

Abstract
AT rich interactive domain 1A (ARID1A) is one of the most commonly mutated genes in a broad variety of tumors. The mechanisms that involve ARID1A in ampullary cancer progression remains elusive. Here, we evaluated the frequency of ARID1A and KRAS mutations in ampullary adenomas and adenocarcinomas and in duodenal adenocarcinomas from two cohorts of patients from Singapore and Romania, correlated with clinical and pathological tumor features, and assessed the functional role of ARID1A. In the ampullary adenocarcinomas, the frequency of KRAS and ARID1A mutations was 34.7% and 8.2% respectively, with a loss or reduction of ARID1A protein in 17.2% of the cases. ARID1A mutational status was significantly correlated with ARID1A protein expression level (P=0.023). There was a significant difference in frequency of ARID1A mutation between Romania and Singapore (2.7% versus 25%, P=0.04), suggestive of different etiologies. One somatic mutation was detected in the ampullary adenoma group. In vitro studies indicated the tumor suppressive role of ARID1A. Our results warrant further investigation of this chromatin remodeller as a potential early biomarker of the disease, as well as identification of therapeutic targets in ARID1A mutated ampullary cancers.
Keywords: Ampullary cancer, Sanger sequencing, ARID1A, KRAS
Introduction
Ampullary cancers form a group of rare neoplasms [1]. These tumors arise in the area known as the ampulla of Vater. This is a unique and complex anatomic region found at the confluence of the common bile duct and main pancreatic duct that opens into the duodenum through the papilla of Vater. The area where the ampulla Vater is located is covered by three types of epithelia: duodenal, pancreatic and biliary [2]. Therefore, neoplasms arising from this region can be classified as either pancreatobiliary, intestinal and mixed or ambiguous type [3,4]. The intestinal subtype originating from the intestinal epithelium lining [5] is associated with a longer overall survival compared to the pancreatobiliary type [6,7]. This may reflect the fact that the intestinal subtype is more related genetically to colorectal cancers, which have a superior prognosis compared to the pancreatobiliary subtypes.
Because of the close anatomical relations in this area, the true origin of some neoplasms is difficult to establish. As a result, the ampullary tumors are sometimes classified together with neoplasm that arise from head of the pancreas, distal common bile duct or periampullary duodenum [8-10], and are termed periampullary cancers. Based on site of origin, the most common periampullary tumors are the pancreatic neoplasms followed by ampullary cancers, tumors of the bile duct, and duodenum [11,12].
ARID1A or AT rich interactive domain 1A is a recently identified putative tumor suppressor gene located on chromosome 1p36.11. The gene encodes for a large protein (ARID1A or BAF250a) which is the variant subunit [13] of the SWI/SNF (SWItch/Surcose NonFermentable) multi-component complex. This is an ATP-dependent chromatin remodeling complex [14] that uses the energy of ATP hydrolysis to slide the DNA around the nucleosome and to alter gene expression in a tissue specific manner [15-17]. ARID1A is considered both a “caretaker” as well as “gatekeeper” [18], and may mediate carcinogenesis by its involvement in cell proliferation, differentiation, and apoptosis [17]. It also modulates cell-cycle genes such as c-myc and is involved in the PI3K pathway [19], with two-hit mutations and/or protein loss demonstrated in mutation-carrying tumors, most notably gastric and ovarian carcinomas [20,21].
ARID1A has been identified as one of the most frequently mutated genes in human cancers by multiple next-generation genomic sequencing studies [22,23] and high incidence of ARID1A somatic mutations have been identified in subtypes of ovarian cancers [20,24], breast cancer [25], gastric cancer [21], clear cell renal carcinomas [27] and hepatocellular carcinoma [28,29].
In pancreatic cancer, one study described ARID1A mutations in 8% of the patients [30], while another study showed that ARID1A mutations by chromosomal deletion were detected in 47% of the tumor samples and cell lines [31]. ARID1A mutations were recently discovered in two ampullary cohorts of patients with a frequency of 11% and 5% (in the discovery screen), respectively [32,33].
The role of ARID1A in ampullary malignancies is only beginning to be uncovered. The prognostic and clinical significance of these mutations remains to be established. In this study, we aimed to determine the frequency of ARID1A and KRAS mutations in two cohorts of patients from Singapore and Romania, to correlate genomic and proteomic data with clinical information and to determine the functional role of ARID1A in ampullary cancers.
Materials and methods
Patients and tissue samples
Samples of ampullary (n=74) and duodenal (n=6) adenocarcinoma, and ampullary adenomas (n=3) from patients who underwent surgical resection were identified and retrieved from two tissue banks: Singapore Tissue Repository, Singapore and Fundeni Clinical Institute Tissue Bank, Romania. Tissue was obtained with informed written consent from each patient. The study was approved by the SingHealth Centralized Institutional Review Board, Singapore and Ethical Committee of Fundeni Clinical Institute, respectively. Paraffin-embedded formalin fixed tissues were identified and retrieved from the Pathology Department of Singapore General Hospital and Fundeni Clinical Institute. Clinical and histopathological data were collected and reviewed by two independent GI pathologists. The site of each tumor’s origin was based on histopathological reports.
Clinical outcomes
Data regarding the duration of follow-up, time to recurrence, survival or censoring was calculated in months from the date of surgery for each patient. Censoring of the overall survival was done at the date of the last follow-up if death did not occur or if the cause of dead was unrelated to the disease. If available, the time to recurrence was calculated in months from date of surgery for each patient until the date of recurrence (either local or distant). Censoring of the time to recurrence was done at the date of last follow-up if recurrence was not observed.
DNA samples
Qiagen Blood and Cell Culture Mini Kit (Hilden, Germany) was used to extract genomic DNA from adjacent benign (when available) and tumor tissue as per manufacturer’s instructions from a total of 58 patients. In brief, the frozen tissue was grounded with a pestle and mortar, then mixed with RNaseA, Buffer G2 and Protease and incubated overnight at 50°C. The genomic DNA was bound to column, washed, eluted and quantified using spectrophotometry. Integrity was checked by running 200 ng of genomic DNA on 0.8% agarose gel.
