data for the fit of kinetic constants of self-assembling process, using the model described in the following section. Errors on kinetic constants were obtained using Monte Carlo analysis.
The steps of the TET2 self-assembly process were defined as follows: ν0, release of monomer from the soluble aggregate (Ag); ν1, sum of steps leading to ensemble of oligomeric (Ol) intermediates from flexible monomeric (Mo) intermediate; and ν2, formation of mature dodecamer (Do). Because of the significant differences in monomer, flexible intermediate, and dodecamer spectra and measured time scales much smaller than the diffusion limit (association rate constant ka << 105), all steps were considered as conformation-limited and therefore modeled as first-order reactions
The changes in concentrations are given by
where Ag, Mo, Ol, and Do represent the concentrations of the soluble aggregate, flexible monomer, ensemble of oligomeric states, and dodecamer, respectively. The dissociation rate constants were assumed negligibly small (k1r ≈ k2r ≈ 0) under consideration of significant dodecamer stability (39) and low probability of oligomer dissociation coupled to unfolding into flexible monomer. The initial ratio of the soluble aggregate and flexible intermediate was included as the fit parameter. In the condition of low concentration of flexible intermediate, the forward disaggregation reaction to flexible intermediate dominates, and therefore, the reverse reaction was neglected and reverse rate constant approximates to zero (k0r ≈ 0). Because the peak intensities depend on the protein correlation time, internal dynamics, and their significant differences between self-assembly intermediates and dodecamer, the fitted intermediate progression curves were linearly scaled to the corresponding data sets of normalized peak intensities within the fitting procedure. The fitted values for apparent constants k0, k1, and k2 were 1.27 × 10−3 ± 0.03 × 10−3 s−1, 2.3 × 10−2 ± 0.2 × 10−2 s−1, and 2.5 × 10−3 ± 0.3 × 10−3 s−1, respectively. The initial equilibrium comprised 34 ± 2% of the flexible intermediate and 66 ± 2% of the soluble aggregate.
Isotopic chase experiments, acquisition of native MS spectra, and their analysis
To perform isotopic chase experiments, the unlabeled TET2 and [U-2H,13C,15N]TET2 were disassembled into monomers and then reassembled as described above. Initially, the self-reassembly of the unlabeled and labeled complexes was performed in separated reactions. After 60 s from the beginning of the reassembly, the labeled and unlabeled samples were mixed in a 1:11 ratio, and they assembled into unlabeled and isotopically hybrid dodecamers (Fig. 2).
The TET2 samples were desalted on Vivaspin columns with a 100-kDa cutoff (Sartorius France SAS). They were analyzed by native MS (40). Protein ions were generated using a nanoflow electrospray (nanoelectrospray ionization) source. Nanoflow platinum-coated borosilicate electrospray capillaries were bought from Thermo Electron SAS. MS analyses were carried out on a quadrupole time-of-flight mass spectrometer (Q-TOF Ultima, Waters Corporation). The instrument was modified for the detection of high masses (41, 42). The following instrumental parameters were used: capillary voltage, 1.2 to 1.3 kV; cone potential, 40 V; RF lens-1 potential, 40 V; RF lens-2 potential, 1 V; aperture-1 potential, 0 V; collision energy, 30 to 140 V; and microchannel plate, 1900 V. All mass spectra were calibrated externally using a solution of cesium iodide (6 mg/ml in 50% isopropanol) and were processed with the MassLynx 4.0 software (Waters Corporation) and Massign (43).
Assignment of the number of labeled subunits in protein complexes by native MS
To determine the number of labeled subunits, the molecular weights of protein complexes were calculated by the analyses of the MS and tandem MS spectra (shown in Fig. 2, B and C). First, the average mass of unlabeled complex was determined (Fig. 2B, blue spectrum). To do this, the m/z values of two consecutive peaks were experimentally measured (for example, m/z and m/z1 9837 and 10,045, respectively). The z values of these two peaks differed only by 1 unit (z1 = z − 1). Taking into account the m/z and m/z1 values (z1 = z − 1), the average mass of the unlabeled species was determined to be 468,275 ± 130 Da, which corresponds to a unlabeled homododecamer. Second, the peaks at m/z 10,650 and 10,980, which were exclusively present in the MS spectrum of the isotopically hybrid dodecamer, were subjected to tandem MS experiments (44). In these experiments, the ions were accelerated in a collision cell full of argon and subjected to collision-induced dissociation. The ions dissociated, ejecting monomers, which were detected at low m/z (mass = 39,022 ± 10 Da), and generated “stripped complexes” (11-mer) at high m/z (m/z range, 15,000 to 22,000; shown in Fig. 2C). The masses of the stripped complexes, determined as described above, allowed us to determine the composition of the peaks. This means that the 10,650 ions generated fully unlabeled 11-mers (mass, 429,280 ± 40 Da). The 10,980 ions dissociated into 11-mers, containing a labeled subunit and 10 unlabeled proteins (433,760 ± 100 Da). This confirmed that the flexible intermediate was a monomer.
