

Abstract
Objective:
To explore the prognostic value of initial clinical and mutational findings in infants with SCN1A mutations.
Methods:
Combining sex, age/fever at first seizure, family history of epilepsy, EEG, and mutation type, we analyzed the accuracy of significant associations in predicting Dravet syndrome vs milder outcomes in 182 mutation carriers ascertained after seizure onset. To assess the diagnostic accuracy of all parameters, we calculated sensitivity, specificity, receiver operating characteristic (ROC) curves, diagnostic odds ratios, and positive and negative predictive values and the accuracy of combined information. We also included in the study demographic and mutational data of the healthy relatives of mutation carrier patients.
Results:
Ninety-seven individuals (48.5%) had Dravet syndrome, 49 (23.8%) had generalized/genetic epilepsy with febrile seizures plus, 30 (14.8%) had febrile seizures, 6 (3.5%) had focal epilepsy, and 18 (8.9%) were healthy relatives. The association study indicated that age at first seizure and frameshift mutations were associated with Dravet syndrome. The risk of Dravet syndrome was 85% in the 0- to 6-month group, 51% in the 6- to 12-month range, and 0% after the 12th month. ROC analysis identified onset within the sixth month as the diagnostic cutoff for progression to Dravet syndrome (sensitivity = 83.3%, specificity = 76.6%).
Conclusions:
In individuals with SCN1A mutations, age at seizure onset appears to predict outcome better than mutation type. Because outcome is not predetermined by genetic factors only, early recognition and treatment that mitigates prolonged/repeated seizures in the first year of life might also limit the progression to epileptic encephalopathy.
The voltage-gated sodium channel SCN1A gene is, among all the known epilepsy genes, the most clinically relevant, with the largest number of epilepsy-related mutations characterized.1 Epilepsy phenotypes associated with SCN1A mutations include familial febrile seizures (FS), GEFS+ (generalized or, as more recently proposed, genetic epilepsy with FS plus), and Dravet syndrome, the last representing by far the most severe phenotype (Online Mendelian Inheritance in Man No. 182389). Observations that in Dravet syndrome, but not in the other SCN1A-associated phenotypes, early normal development is followed by severe cognitive impairment and additional neurologic features2 suggest that early epileptic activity contributes to impaired brain function, resulting in an epileptic encephalopathy.3 This causal link has not yet been demonstrated, however.
At present, most pediatric epilepsy specialists suggest mutation screening of the SCN1A gene soon after an infant experiences prolonged/repeated fever-associated seizures because they suspect that these seizures may represent the initial manifestations of Dravet syndrome.4 However, early detection of an SCN1A mutation leaves important practical questions unanswered concerning management, prognosis, and counseling in that genotype-phenotype correlations are loose and after a common early clinical presentation the phenotypic spectrum may vary considerably in severity.
We studied 200 individuals with SCN1A mutations and explored the prognostic value of mutational data and early clinical findings that may help clinicians to set up management choices adapted to individuals at higher risk of progressing to Dravet syndrome, without delaying them until the epileptic encephalopathy has become obvious.
METHODS
We retrospectively analyzed 200 consecutive individuals with mutations in the SCN1A gene. Patients were enrolled from 6 Italian tertiary clinical centers with pediatric epilepsy expertise as part of a pilot study we conducted to preliminarily test the accuracy and feasibility of the data entry online form to adopt for the Italian National Registry for Dravet Syndrome and SCN1A-Related Conditions (RESIDRAS; http://www.residras.com). We included all patients and healthy carriers with SCN1A mutations who were consecutively observed in the participating centers and were >24 months of age when last seen because this is the age at which Dravet syndrome can usually be diagnosed.5 Clinical data were collected through a standardized form including demographic data, family and personal history, age at/duration of first seizure, presence of fever, and neurologic and neuropsychological outcome (figure e-1 at Neurology.org). The epilepsy phenotype was classified according to the International League Against Epilepsy criteria.6 However, considering that such criteria predate the identification of SCN1A as the causative gene for Dravet syndrome and that some authors have subsequently described mutated patients with Dravet syndrome and seizure onset beyond the first year of life,7–9 we did not firmly predefine age at first seizure as a cutoff time for diagnosis but relied on the clinical severity. We maintained the distinction between GEFS+ and focal epilepsy because some patients manifested focal seizures only and represented, in our opinion, a distinctive subgroup. We identified the following clinical subgroups: (1) Dravet syndrome (including the so-called borderline forms), (2) GEFS+, (3) focal epilepsy, (4) FS, and (5) SCN1A mutation carriers who had never experienced seizures. Definitions of the different clinical subgroups are provided in appendix e-1. To explore the value of early available parameters as prognostic indicators, we focused on patients' characteristics at seizure onset. Because Dravet syndrome is the most severe SCN1A-associated phenotype, with a constantly unfavorable outlook and requiring the most complex management choices, we analyzed data with patients divided into 2 groups: Dravet (also including borderline forms) and non-Dravet (including FS, GEFS+, focal epilepsy). Patients in the non-Dravet group were all free of seizures at last follow-up and exhibited normal or slightly delayed cognitive development.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained for each individual. The study was approved by the Pediatric Ethics Committee of the Tuscany Region, in the context of both the EU Project DESIRE–602531 and the RESIDRAS initiative.