Evaluation of ARID1A and KRAS mutations in tumors and matched normal specimens
Genomic DNA from tumor and matched normal samples were amplified with Illustra GenomPhi HY DNA Amplification Kit (GE healthcare, Buckinghamshire, UK). PCR was performed with Platinum Taq Polymerase (Life technologies, Carlsbad, USA) and cycled at 95°C for 10 minute; 39 cycles of 95°C for 30 seconds; 58/60°C for 30 seconds, 72°C for 1 minute and a final extension of 72°C for 10 min. Purified PCR products were sequenced with ABI BigDye Terminator v3.1 (Life technologies, Carlsbard, USA) following manufacturer’s protocol and ran on an ABI 3730 genetic analyzer. The results were analyzed using Seqman II DNAstar (v.5.05) (Madison, Wisconsin, USA) and Chromas 2.4.3 (South Brisbane, Australia). The primers used for ARIDIA and KRAS to perform PCR and Sanger sequencing are the ones described previously [24]. Additionally the primer sequences can be requested from the corresponding author.
ARID1A immunohistochemistry and subtyping
Staining of ARID1A (Sigma, HPA005456, dilution 1:50) was performed using citrate buffer (pH 6.0) with pressure cooking (3 min) for antigen retrieval. Primary antibody incubation was for 1 h at room temperature. Manual DAKO Envison KIT was used for visualization. Normal epithelial and lymphoid cell nuclei were used as positive internal controls. Results were interpreted as loss if complete nuclear and/or cytoplasmatic staining was observed, weak staining if partial nuclear and/or cytoplasmatic staining was observed or as positive if strong nuclear and/or cytoplasmatic staining was observed. Immunohistochemistry staining for ARID1A was done in a total of 64 patients.
For subtyping, available paraffin embedded tissues (n=63, out of 83) were used for assembly of TMA (tissue microarray) blocks with two cores per tumor. Serial section were cut and stained for MUC1 (Novocastra, NCL-MUC01, dilution 1:150), MUC2 (Novocastra, NCL-MUC-2-CE, dilution 1:100), CDX2 (Dako, M3636, dilution 1:100) and CK20 (Dako, M7019, dilution 1:50) on a Leica BOND-MAX III automated IHC stainer, in order to establish the type of differentiation: intestinal, pancreatobiliary or ambiguous type [4].
The immunohistochemistry staining for these markers was evaluated as positive if more than 50% of the cells were positively stained, as weak to moderate if less than 50% of the cells were positively stained and as negative if complete loss of staining was noted.
SNP array
For analysis of copy number and loss of heterozygosity the HumanOmni Express 24 v1.1 beadchip from Illumina was used according to manufacturer instruction. Seven pairs of samples with ARID1A somatic mutation were genotyped and loss of heterozygosity for chromosome 1p was evaluated by ASCAT 2.0 software (Allele-Specific Copy number Analysis of Tumors). The software provided an estimation of the B-allele frequency and log2R ratio. LOH was given by B allele frequency where values deviating from 0.5 indicated allelic imbalance or LOH. Copy-number alterations were identified based on the allele-specific copy-number counts and average ploidy.
Cell cultures
Two ampulla of Vater cell lines, SNU-478 and SNU-869, were purchased from Korean Cell Line Bank and were maintained in RPMI-1640 supplemented with 300 mg/L L-glutamine, 10% fetal bovine serum and penicillin/streptomycin at 37°C, 5% CO2. Media was changed at least twice a week. Cell line genotypes were obtained from published studies [34,35], COSMIC database [36] and CCLE database [37].
Cell transfection
SNU-478 was forward transfected and SNU-869 was reversed transfected with siRNA against ARID1A (ON-Target Plus smart pool Human ARID1A: L-017263-00, ON-Target Plus siRNA Human ARID1A: J-017263-06, ON-Target Plus siRNA Human ARID1A: J-017263-07, Dharmacon, IL) or with a non-targeted control (ON-TARGET plus Non-targeting Pool: D-001810-10, Dharmacon, IL) at a final concentration of 50 nM using Lipofectamine RNAiMAX (Invitrogen, CA) according to the manufacturer’s protocol.
Cell proliferation assay
Forty-eight hours after transfection, 2×10^3 SNU-478 cells and 4×10^3 SNU-869 cells were plated in 96-well plates in triplicate. Cell proliferation was monitored every 24 hours for 4 days using Cell Titer Glo assay (Promega, WI) according to the manufacturer’s instructions. Relative light units were measured with a PerkinElmer plate reader. Three independent experiments were performed, and results are represented as the average normalized to the control at each time point (mean ± s.e.m).
Real time PCR
Forty-eight hours post-transfection total RNA was extracted with Trizol Reagent (Life Technologies, NY) followed by purification using a column based kit (RNeasy mini kit, Qiagen). 1000 ng of total RNA was reverse-transcribed using iScript cDNA Synthesis kit (Bio-Rad, CA). Sso Fast Eva Green Super-Mix using Bio-Rad CFX 96 Real Time Detection System (Bio-Rad, CA) were used to quantify the expression of ARID1A and GAPDH, as an endogenous control, by quantitative real time-PCR. Relative mRNA expression was calculated using 2^(-ΔΔCt) method and normalized to GAPDH expression. Primers used can be requested from the corresponding author.
Western blot
Cells were lysed 48 hours post-transfection with lysis buffer [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate and 1% NP-40 (Igepal)] in the presence of freshly added protease inhibitor and PhoSTOP (Roche). Cell lysate were disrupted and centrifuged at 14000 rpm for 15 min, loaded on gel and transferred, after separated on SDS-PAGE gel, to PVDF or nitrocellulose membranes (Bio-Rad, CA). Membranes were blocked in PBS with 5% skim milk and 0.1% Tween-20, probed with antibodies to ARID1A (1:1000 dilution, Cell Signalling Technology, 12354) and β-actin (1:100,000 dilution, Sigma Aldrich, MO, A1978), incubated overnight at 4°C, washed and incubated with HRP-conjugated secondary antibodies (IgG anti-mouse NA931 or IgG anti-rabbit NA934, Amersham). Signals were visualized using either SuperSignal West Pico chemiluminescent substrate, (Thermo Scientific) or ECL prime Western Blotting Detection Reagent RPN2236 (Amersham) and Kodak BioMax XAR film (VWR International).
Statistical analysis
Categorical variables were evaluated using Fisher’s exact test for two-by-two comparison or Pearson’s χ2 for comparison that exceeded the two-by-two condition. Differences between groups were evaluated by means of nonparametric Mann-Whitney or Kruskal-Wallis test. Survival analyses were performed using the Kaplan-Meier curves and the differences in survival curves were assessed by Log rank Mantel Cox test. A P≤0.05 was considered statistically significant and noted as: *P≤0.05, **P≤0.01, ***P≤0.001. All tests were performed with Graph-Pad Prism 5 (GraphPad Software Inc, San Diego, CA).