EM data acquisition and analysis
Four microliters of sample at approximately 0.1 mg/ml was adsorbed onto the clean face of a carbon film on a mica sheet (carbon/mica interface) and negatively stained with 2% (w/v) ammonium molybdate (pH 7.4) in a time course experiment every 30 s. Micrographs were taken under low-dose conditions with a CM12 LaB6 electron microscope working at 120 kV and with nominal magnifications of ×60,000 using a Gatan Orius SC1000 charge-coupled device camera.
Supplementary Material
Acknowledgments
We would like to thank R. Awad, I. Ayala, A. Favier, C. Mas, A. Hessel, J. Perard, and L. Signor for technical help on the Integrated Structural Biology, Grenoble (ISBG) platforms and B. Brutscher, K. Embrey, P. Gans, M. Plevin, and P. Schanda for comments and stimulating discussions. Funding: The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme FP7/2007-2013 grant agreement no. 260887. C.M. benefited from a Grenoble Alliance for Integrated Structural Cell Biology (GRAL) fellowship. This work used the biophysics, high-field NMR, EM, isotopic labeling, and MS facilities at the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from the French Infrastructure for Integrated Structural Biology (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology. Author contributions: P.M., R.K., C.A., and J.B. designed the experiments. P.M. performed and analyzed the NMR experiments. R.K. and E.C. prepared the samples. E.B.E. acquired and analyzed the MS spectra. C.M. and G.S. performed EM data acquisition and analysis. P.M., R.K., E.B.E., G.S., C.A., and J.B. discussed the results and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1601601/DC1
fig. S1. TET2 structure, schematics of the NMR in situ kinetics experiment and disassembly, and re-assembly of TET2.
fig. S2. Subtraction of the dodecamer evolution from the kinetic spectral series for deconvolution of the flexible monomeric intermediate evolution from the dodecamer buildup.
fig. S3. Fit of dodecamer and dodecamer-like species buildup curve.
fig. S4. EM images of TET2 self-assembly including the zooms of the red-squared regions.
fig. S5. EM images of TET2 decamer intermediate.
fig. S6. Analysis of TET2 oligomeric interfaces using PISA (45).
Reference (45)
REFERENCES AND NOTES
- 1.Whitesides G. M., Grzybowski B., Self-assembly at all scales. Science 295, 2418–2421 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Schreiber G., Kinetic studies of protein–protein interactions. Curr. Opin. Struct. Biol. 12, 41–47 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Balbach J., Forge V., van Nuland N. A., Winder S. L., Hore P. J., Dobson C. M.. Following protein folding in real time using NMR spectroscopy. Nat. Struct. Biol. 2, 865–870 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Reining A., Nozinovic S., Schlepckow K., Buhr F., Fürtig B., Schwalbe H., Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 499, 355–359 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Smith M. J., Marshall C. B., Theillet F.-X., Binolfi A., Selenko P., Ikura M., Real-time NMR monitoring of biological activities in complex physiological environments. Curr. Opin. Struct. Biol. 32, 39–47 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Mulder A. M., Yoshioka C., Beck A. H., Bunner A. E., Milligan R. A., Potter C. S., Carragher B., Williamson J. R., Visualizing ribosome biogenesis: Parallel assembly pathways for the 30S subunit. Science 330, 673–677 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen T. H. D., Galei W. P., Bai X.-c., Oubridge C., Newman A. J., Scheres S. H. W., Nagai K., Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 Å resolution. Nature 530, 298–302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer N., Konevega A. L., Wintermeyer W., Rodnina M. V., Stark H., Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature 466, 329–333 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Morgner N., Montenegro F., Barrera N. P., Robinson C. V., Mass spectrometry—From peripheral proteins to membrane motors. J. Mol. Biol. 423, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appolaire A., Colombo M., Basbous H., Gabel F., Girard E., Franzetti B., TET peptidases : A family of tetrahedral complexes conserved in prokaryotes. Biochimie 122, 188–196 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Borissenko L., Groll M., Crystal structure of TET protease reveals complementary protein degradation pathways in prokaryotes. J. Mol. Biol. 346, 1207–1219 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Sprangers R., Kay L. E., Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature 445, 618–622 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Appolaire A., Rosenbaum E., Durá M. A., Colombo M., Marty V., Savoye M. N., Godfroy A., Schoehn G., Girard E., Gabel F., Franzetti B., Pyrococcus horikoshii TET2 peptidase assembling process and associated functional regulation. J. Biol. Chem. 288, 22542–22554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tugarinov V., Kanelis V., Kay L. E., Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat. Protoc. 