Genetic analysis.
Methods for genetic analysis are provided in appendix e-1.
Statistical analysis.
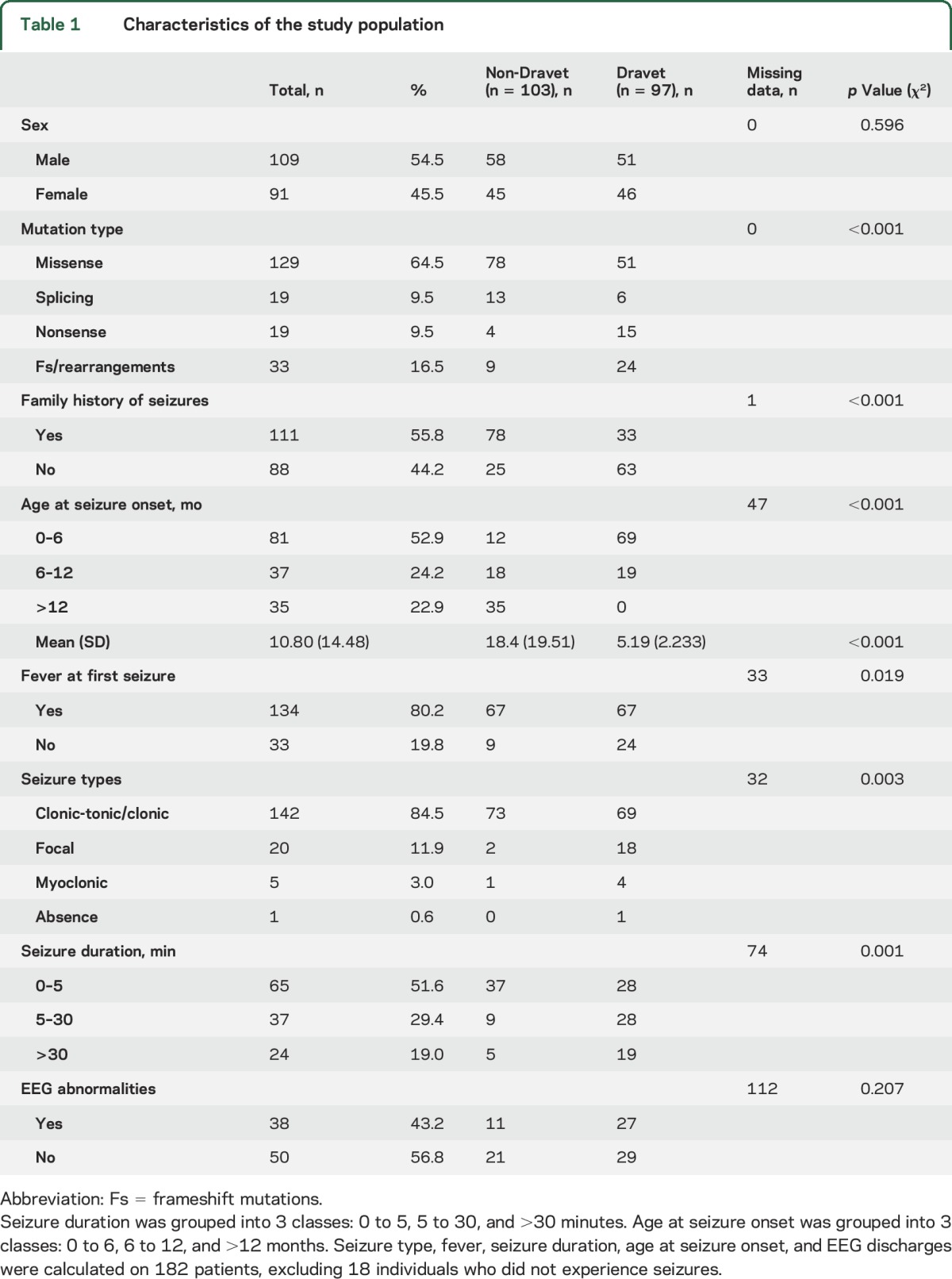
We used the STATA 13 (T Stat s.r.l.) for statistical analysis and descriptive statistics to describe the participants' main variables. For each Dravet/non-Dravet outcome, we performed the Pearson χ2 test of independence on tables of frequency for each categorical variable of interest (mutation type, sex, type of first seizure, age at seizure onset, presence/absence of fever at onset, first seizure duration, familial epilepsy, EEG discharges) and Student t test for unequal variance for each continuous variable. Age at seizure onset was analyzed both as a continuous and as a categorical variable with individuals grouped into 3 classes: 0 to 6, 6 to 12, or >12 months at onset (table 1).