Additional Material and Methods details can be requested to the corresponding author.
Results
Patient characteristics
The clinico-pathologic characteristics of all patients and the flowchart summarizing the experiments within our study are detailed in Table 1 and Figure 1. Of the 83 tumors included in the study, 74 (89.1%) were ampullary adenocarcinomas. Statistically significant differences between tumor groups based on patients’ clinicopathologic characteristics are shown in Table 1.
Table 1.
Summary of clinico-pathological features of patients included in the study
| Characteristics | Ampullary tumors (n=74) | Dudodenal tumorsa (n=6) | Ampullary adenomaa (n=3) | p-valueb | |
|---|---|---|---|---|---|
|
| |||||
| Total | Cohort 1 (n=49) | Cohort 1 | Cohort 1 | ||
|
| |||||
| Cohort 2 (n=25) | |||||
| Age at surgery (yrs), median [range] | 65 [38-79] | 62 [40-78] | 54 [36-74] | 69 [46-75] | 0.33 |
| 67 [38-79] | |||||
| Gender (n, %) | 0.06 | ||||
| Female | 33 (44.6%) | 22 (29.7%) | 1 (16.7%) | 3 (100.0%) | |
| 11 (14.9%) | |||||
| Male | 41 (55.4%) | 27 (36.5%) | 5 (83.3%) | 0 (0.0%) | |
| 14 (18.9%) | |||||
| Differentiation degree (n, %) | 0.43 | ||||
| G1, G1-G2 | 27 (36.5%) | 22 (29.7%) | 3 (50.0%) | NA | |
| 5 (6.8%) | |||||
| G2, G2-G3 | 36 (48.6%) | 20 (27.0%) | 1 (16.7%) | NA | |
| 16 (21.6%) | |||||
| G3 | 9 (12.2%) | 5 (6.8%) | 1 (16.7%) | NA | |
| 4 (5.4%) | |||||
| Unknownc | 2 (2.7%) | 2 (2.7%) | 1 (16.7%) | NA | |
| 0 (0.0%) | |||||
| pN (n, %) | NA | 1.0 | |||
| N0 | 48 (64.9%) | 30 (40.5%) | 4 (66.7%) | NA | |
| 18 (24.3%) | |||||
| N1 | 25 (33.8%) | 18 (24.3%) | 2 (33.3%) | NA | |
| 7 (9.5%) | |||||
| Unknownc | 1 (1.4%) | 1 (1.4%) | 0 (0.0%) | NA | |
| 0 (0.0%) | |||||
| pT (n, %) | <0.0001 | ||||
| T1 | 13 (17.6%) | 9 (12.2%) | 0 (0.0%) | NA | |
| 4 (5.4%) | |||||
| T2 | 28 (37.8%) | 18 (24.3%) | 1 (16.7%) | NA | |
| 10 (13.5%) | |||||
| T3 | 28 (37.8%) | 19 (25.7%) | 1 (16.7%) | NA | |
| 9 (12.2%) | |||||
| T4 | 4 (5.4%) | 2 (2.7%) | 4 (66.7%) | NA | |
| 2 (2.7%) | |||||
| Unknownc | 1 (1.4%) | 1 (1.4%) | 0 (0.0%) | NA | |
| 0 (0.0%) | |||||
| Tumor size (cm), median [range] | 2.0 [0.4-10.0] | 2.0 [0.4-10.0] | 4 [2.8-6.0] | 5 [2.0-7.0] | 0.077 |
| 2.05 [1.0-7.0] | 0.008 d | ||||
| Status (n, %) | 0.47 | ||||
| Alive | 38 (51.4%) | 20 (27.0%) | 0 (0.0%) | 1 (33.3%) | |
| 18 (24.3%) | |||||
| Dead | 34 (45.9%) | 27 (36.5%) | 1 (16.7%) | 2 (66.7%) | |
| 7 (9.5%) | |||||
| Unknownc | 2 (2.7%) | 2 (2.7%) | 5 (83.3%) | 0 (0.0%) | |
| 0 (0.0%) | |||||
| Overall survival (months), median [range] | 26.33 [0.39-160.3] | 27.5 [0.76-160.3] | - | 121.0 [109.8-132.3] | 0.64 |
| 19.2 [0.39-57.7] | |||||
| Morphological subtypee | 0.26 | ||||
| Pancreatobilliary | 24 (42.1%) | 12 (21.1%) | 2 (66.7%) | 0 (0.0%) | |
| 12 (21.1%) | |||||
| Intestinal | 23 (40.4%) | 13 (22.8%) | 1 (33.3%) | 3 (100.0%) | |
| 10 (17.5%) | |||||
| Ambigous | 10 (17.5%) | 7 (12.3%) | 0 (0.0%) | 0 (0.0%) | |
| 3 (5.3%) | |||||
| Nationality (n, %) | 0.07 | ||||
| Romania | 37 (50.0%) | 37 (50.0%) | 5 (83.3%) | 3 (100.0%) | |
| 0 (0.0%) | |||||
| Singapore | 37 (50.0%) | 12 (16.2%) | 1 (16.7%) | 0 (0.0%) | |
| 25 (33.8%) | |||||
Cohort 1-analyses performed: Sanger sequencing and SNP array (n=7), immunohistochemistry for ARID1A, and classification into pancreatobilliary, intestinal or mixed sub-type; Cohort 2-analysis performed: immunohistochemistry for ARID1A, and classification into pancreatobilliary, intestinal or mixed sub-type.
all the duodenal tumors and ampullary adenocarcinoma are included in Cohort 1 only;
p-values were calculated between all three group (the group designated as total ampullary tumors vs duodenal tumors vs ampullary adenoma) unless otherwise specified or if one group had NA or missing information; differences between age and tumor sizes were calculated by means of Kruskal-Wallis test; differences in overall survival were calculated by means of Log-Rank Mantel Cox test; the rest were tested by Fisher exact t-test or Chi-square test;
not included in p-value calculation;
p-value calculated between ampullary and duodenal tumors groups only;
only samples assessed for morphological subtype were included.
Figure 1.

Flow chart summarizing the patients groups, experiments and analysis performed within the study.