1, 749–754 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Sprangers R., Velyvis A., Kay L. E., Solution NMR of supramolecular complexes: Providing new insights into function. Nat. Methods 4, 697–703 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Kerfah R., Plevin M. J., Sounier R., Gans P., Boisbouvier J., Methyl-specific isotopic labeling: A molecular tool box for solution NMR studies of large proteins. Curr. Opin. Struct. Biol. 32, 113–122 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Tugarinov V., Hwang P. M., Ollerenshaw J. E., Kay L. E., Cross-correlated relaxation enhanced 1H−13C NMR Spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J. Am. Chem. Soc. 125, 10420–10428 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Amero C., Schanda P., Durá M. A., Ayala I., Marion D., Franzetti B., Brutscher B., Boisbouvier J., Fast two-dimensional NMR spectroscopy of high molecular weight protein assemblies. J. Am. Chem. Soc. 131, 3448–3449 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Lapinaite A., Simon B., Skjaerven L., Rakwalska-Bange M., Gabel F., Carlomagno T., The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature 502, 519–523 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Rosenzweig R., Kay L. E., Solution NMR spectroscopy provides an avenue for the study of functionally dynamic molecular machines: The example of protein disaggregation. J. Am. Chem. Soc. 138, 1466–1477 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Cornilescu G., Delaglio F., Bax A., Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Ayala I., Sounier R., Usé N., Gans P., Boisbouvier J., An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J. Biomol. NMR 43, 111–119 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Schanda P., Forge V., Brutscher B., Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 104, 11257–11262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahakyan A. B., Vranken W. F., Cavalli A., Vendruscolo M., Structure-based prediction of methyl chemical shifts in proteins. J. Biomol. NMR 50, 331–346 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Sharon M., Structural MS pulls its weight. Science 340, 1059–1060 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Levy E. D., Boeri Erba E., Robinson C. V., Teichmann S. A., Assembly reflects evolution of protein complexes. Nature 453, 1262–1265 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy E. D., Pereira-Leal J. B., Chothia C., Teichmann S. A., 3D complex: A structural classification of protein complexes. PLOS Comput. Biol. 2, e155 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Appolaire A., Girard E., Colombo M., Durá M. A., Moulin M., Härtlein M., Franzetti B., Gabel F., Small-angle neutron scattering reveals the assembly mode and oligomeric architecture of TET, a large, dodecameric aminopeptidase. Acta Crystallogr. D Biol. Crystallogr. 70, 2983–2993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemberg M., Yaliraki S. N., Barahona M., Stochastic kinetics of viral capsid assembly based on detailed protein structures. Biophys. J. 90, 3029–3042 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson A. J., Hu Y. H. E., Keinan E., Chemical mimicry of viral capsid self-assembly. Proc. Natl. Acad. Sci. U.S.A. 104, 20731–20736 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaenicke R., Lilie H., Folding and association of oligomeric and multimeric proteins. Adv. Protein Chem. 53, 329–401 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H., DNA origami with complex curvatures in three-dimensional space. Science 332, 342–346 (2011). [DOI] [PubMed] [Google Scholar]
- 33.King N. P., Sheffler W., Sawaya M. R., Vollmar B. S., Sumida J. P., André I., Gonen T., Yeates T. O., Baker D., Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336, 1171–1174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Helmus J. J., Jaroniec C. P., Nmrglue: An open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 355–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.A. B. Downey, J. Elkner, C. Meyers, How to think like a computer scientist, in Think Python (O’Reilly Media, 2012). [Google Scholar]
- 37.E. Jones, T. Oliphant, P. Peterson et al, SciPy: Open source scientific tools for python (2001); www.scipy.org.
- 38.Amero C., Durá M. A., Noirclerc-Savoye M., Perollier A., Gallet B., Plevin M. J., Vernet T., Franzetti B., Boisbouvier J., A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J. Biomol. NMR 50, 229–236 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Rosenbaum E., Gabel F., Durá M. A., Finet S., Cléry-Barraud C., Masson P., Franzetti B., Effects of hydrostatic pressure on the quaternary structure and enzymatic activity of a large peptidase complex from Pyrococcus horikoshii. Arch. Biochem. Biophys. 517, 104–110 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Boeri Erba E., Petosa C., The emerging role of native mass spectrometry in characterising the structure and dynamics of macromolecular complexes. Protein Sci. 24, 1176–1192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sobott F., Hernández H., McCammon M. G., Tito M. A., Robinson C. V., A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 74, 1402–1407 (2002). [DOI] [PubMed] [Google Scholar]
- 42.van den Heuvel R. H. H., van Duijn E., Mazon H., Synowsky S. A., Lorenzen K., Versluis C., Brouns S. J. J., Langridge D., van der Oost J., Hoyes J., Heck A. J. R., Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal. Chem. 78, 7473–7483 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Morgner N., Robinson C. V., Massign: An assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal. Chem. 84, 2939–2948 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Hernández H., Robinson C. V., Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2, 715–726 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1601601/DC1
fig. S1. TET2 structure, schematics of the NMR in situ kinetics experiment and disassembly, and re-assembly of TET2.
fig. S2. Subtraction of the dodecamer evolution from the kinetic spectral series for deconvolution of the flexible monomeric intermediate evolution from the dodecamer buildup.
fig. S3. Fit of dodecamer and dodecamer-like species buildup curve.
fig. S4. EM images of TET2 self-assembly including the zooms of the red-squared regions.
fig. S5. EM images of TET2 decamer intermediate.
fig. S6. Analysis of TET2 oligomeric interfaces using PISA (45).
Reference (45)