Table 1.
Characteristics of the study population
The studied population also included probands' relatives, as ascertained by familial segregation of clinical manifestations and mutations. Because of the hierarchical structure of data, we fit a standard logistic regression model that was amended to have random effects for each family. More formally put, we used a sandwich estimator for the variance-covariance matrix.10 We performed a sensitivity analysis for a multilevel logistic model using a stepwise method.
To assess and compare the diagnostic accuracy of all the parameters we chose for discriminating at first seizure those patients who would develop Dravet syndrome from those who would face a less severe outcome, we calculated sensitivity, specificity, receiver operating characteristic (ROC) curves, diagnostic odds ratios (DORs), and positive and negative predictive values. Finally, we combined information from the different parameters. When appropriate, confidence intervals (CIs) were calculated with the use of exact likelihood.11 Level of significance was set at 5% 2 sided.
RESULTS
Patients.
We analyzed 200 consecutive individuals carrying SCN1A mutations (109 male, 91 female carriers) with an average age of 18.58 years at last follow-up (SD 18.08, range 2.06–81.06 years ). Seizures were the presenting symptom in 182 patients belonging to 139 unrelated families. In 33 instances, an SCN1A mutation was present in more than one family member. Of the 200 mutation carriers, 97 (48.5%) had Dravet syndrome, including borderline forms. In the non-Dravet group, distribution of phenotypes included 49 patients (23.8%) with GEFS+, 30 (14.8%) with FS, and 6 (3.5%) with focal seizures and 18 (8.9%) healthy individuals >18 years old who had undergone genetic testing during family studies for mutation confirmation and inheritance determination. The overall penetrance was 77%. Pedigrees with incomplete penetrance are shown in figure 1.
Figure 1. Schematic representation of SCN1A mutations identified in this study.

Nomenclature of mutations followed recommendations of the Human Genome Variation Society.
Association of clinical and mutational data with Dravet syndrome.
We used the Pearson χ2 independence test to analyze the distribution of categorical clinical variables in the Dravet and non-Dravet groups and the Student t test for unequal variance for each continuous variable of interest. We analyzed mutation type and 7 parameters that are usually available at clinical presentation, including sex, family history of epilepsy, age at seizure onset, type and duration of first seizure, fever at first seizure, and epileptiform discharges (sharp waves, spikes, spikes and waves) at first EEG. Six of the analyzed parameters, with the exception of sex and EEG abnormalities, had a significantly different distribution in the 2 groups (table 1). The Student t test showed a significantly different age at seizure onset in patients with Dravet syndrome vs individuals without Dravet (5.19 ± 2.23 vs 18.4 ± 19.51 months; p < 0.001).