A total of 63 patients with available paraffin blocks were analyzed by immunohistochemistry for CK20, CDX2, MUC1 and MUC2 for subtype classification into pancreatobiliary, intestinal or ambiguous subtype (Figures 2A and 5A). Fifty-seven out of the 63 patients (90.4%) had ampullary adenocarcinomas, and 42.1% of samples in this group showed a pancreatobiliary profile by IHC staining, 40.4% showed IHC intestinal staining and 17.5% of ambiguous staining. In the duodenum tumor group, 2 out of 3 analyzed samples (66.7%) showed pancreatobiliary staining, while the adenoma group showed in all 3 cases (100%) intestinal staining (Figure 2A).
Figure 2.

Analysis of ARID1A and KRAS non-synonymous somatic mutations. A: Plot summarizing samples with non-synonymous somatic mutations in ARID1A and KRAS based on tumor site and immunohistochemistry subtype classification. B: Graphic representation of ARID1A and KRAS somatic non-synonymous mutations (modified from cBio portal). C: Type of KRAS non-synonymous somatic mutations based on tumor site and percentage of KRAS mutations in the whole cohort. D: Survival curves of ampullary adenocarcinoma cohort on mutational status of ARID1A and KRAS (in the wild type group were included patients with somatic or germline synonymous mutations but non-synonymous germline mutations were excluded.
Figure 5.
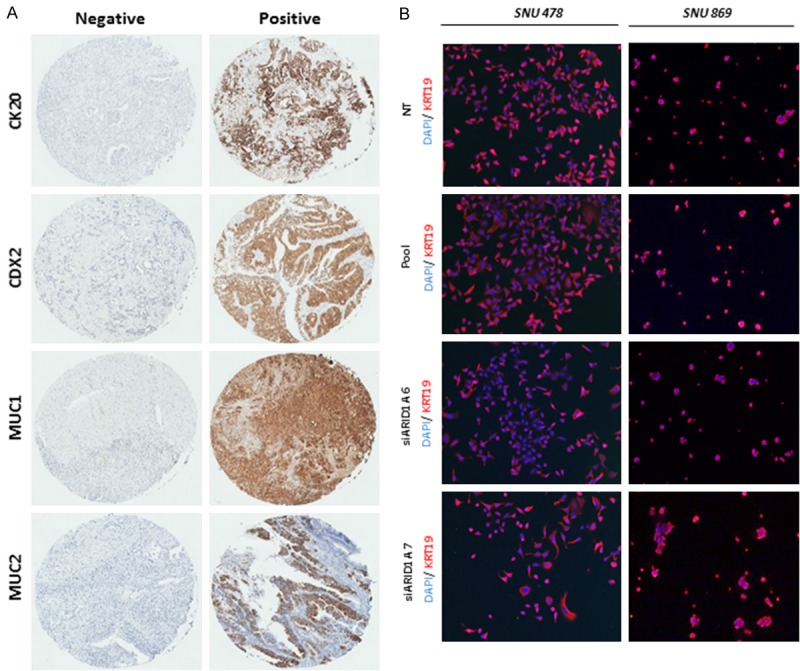
A: Immunohistochemistry of representative cases stained for CK20, CDX2, MUC1 and MUC2 as either negative or positive. B: Immunofluorescence of KRT19 in SNU-478 and SNU-869 cell lines (picture taken with 4× objective).
Analysis of the ampullary adenocarcinoma group alone showed that the tumor size was different in the analyzed subgroups and had medians ranging from 2.0 cm in the pancreatobiliary subgroup, 3.0 cm in the intestinal subgroup to 1.75 cm in the ambiguous subtype.
ARID1A and KRAS mutational analysis
A total of 49 consecutive cases of ampullary adenocarcinoma were analyzed for mutations in ARID1A (37 cases from Romania and 12 cases from Singapore). Given the high frequency of KRAS gene as an oncogenic driver in pancreatic adenocarcinoma, and that some of the ampullary cancers develop from the pancreatic lining, we also studied the mutations in the KRAS gene. As a comparison, we also analyzed a group of 6 duodenal adenocarcinomas (5 cases from Romania and 1 case from Singapore). Furthermore, to determine the involvement of ARID1A mutations in premalignant stages, a group of 3 ampullary adenomas (all from Romania) were also included in the analysis (Figure 2A).
Sequencing the 20 exons of ARID1A lead to the identification of 4 somatic ARID1A mutations in the ampullary adenocarcinoma group (8.2%), 2 somatic ARID1A mutations in the duodenal tumor group (33%), and 1 somatic mutation in the ampullary adenoma (33%) (Figure 2A). These mutations included two nonsense mutations (p.Q172X and p.R1276X), four frameshift insertion-deletion (p.G276f.s, p.P599fs, p.L2136 f.s, p.A2241fs) and one splice site (Figure 2B and Table 2), indicating functional loss of ARID1A in these samples and a reminiscent tumor suppressive role.
Table 2.
Detailed description of the ARID1A non-synonymous somatic mutations and associated protein expression
| Sample | Country | Histologic diagnostic/tumor location | Mutation type/Affected exon | Nucleotide changea | Protein Change | IHC | MSI | Predicted ARID1A status by SNP array | Amplification/deletion at gene locus | KRAS status | In COSMIC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 37417607 | SG | Adenocarcinoma with signet ring morphology/Ampullary carcinoma group | Deletion (exon 20) | c.6407_6423 del TGGCCACACCCCCCTTC | fs | Weak staining | Normal | LOH | Deletion | WT | No |
| 2000368 | SG | Adenocarcinoma/Ampullary carcinoma group | Deletion (exon 1) | c.827 del G | fs | Loss | MSH6 loss | LOH | No | p.G12V (s) | Yes |
| 20020287 | SG | Papillary adenocarcinoma mucinous type/Ampullary carcinoma group | Nonsense (exon 1) | c.514C>T | p.Q172X | Weak staining | MSH6 loss PMS loss | No | No | p.G12D (g) | No |
| 1346 | RO | Adenocarcinoma/Ampullary carcinoma group | Deletion (exon 20) | c.6723_6733 del GGCCAAGGTGG | fs | NA | NA | LOH | No | WT | No |
| 11158260 | SG | Adenocarcinoma/duodenum carcinomagroup | Nonsense (exon 15) | c.3826C>T | p.R1276X | Weak staining | MSH6 loss | No | No | p.G12D (s) | Yes |
| 1303 | RO | Adenocarcinoma/duodenum carcinoma group | Insertion (exon 3) | c.1795_1796 ins CACC | fs | NA | NA | LOH | Deletion | p.G12V (s) | No |
| 0986 | RO | Tubular adenoma low grade/Ampullary adenoma group | Splice site (exon 2) | Splice site+1G>T | Splice_site | Normal | Normal | No | No | WT | No |
SG-Singapore; RO-Romania; LOH-loss of heterozigosity at chromosome 1p, ARID1A locus; f.s-frame shift; WT-wild-type; s-somatic; g-germline; NA-insufficient/unavailable/not assessed sample.