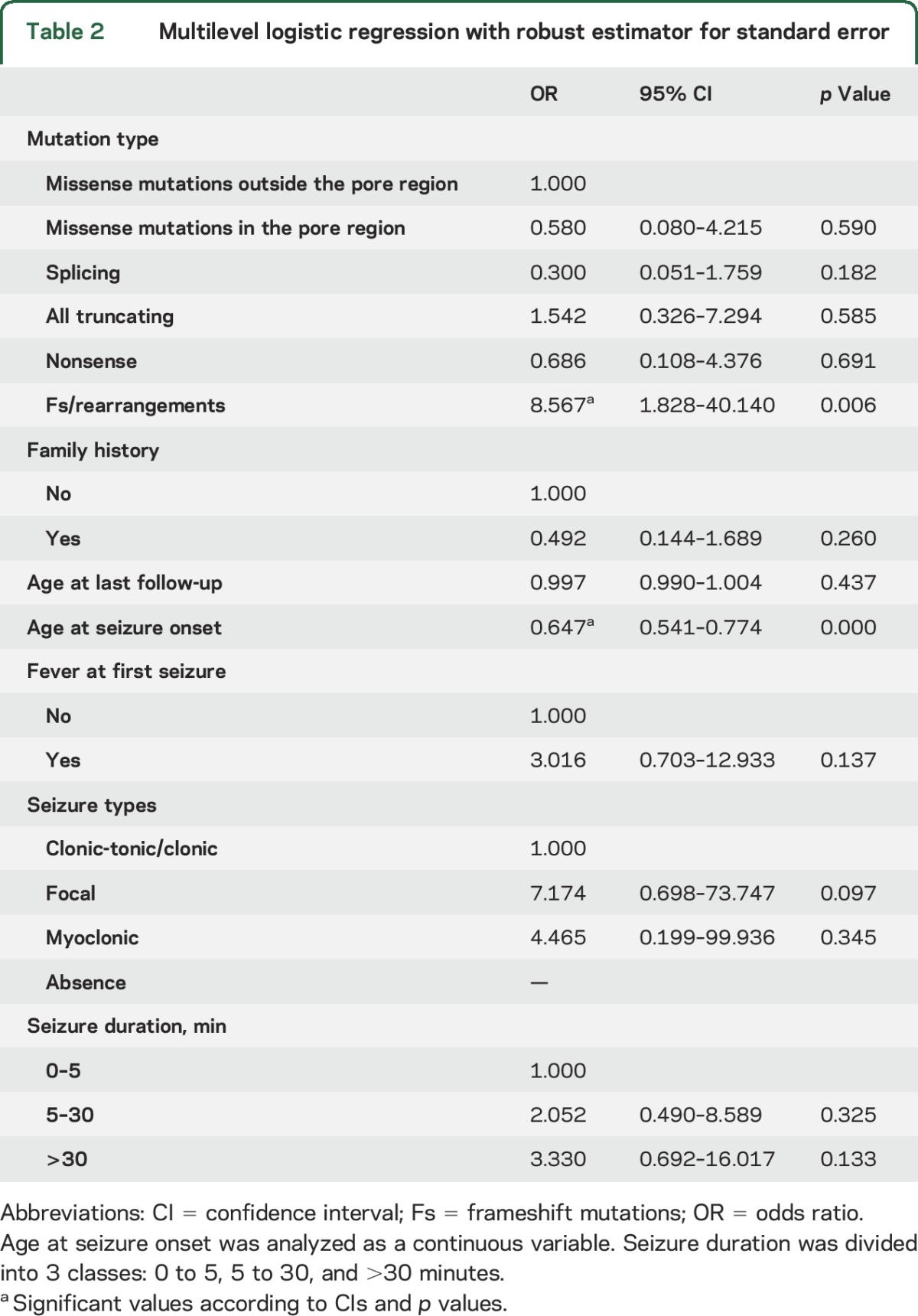
We fit a multilevel logistic model with the significant parameters and adjusted for age at last follow-up (table 2). The multivariate model showed that age at seizure onset (OR = 0.647, 95% CI 0.541–0.774, p < 0.001) and frameshift vs missense mutations (OR = 8.567, 95% CI 1.828–40.140, p = 0.006) were significantly associated with Dravet syndrome. The sensitivity analysis confirmed the results obtained in the multilevel logistic models (table 2).
Table 2.
Multilevel logistic regression with robust estimator for standard error
Association of age at seizure onset with Dravet syndrome.
Age at seizure onset, analyzed as both a continuous and a categorical variable, was distributed differently in patients with Dravet and those without Dravet syndrome. Mean age at first seizure was 5.19 months for patients with Dravet and 18.4 months for those without Dravet syndrome (table 1). Multilevel logistic regression analysis showed an OR of 0.647, meaning that an older age at seizure onset represents a protective factor against the risk of developing Dravet syndrome (table 2). None of the patients who experienced their first seizure after 12 months of age developed Dravet syndrome.
Association of mutations with Dravet syndrome.
We found 123 different mutations, 65 of which were novel, in 200 individuals (table e-1). Fifty-four mutations (42.2% of 128 individuals for whom heritability was tested) were de novo and 74 (57.8%) were inherited, 6 of which were from a parent with somatic mosaicism. Mutations were distributed throughout the gene (figure 2), and except for familial cases, only 11 were observed in more than one individual.
Figure 2. Pedigrees of families exhibiting incomplete penetrance of SCN1A mutations.

 = Dravet syndrome;
= Dravet syndrome;  = FS;
= FS;  = generalized/genetic epilepsy with febrile seizures plus;
= generalized/genetic epilepsy with febrile seizures plus;  = focal epilepsy; +/− = heterozygous SCN1A mutation; +/+ = absence of the SCN1A mutation.
= focal epilepsy; +/− = heterozygous SCN1A mutation; +/+ = absence of the SCN1A mutation.
We evaluated the effect of missense mutations using different bioinformatic tools (appendix e-1) based on functional prediction scores and conservation scores. We deemed as damaging all substitutions predicted to be deleterious by at least 2 conservation and 2 prediction algorithms and all the mutations resulting in loss of function (nonsense, frameshift, splicing, and genomic rearrangements). To estimate the allelic frequency of the mutations, we interrogated public frequency databases (appendix e-1). We identified 71 different missense mutations, of which 62 were not present in databases, 6 were reported in the Exome Aggregation Consortiumto have a frequency of <0.0001%, and 3 (p.Thr1250Met, p.Arg542Gln, and p.Arg604His) with a frequency of ≥0.0001% (table e-1). We grouped mutations into 5 classes: missense, missense falling into the pore-forming region, splicing, nonsense, and frameshift (including rearrangements of entire exons). Multilevel logistic regression revealed that frameshift mutations and rearrangements confer a significantly higher risk of developing Dravet syndrome (OR = 8.567, 95% CI 1.828–40.140, p = 0.006, table 2) with respect to missense mutations.
Diagnostic test.
To compare the diagnostic accuracy of all parameters, we estimated sensitivity, specificity, ROC area, DOR, and positive and negative predictive values (table 3). We performed this analysis on the 119 patients for whom information on all 8 phenotypic and genotypic items was available (table 1). Patients carrying variants of uncertain significance (p.Thr1250Met, p.Arg542Gln, and p.Arg604His) were excluded from this analysis. ROC analysis showed age at seizure onset to accurately recognize patients with Dravet syndrome. On the basis of this parameter, we identified 3 subgroups within which the probability of developing Dravet syndrome was significantly different: 85% in the 0- to 6-month seizure-onset group, 51% in the 6- to 12-month group, and none after the 12th month of age. The optimal diagnostic cutoff was 6 months of age (sensitivity = 83.3%, 95% CI 72.7–91.1; specificity = 76.60%, 95% CI 62.0–87.7, table 3).
Table 3.
Diagnostic accuracy of analyzed parameters
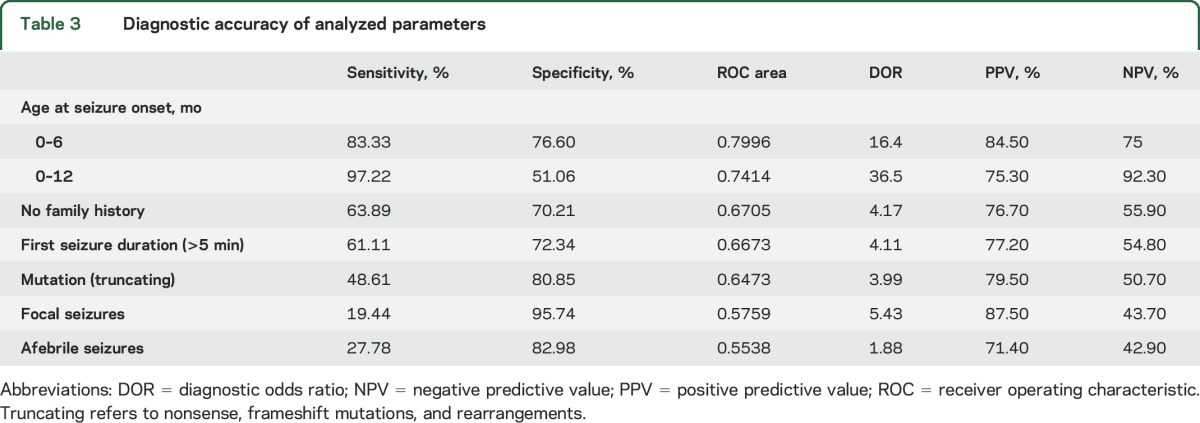
On the basis of the results shown in table 3, we explored a combination of parameters that, however, did not yield higher significativity than the test based on age at first seizure.