Reference sequence CCDS285.1.
We further analyzed the loss of heterozygosity (LOH) in the 7 matched pairs of normal and tumoral samples that harbored mutations. The results were analyzed by ASCAT (Allele-Specific Copy number Analysis of Tumors) that indicated that 3 out of 4 analyzed cases from the ampullary group presented LOH at the gene locus (Table 2; Figure 3). In both ampullary and duodenal tumors LOH was identified in patients concomitantly harboring somatic frame shift indels suggesting loss of the functional protein.
Figure 3.
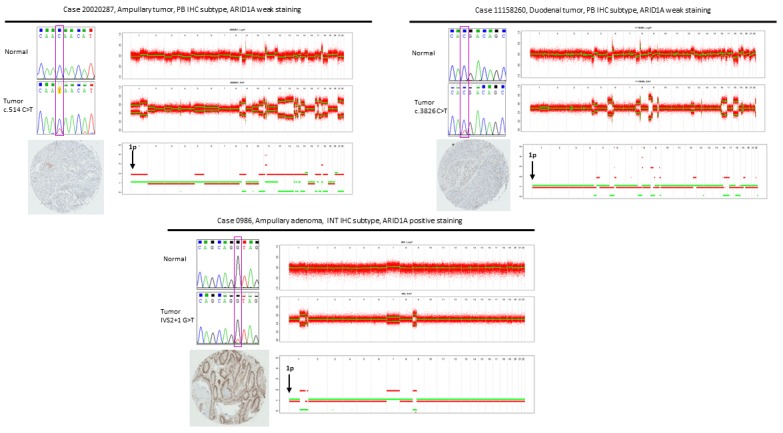
ARID1A genomic and proteomic characterization in representative cases with mutations. For each case, three panels are presented: upper left-sequencing chromatograms for normal and tumoral sample, bottom-left: immunohistochemistry staining, right-analysis of copy number alterations by ASCAT in tumoral sample (top panel-Log R ratio, middle panel: B allele frequency, bottom panel: allele specific copy numbers).
Collectively, the mutational data and LOH analysis support the tumor suppressor role of ARID1A in ampullary and duodenal adenocarcinomas.
Of note, the frequency of ARID1A somatic mutations in the ampullary group was distinct between Singapore and Romania cohort of 25% and 2.7%, respectively (P=0.04, Table 3). This observation suggests that the same cancer from different geographic regions might have different etiologies.
Table 3.
Non-synonymous somatic mutation frequencies of ARID1A and KRAS in Singapore and Romania cohorts
| Singapore | Romania | Total | p-value | q-valuea | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Ampullary carcinoma (n=12) | Duodenal carcinoma (n=1) | Ampullary carcinoma (n=37) | Duodenal carcinoma (n=5) | Ampullary carcinoma (n=49) | Duodenal carcinoma (n=6) | |||
| ARID1A | 25% (3) | 100% (1) | 2.7% (1) | 20% (1) | 8.16% (4) | 33.3% (2) | 0.04 | 0.08 |
| KRAS | 41.7% (5) | 100% (1) | 32.43% (12) | 40% (2) | 34.7% (17) | 50% (3) | 0.72 | 0.72 |
Benjamini-Hochberg correction.
Analysis on clinico-pathologic features of the patients showed that there was a statistical significant difference based on the degree of differentiation between ARID1A mutant and ARID1A wild-type group (P=0.01). Some of ARID1A mutant cases (2/4, 50%) had a poorly tumor grade (G3) that might suggest a more aggressive clinical phenotype of these cases.
The mutational status of ARID1A was shown to be statistically significant correlated with protein expression (P=0.023, Table 4) that points to the possible use of protein expression as a surrogate marker for ARID1A mutations. Analysis between the mutational status and overall survival (Figure 2D) or tumor recurrence showed no statistically significant association.
Table 4.
Analysis on ARID1A mutational status in the ampullary tumor group
| Characteristics | ARID1A status | KRAS status | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mutant (n=4) | Wild-typea (n=42) | p-valueb | Mutant (n=17) | Wild-typea (n=31) | p-valueb | |
| Age at surgery (yrs), median [range] | 59 [44-71] | 61 [40-78] | 0.52 | 66 [48-77] | 60 [40-78] | 0.4 |
| Gender (n, %) | 0.31 | 0.77 | ||||
| Female | 3 (75.0%) | 18 (42.9%) | 8 (47.1%) | 13 (41.9%) | ||
| Male | 1 (25.0%) | 24 (57.1%) | 9 (52.9%) | 18 (58.1%) | ||
| Differentiation degree (n, %) | 0.01 | 0.37 | ||||
| G1, G1-G2 | 1 (25.0%) | 20 (47.6%) | 6 (35.3%) | 16 (51.6%) | ||
| G2, G2-G3 | 1 (25.0%) | 18 (42.9%) | 7 (41.2%) | 12 (38.7%) | ||
| G3 | 2 (50.0%) | 2 (4.8%) | 3 (17.6%) | 2 (6.5%) | ||
| Unknownc | 0 (0.0%) | 2 (4.8%) | 1 (5.9%) | 1 (3.2%) | ||
| pN (n, %) | 1.0 | 0.21 | ||||
| N0 | 3 (75.0%) | 27 (64.3%) | 8 (47.1%) | 21 (67.7%) | ||
| N1 | 1 (25.0%) | 15 (35.7%) | 9 (52.9%) | 9 (29.0%) | ||
| Unknown | 0.0 (0.0%) | 0.0 (0.0%) | 0.0 (0.0%) | 1 (3.2%) | ||
| pT (n, %) | 0.7 | 0.85 | ||||
| T1 | 0 (0.0%) | 9 (21.4%) | 4 (23.5%) | 5 (16.1%) | ||
| T2 | 2 (50.0%) | 16 (38.1%) | 5 (29.4%) | 12 (38.7%) | ||
| T3 | 2 (50.0%) | 15 (35.7%) | 7 (41.2%) | 12 (38.7%) | ||
| T4 | 0 (0.0%) | 2 (4.8%) | 1 (5.9%) | 1 (3.2%) | ||
| Unknownc | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 1 (3.2%) | ||
| Tumor size (cm), median [range] | 1.75 [0.5-3.0] | 2.0 [0.4-10] | 0.54 | 2.5 [0.5-10] | 1.75 [0.4-6] | 0.4 |
| Status (n, %) | 1.0 | 0.12 | ||||
| Alive | 2 (50.0%) | 16 (38.1%) | 4 (23.5%) | 16 (51.6%) | ||
| Dead | 2 (50.0%) | 24 (57.1%) | 11 (64.7%) | 15 (48.4%) | ||
| Unknownc | 0 (0.0%) | 2 (4.8%) | 2 (11.8%) | 0 | ||
| Overall survival (months), median [range] | 39.21 [1.64-116.3] | 29.8 [0.76-160.3] | 0.48 | 21.96 [0.76-112.0] | 58.82 [1.25-160.31] | 0.087 |
| Kras mutational status (n, %) | 1.0 | |||||
| Kras mutant | 1 (25.0%) | 14 (33.3%) | _ | _ | _ | |
| Kras wild type | 3 (75.0%) | 28 (66.7%) | _ | _ | ||
| ARID1A IHC subtyped (n, %) | 0.72 | 0.1 | ||||
| Pancreatobilliary | 1 (50.0%) | 10 (35.7%) | 5 (38.5%) | 6 (33.3%) | ||
| Intestinal | 1 (50.0%) | 11 (39.3%) | 3 (23.1%) | 10 (55.6%) | ||
| Ambigous | 0 (0.0%) | 7 (25.0%) | 5 (38.5%) | 2 (11.1%) | ||
| ARID1A IHCd (n, %) | 0.023 | 0.7 | ||||
| ARID1A loss/weak staining | 3 (100.0%) | 6 (23.1%) | 4 (28.6%) | 4 (22.2%) | ||
| ARID1A positive | 0 (0.0%) | 20 (76.9%) | 10 (71.4%) | 14 (77.8%) | ||
only wild-type patients and patients with synonymous mutations were included; germline mutations were excluded;
p-values were calculated between ARID1A mutant vs wild-type and KRAS mutant vs wild-type; differences between age and tumor sizes were calculated by means of Mann-Whitney test; differences in overall survival were calculated by means of Log-Rank Mantel Cox test; the rest were tested by Fisher exact t-test or Chi-square test;
not included in p-value calculation;
only samples with available data were included.
KRAS gene sequencing identified 22 missense non-synonymous mutations, 17 in the ampullary adenocarcinoma group (34.7%), 3 in the duodenal tumors group (50%) and 2 in the ampullary adenoma group (66%) (Figure 2A, 2B). In the ampullary adenocarcinoma cohort, the majority of KRAS mutation occurred in the pancreatobiliary and ambiguous subtype (5/13, 38.5%) followed by the intestinal subtype (3/13, 23.1%). The frequency of KRAS mutations is much lower than the frequency reported in pancreatic cancer, but in line with previous reported results in ampullary adenocarcinoma [32,33].
In our cohort, it is possible that, from the point of view of the KRAS mutations, the ambiguous subtype is more related to the pancreatobiliary subtype than to the intestinal one. In the whole cohort, the majority of the mutations occurred in codon 12 (19/22 cases, 86.4%) with the most frequent mutation being c.35G>A (p.G12D) (12/22, 55%) (Figure 2C). Survival analysis in ampullary tumor group showed a significant statistically difference in the overall survival when comparing the patients with c.35G>A (p.G12D) mutations versus wild-type patients (P=0.028, Figure 2D). These findings support the prognostic implication of c.35G>A (p.G12D) KRAS mutations in ampullary cancers.
ARID1A protein expression is commonly weaker expressed or loss in cases with mutations
Given the relatively high rate of mutations (8.2%) seen in the subgroup of ampullary adenocarcinoma, immunohistochemistry staining was performed on whole slides or on TMA prepared from tumor blocks with a commercially available antibody (Figure 3). A total of 58 ampullary adenocarcinoma cases were stained for ARID1A. A loss or reduction of ARID1A expression was observed in 10 cases (17.2%), while the rest of 48 showed normal staining for ARID1A. Analysis of this group showed that loss or weak staining of ARID1A is more common in younger patients (P=0.04, Table 5).
Table 5.
Analysis on ARID1A immunohistochemistry status in the ampullary tumors group
| Characteristics | ARID1A loss or weak staining (n=10) | ARID1A positive staining (n=48) | p-valuea |
|---|---|---|---|
| Age at surgery (yrs), median [range] | 54.0 [38.0-71.0] | 67.5 [41.0-79.0] | 0.04 |
| Gender (n, %) | |||
| Female | 6 (60.0%) | 19 (39.6%) | 0.30 |
| Male | 4 (40.0%) | 29 (60.4%) | |
| Differentiation degree (n, %) | 0.29 | ||
| G1, G1-G2 | 2 (20.0%) | 11 (22.9%) | |
| G2, G2-G3 | 5 (50.0%) | 30 (62.5%) | |
| G3 | 3 (30.0%) | 5 (10.4%) | |
| Unknownb | 0 (0.0%) | 2 (4.2%) | |
| pN (n, %) | 0.29 | ||
| N0 | 5 (50.0%) | 33 (68.75%) | |
| N1 | 5 (50.0%) | 15 (31.25%) | |
| pT (n, %) | 0.87 | ||
| T1 | 1 (10.0%) | 7 (14.6%) | |
| T2 | 5 (50.0%) | 19 (39.6%) | |
| T3 | 3 (30.0%) | 19 (39.6%) | |
| T4 | 1 (10.0%) | 3 (6.3%) | |
| Tumor size (cm), median [range] | 2.5 [1.0-3.0] | 2.0 [0.4-10.0] | 0.57 |
| Status (n, %) | |||
| Alive | 5 (50.0%) | 27 (56.2%) | 1.0 |
| Dead | 4 (40.0%) | 21 (43.8%) | |
| Unknownb | 1 (10.0%) | 0 (0.0%) | |
| Overall survival (months), median [range] | 39.2 [1.64-127.9] | 24.9 [0.39-160.3] | 0.37 |
| ARID1A IHC subtype (n, %) | |||
| Pancreatobilliary | 3 (30.0%) | 21 (43.8%) | 0.59 |
| Intestinal | 5 (50.0%) | 18 (35.7%) | |
| Ambigous | 1 (10.0%) | 9 (18.8%) | |
| Unknownb | 1 (10.0) | 0 (0.0%) |
differences between age and tumor sizes were calculated by means of Mann-Whitney test; differences in overall survival were calculated by means of Log-Rank Mantel Cox test; the rest were tested by Fisher exact t-test or Chi-square test;
not included in p-value calculation.