DISCUSSION
Previous studies have examined the spectrum of SCN1A mutations associated with Dravet syndrome8,12 and suggested clinical criteria for SCN1A screening based on early seizure characteristics.13 These studies, performed on patient populations whose clinical characteristics were highly suggestive of Dravet syndrome, have contributed to delineate its genotypic and phenotypic spectra. The approaches used, however, could not address the opposite perspective of defining the risk of divergent outcomes in a population of mutation-positive patients whose clinical presentation is relatively similar at seizure onset. Considering that SCN1A screening is widely performed soon after early prolonged/repeated FS appear, at a stage when either benign or ominous outcomes are still possible, we attempted to identify reliable early clinical and mutational outcome predictors that can help clinicians to deal with a frequently encountered dilemma.
To gather a sample population that was representative of the spectrum of SCN1A-associated phenotypes, as a first step, we collected clinical details on 200 individuals with damaging SCN1A mutations, including healthy relatives of mutation carriers. Of the 182 patients with seizures, 53.6% had Dravet syndrome and 46.4% had milder conditions, including GEFS+ (27%), FS (16.2%), and focal seizures (3.2%). Eighteen remaining relatives of probands (9% of the whole sample) were mutation carriers who never experienced seizures. We then analyzed 7 clinical parameters usually available at seizure onset—sex, family history of epilepsy, age at/fever at/type of/duration of first seizure, and abnormalities at first EEG—and found 5 of them to exhibit a significantly different distribution in the Dravet and non-Dravet groups. While the Dravet group had a higher frequency of onset within the sixth month (p < 0.001), of focal seizures (p = 0.003), and of seizures lasting >5 minutes (p = 0.001), family history (p < 0.001) and evidence of fever at seizure onset (p = 0.019) were more frequent in the non-Dravet group. Multilevel logistic regression showed that age at seizure onset was the more relevant parameter associated with Dravet syndrome (OR = 0.647, 95% CI 0.541–0.774, p < 0.001). The diagnostic ROC analysis identified age at seizure onset as the main and statistically relevant indicator, with the optimal diagnostic cutoff for age being 6 months (sensitivity = 83.33%; specificity = 76.60%). The risk for Dravet syndrome declined progressively according to the age group at seizure onset, being 85% in the 0- to 6-month group, 51% in the 6- to 12-month group, and 0% after the 12th month.
We observed 123 different SCN1A mutations. As previously reported,14,15 truncating mutations were more represented in patients with Dravet than in those without Dravet syndrome (40% vs 13%). We also observed 13 truncating mutations associated with mild phenotypes and 37 missense mutations outside the pore-forming region in Dravet syndrome, thus confirming the variable phenotypic consequences of SCN1A mutations.1 Although genotype-phenotype studies have pointed out that less severe phenotypes are more common with missense than truncating mutations16–18 and that missense mutations in the pore-forming region tend to be associated with more severe phenotypes,19 correlations between specific mutations and specific phenotypes are weak.20 A meta-analysis of 155 missense SCN1A mutations indicated that the physicochemical properties of amino acid changes influence the epilepsy phenotype and could be used to predict the phenotype associated with each mutation.21 Similarly, an attempt to correlate the Grantham score of missense mutations, a measure of physicochemical differences between amino acids, with phenotypic outcome demonstrated that the type of amino acidic substitution does not independently predict different phenotypes within the spectrum of SCN1A-related epilepsies.15 Our estimation through ROC analysis of the value of different mutation types to predict the risk of developing Dravet syndrome sets the best cutoff grouping truncating mutations vs splicing and missense mutations but could not predict with high confidence Dravet syndrome vs milder phenotypes (sensitivity = 48.61%, specificity = 80.85%). Therefore, localization and type of SCN1A mutations are, on their own, less accurate predictors of outcome than assessment based on age at seizure onset. Combining mutational data with clinical parameters did not significantly improve the discrimination performance of the test.