The loss/weak staining of ARID1A protein in the cases without detectable mutation or with germline mutation in the tumor might suggest that other mechanisms such as epigenetic silencing of the protein may be involved in down regulating ARID1A in ampullary cancers. In addition, the duodenal tumors showed reduced staining, while the ampullary adenoma cases showed no loss (normal) of staining for ARID1A.
Functional analyses
In order to investigate ARID1A function in vitro, we used two immortalized cell lines derived from ampulla of Vater tumors, SNU-478 and SNU-869 with wild-type ARID1A. ARID1A knockdown was confirmed by qPCR and by Western blotting at 48 hours post-transfection. Cell proliferation was significantly promoted after siRNA treatment in both cell lines (Figure 4A, 4B). In order to test if ARID1A knockdown also induces a pro-invasive phenotype, we assessed by qPCR the expression of a panel of markers associated with epithelial to mesenchymal transition (EMT). KRT19, MMP1 and SOX4 were tested in both cell lines, and CDH1 (E-Cadherin) and vimentin were tested only in SNU-869 cells. In both knock-down cell lines (SNU-478 and SNU-869) SOX4 was found to be up-regulated, while vimentin and TWIST1 were found up-regulated only in the SNU-869 knock-down cell lines. Collectively, the functional analysis data indicate that ARID1A knock-down induces an increase in cell proliferation in ampullary cancer cells, indicative for the tumor suppressive nature of the gene, and promote the expression of markers associated with EMT phenotype (Figures 4C and 5B).
Figure 4.
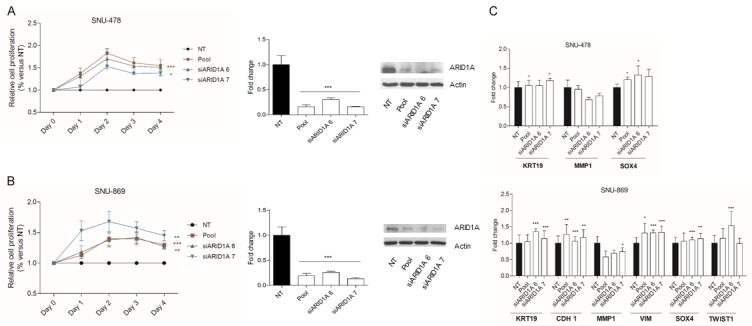
Functional studies in SNU-478 and SNU-869 cell lines. A: Relative proliferation curves, qPCR and western blot in SNU-478 cell line treated with non-targeting siRNA or Pool siRNA, siARID1A6 and siARID1A7. B: Relative proliferation curves, qPCR and Western blot in SNU-869 cell line treated with non-targeting siRNA or Pool siRNA, siARID1A6 and siARID1A7. C: Real time PCR analysis of EMT markers in SNU-478 and SNU-869 cell lines (in SNU-478, no amplification was detected for VIM and TWIST1 while CDH1 was reported to be hypermethylated (ref 34, 35).
Discussion
ARID1A has emerged from the whole exome and genome studies as one of the most commonly mutated gene in human cancers [23]. Loss of its expression is due to nonsense and frame-shift mutations in the gene-coding region that leads to mRNA decay and protein truncation [17].
It has been suggested that ARID1A mutations carry prognostic significance, but the results are inconsistent [21,38,39,40]. In liver cancer cell lines, ARID1A knock down promoted migration and invasion and had been suggested that loss-of-function ARID1A mutations may be a crucial event in hepatocellular carcinoma invasion and metastasis [29].
In our study, we found a rather high frequency of ARID1A somatic mutations (8.2%) while loss or weak ARID1A staining confirmed by immunohistochemistry was detected in 17.2% of the ampullary cancers. The presence of mutations was correlated with loss/weak staining of the protein. Further analysis showed that there is a significant correlation between degree of differentiation of the ampullary adenocarcinoma and ARID1A mutational status.
Our mutational screening extended to a group of duodenal tumors and ampullary adenomas identified two and respectively one additional ARID1A mutations in these groups. These results may be indicative of ARID1A involvement in early stages of carcinogenesis. Previous studies had also identified ARID1A alterations in non-cancerous lesions either as mutations, in a colon adenomas with moderate dysplasia and with APC mutations [41] or as loss of ARID1A protein in ovarian clear cell carcinoma in precursor lesions like non-atypical endometriosis adjacent to carcinoma or benign clear-cell adenofibroma [42]. In our study, protein expression analysis showed that the weak staining was observed in the duodenal tumor group but not in the ampullary adenoma group.
Given the degree of correlation between the presence of mutations and loss/weak staining in ampullary cancers, our data indicate that IHC might be a reliable method for the detection of ARID1A mutations in ampullary carcinoma. On the other hand, there were samples that had no loss of staining of ARID1A indicating that additional unknown mechanisms are involved in the retention of ARID1A protein.
A better overall survival of 39.2 months was observed in the group of ampullary adenocarcinomas with mutant ARID1A compared to wild-type ARID1A that had an overall survival of 29.8 months. The same trend was observed when comparing patients with ARID1A evaluated by immunohistochemistry as loss or weak and negative staining (39.2 versus 24.9 months). Additionally loss or weak ARID1A protein expression was observed in younger patients.
Analysis on KRAS mutations showed a worse overall survival between the ampullary cancer patients with c.35G>A (G12D) mutations compared to wild type ones. As KRAS is a well-known driver of pancreatic cancer and ampullary adenocarcinoma might emerge from the pancreatic lining there is the possibility that a sub-group of patients with a poor survival exists among ampullary cancer group.