Of the 33 families included in our cohort, all 20 families exhibiting complete penetrance carried truncating mutations, while the remaining 13 exhibiting incomplete penetrance carried either splicing or missense mutations (figure 2). Overall, penetrance was highly mutation-dependent, reaching 100% in families with truncating mutations and 77% in those with segregating missense mutations. Nonpenetrance for SCN1A mutations, observed in 9% of our sample, has already been reported in GEFS+22 and less frequently in Dravet syndrome,23 but its frequency had not been assessed on large series before. We also observed wide intrafamilial phenotypic variability, with only 5 of 33 families exhibiting the same phenotype in all affected members. High phenotypic variability within the same family15,23–27 is interpreted as a consequence of epistasis.
From a pathophysiologic perspective, there seems to be an age-at-seizure-onset–dependent response of the brain carrying an SCN1A mutation to early epileptogenesis whereby onset within the sixth month almost regularly progresses as an epileptic encephalopathy, while onset after the 12th month never does. It is unlikely that this course was influenced by treatment choices in the population studied because no uniform attitude or protocol exists, with either immediate treatment of seizures or long-term medication currently being started on the basis of individual preferences concerning drug(s) and timing.
It remains to be clarified whether the worst prognosis related to a younger age at seizure onset can be entirely explained by genetic factors, with earlier onset just being an expression of a lower seizure threshold prompted by the most damaging mutations. Earlier onset of seizure activity can actually by itself induce changes that permanently lower seizure threshold and cause cognitive impairment28 above and beyond what is caused by the underlying mutation. Studies on animal models have demonstrated that the deleterious consequences of seizures strongly depend on the developmental stage at which they occur: immature neurons having few synapses and more developed neurons that express a multitude of functional synapses endure different consequences.28,29 Long-lasting effects of seizures may derive from seizure-induced transformation of a naive network to one that has increased seizure susceptibility. In particular, converging evidence has been gathered that in the rat brain, early/prolonged hyperthermic seizures cause permanent changes resulting in long-standing increased excitability.28 Although it is still unclear whether similar changes occur in the human brain, analogies with the observations that Dravet syndrome develops only when early/prolonged hyperthermic seizures appear in the first year of life, particularly in the first 6 months, are strong.
In pediatric epilepsy practice, young infants with prolonged/repeated fever-related seizures and SCN1A mutations pose considerable concerns in terms of their risk of developing Dravet syndrome, a risk that our study sets at ≈50% overall but at 0% in those with seizure onset after the 12th month. It is of primary importance to discern those at higher risk of severe outcomes and to promptly organize management accordingly. Because this study indicates that age at seizure onset is a reliable indicator of outcome, we suggest that seizure onset within the first year of life should prompt appropriate treatment choices, for example, introduction of the stiripentol-clobazam combination30 before the epileptic encephalopathy is established. Of course this suggestion is valid provided no precious time is lost delaying mutation analysis.4 Although no controlled evidence exists that more appropriate earlier treatment can limit the progression toward the severe end of the SCN1A spectrum, this possibility should be explored through a dedicated trial, which might use the results of this study as a comparator.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the patients and their relatives for participating in this study.
GLOSSARY
- CI
confidence interval
- DOR
diagnostic odds ratio
- FS
febrile seizures
- GEFS+
generalized/genetic epilepsy with febrile seizures plus
- OR
odds ratio
- RESIDRAS
Italian National Registry for Dravet Syndrome and SCN1A-Related Conditions
- ROC
receiver operating characteristic
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept and design: V. Cetica, R. Guerrini. Patient collection: R. Guerrini, C. Marini, S. Chiari, A. Ferrari, F. Sicca, N. Specchio, M. Trivisano, D. Battaglia, I. Contaldo, N. Zamponi, C. Petrelli, T. Granata, F. Ragona, G. Avanzini. Mutation screening and data analysis: V. Cetica, S. Chiari, D. Mei, E. Parrini, D. Pucatti, R. Guerrini. Statistical analysis: L. Grisotto. Drafting of the manuscript: V. Cetica, S. Chiari, R. Guerrini. Critical revision of the manuscript for important intellectual content: R. Guerrini. Obtained funding: R. Guerrini.
STUDY FUNDING
This study was funded by the EU 7th Framework Programme (FP7) under the project DESIRE grant N602531 (to R.G.) and RESIDRAS.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. [DOI] [PubMed] [Google Scholar]
- 2.Guerrini R, Striano P. Dravet syndrome: not just epilepsy. Neurology 2016;87:245–246. [DOI] [PubMed] [Google Scholar]
- 3.Guerrini R, Dravet C. Severe epileptic encephalopathies of infancy, other than West syndrome. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Vol 3. Philadelphia: Lippincott; 1997:2285–2302. [Google Scholar]
- 4.Hirose S, Scheffer IE, Marini C, et al. ; Genetics Commission of the International League Against Epilepsy. SCN1A testing for epilepsy: application in clinical practice. Epilepsia 2013;54:946–952. [DOI] [PubMed] [Google Scholar]
- 5.Guerrini R, Marini C, Mantegazza M. Genetic epilepsy syndromes without structural brain abnormalities: clinical features and experimental models. Neurotherapeutics 2014;11:269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389–399. [DOI] [PubMed] [Google Scholar]
- 7.Kearney JA, Wiste AK, Stephani U, et al. Recurrent de novo mutations of SCN1A in severe myoclonic epilepsy of infancy. Pediatr Neurol 2006;34:116–120. [DOI] [PubMed] [Google Scholar]
- 8.Depienne C, Trouillard O, Saint-Martin C, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 2009;46:183–191. [DOI] [PubMed] [Google Scholar]
- 9.Catarino CB, Liu JY, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 2011;134:2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White H. Maximum likelihood estimation of misspecified models. Econometrica 1982;50:1–25. [Google Scholar]
- 11.Van Belle G, Fisher LD, Heagerty PJ, Lumley T, editors. Biostatistics: A Methodology for the Health Sciences. 2nd ed. New York: Wiley; 2004. [Google Scholar]
- 12.Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012;135:2329–2336. [DOI] [PubMed] [Google Scholar]
- 13.Hattori J, Ouchida M, Ono J, et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia 2008;49:626–633. [DOI] [PubMed] [Google Scholar]
- 14.Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–1685. [DOI] [PubMed] [Google Scholar]
- 15.Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011;76:594–600. [DOI] [PubMed] [Google Scholar]
- 16.Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 2002;58:1122–1124. [DOI] [PubMed] [Google Scholar]
- 17.Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol 2004;30:236–243. [DOI] [PubMed] [Google Scholar]
- 18.Brunklaus A, Zuberi SM. Dravet syndrome: from epileptic encephalopathy to channelopathy. Epilepsia 2014;55:979–984. [DOI] [PubMed] [Google Scholar]
- 19.Kanai K, Hirose S, Oguni H, et al. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology 2004;63:329–334. [DOI] [PubMed] [Google Scholar]
- 20.Parihar R, Ganesh S. The SCN1A gene variants and epileptic encephalopathies. J Hum Genet 2013;58:573–580. [DOI] [PubMed] [Google Scholar]
- 21.Kanai K, Yoshida S, Hirose S, et al. Physicochemical property changes of amino acid residues that accompany missense mutations in SCN1A affect epilepsy phenotype severity. J Med Genet 2009;46:671–679. [DOI] [PubMed] [Google Scholar]
- 22.Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus and prevalence of variants in patients with epilepsy. Am J Hum Genet 2001;68:866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nabbout R, Gennaro E, Dalla Bernardina B, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 2003;60:1961–1967. [DOI] [PubMed] [Google Scholar]
- 24.Marini C, Mei D, Cross JH, Guerrini R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 2006;47:1737–1740. [DOI] [PubMed] [Google Scholar]
- 25.Guerrini R, Cellini E, Mei D, et al. Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene. Epilepsia 2010;51:2474–2477. [DOI] [PubMed] [Google Scholar]
- 26.Depienne C, Trouillard O, Gourfinkel-An I, et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet 2010;47:404–410. [DOI] [PubMed] [Google Scholar]
- 27.Suls A, Velizarova R, Yordanova I, et al. Four generations of epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology 2010;75:72–76. [DOI] [PubMed] [Google Scholar]
- 28.Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol 2006;5:1055–1063. [DOI] [PubMed] [Google Scholar]
- 29.Haut SR, Veliskova J, Moshe SL. Susceptibility of immature and adult brains to seizure effects. Lancet Neurol 2004;3:608–617. [DOI] [PubMed] [Google Scholar]
- 30.Kassaï B, Chiron C, Augier S, et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta-analysis of individual patient data. Epilepsia 2008;49:343–348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.