The patients with concurrent ARID1A and KRAS mutations all had a pT stage 3 or 4 and tumor recurrence. It is not yet clear if in ampullary cancers ARID1A potentiate KRAS mutations or KRAS mutations overcome ARID1A mutations.
Interestingly, in our study there was a difference in the frequency of ARID1A mutation in ampullary adenocarcinoma in the Asian versus European patients. This difference might be due to different racial genetic differences and different exposure to environmental factors, as we have previously shown for cholangiocarcinoma [43].
The molecular differences in ampullary cancers between intestinal and pancreatobiliary type were shown by the different frequencies of KRAS and APC mutations with more frequent KRAS mutation detected in pancreatobiliary subtype [44]. This observation is in line with our study where we found 38.5% KRAS alterations in pancreatobiliary subtype versus 23.1% in intestinal subtype. The fact that we also found 38.5% KRAS mutations in the ambiguous subtype suggest that these tumors may be more related to the pancreatobiliary subtype.
Functional analysis of the ARID1A alteration in ampullary cancer by means of siRNA knock-down approaches, were consistent with a tumors suppressive function of the gene. Knock-down experiments in two cell lines with intact ARID1A promoted some EMT related genes phenotype with SOX4 consistently up-regulated in both cell lines. SOX4 was shown to be critical for EMT in breast cancer and to regulates EMT-relevant genes, among them Ezh2 [45,46]. Moreover, knock-down of ARID1A, ARID1B and SMARCA4 in HPDE pancreatic cell line lead to down-regulation of genes that are up-regulated with knock-down of EZH2 or HDAC. As EZH2 is the catalytic subunit of PRC2 complex (polycomb repressive complex 2), these results lead to the conclusion that SWI/SNF might oppose PRC2 in PDAC cell lines [31] a process that might be mediated by SOX4. As EZH2 is a therapeutically duggable target this results might have direct clinical implication.
The relatively high mutation rate of ARID1A in ampullary cancer shows the critical genomic and epigenomic interplay that leads to tumor development. As recent studies are focusing in targeting ARID1A mutations by showing various synthetic lethality synergies [47,48], tailoring treatment based on ARID1A and KRAS mutational status might soon prove to be an effective therapeutic strategy in ampullary cancers.
In summary, the data presented in this study highlight a potential involvement of ARID1A in early stages of carcinogenesis, together with other factors that are essential for tumor transformation, and show a possible influence of different factors in ampullary cancers tumorigenesis based on geographic regions.
Acknowledgements
This work was supported by project “Hepatocellular carcinoma stratification based on noninvasive markers” (HEPMARK), EEA-JRP-Romania-Norway no. 4SEE.). We thank Liang Kai Koh for his support. D.G.D.’s research is supported by the National Institutes of Health Grants P01-CA080124, R01-CA159258 and by the Warshaw Institute for Pancreatic Cancer Research.
Disclosure of conflict of interest
None.
References
- 1.Demeure MJ, Craig DW, Sinari S, Moses TM, Christoforides A, Dinh J, Izatt T, Aldrich J, Decker A, Baker A, Cherni I, Watanabe A, Koep L, Lake D, Hostetter G, Trent JM, Von Hoff DD, Carpten JD. Cancer of the ampulla of vater: analysis of the whole genome sequence exposes a potential therapeutic vulnerability. Genome Med. 2012;4:56. doi: 10.1186/gm357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang DK, Jamieson NB, Johns AL, Scarlett CJ, Pajic M, Chou A, Pinese M, Humphris JL, Jones MD, Toon C, Nagrial AM, Chantrill LA, Chin VT, Pinho AV, Rooman I, Cowley MJ, Wu J, Mead RS, Colvin EK, Samra JS, Corbo V, Bassi C, Falconi M, Lawlor RT, Crippa S, Sperandio N, Bersani S, Dickson EJ, Mohamed MA, Oien KA, Foulis AK, Musgrove EA, Sutherland RL, Kench JG, Carter CR, Gill AJ, Scarpa A, McKay CJ, Biankin AV. Histomolecular phenotypes and outcome in adenocarcinoma of the ampulla of vater. J. Clin. Oncol. 2013;31:1348–1356. doi: 10.1200/JCO.2012.46.8868. [DOI] [PubMed] [Google Scholar]
- 3.Kimura W, Ohtsubo K. Incidence, sites of origin, and immunohistochemical and histochemical characteristics of atypical epithelium and minute carcinoma of the papilla of vater. Cancer. 1988;61:1394–1402. doi: 10.1002/1097-0142(19880401)61:7<1394::aid-cncr2820610720>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 4.Ang DC, Shia J, Tang LH, Katabi N, Klimstra DS. The utility of immunohistochemistry in subtyping adenocarcinoma of the ampulla of vater. Am J Surg Pathol. 2014;38:1371–1379. doi: 10.1097/PAS.0000000000000230. [DOI] [PubMed] [Google Scholar]
- 5.Chung YE, Kim MJ, Park MS, Choi JY, Kim H, Kim SK, Lee M, Kim HJ, Choi JS, Song SY, Kim KW. Differential features of pancreatobiliary- and intestinal-type ampullary carcinomas at MR imaging. Radiology. 2010;257:384–393. doi: 10.1148/radiol.10100200. [DOI] [PubMed] [Google Scholar]
- 6.Adsay V, Ohike N, Tajiri T, Kim GE, Krasinskas A, Balci S, Bagci P, Basturk O, Bandyopadhyay S, Jang KT, Kooby DA, Maithel SK, Sarmiento J, Staley CA, Gonzalez RS, Kong SY, Goodman M. Ampullary region carcinomas: definition and site specific classification with delineation of four clinicopathologically and prognostically distinct subsets in an analysis of 249 cases. Am J Surg Pathol. 2012;36:1592–1608. doi: 10.1097/PAS.0b013e31826399d8. [DOI] [PubMed] [Google Scholar]
- 7.Overman MJ, Zhang J, Kopetz S, Davies M, Jiang ZQ, Stemke-Hale K, Rummele P, Pilarsky C, Grutzmann R, Hamilton S, Hwang R, Abbruzzese JL, Varadhachary G, Broom B, Wang H. Gene expression profiling of ampullary carcinomas classifies ampullary carcinomas into biliary-like and intestinal-like subtypes that are prognostic of outcome. PLoS One. 2013;8:e65144. doi: 10.1371/journal.pone.0065144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeo CJ, Sohn TA, Cameron JL, Hruban RH, Lillemoe KD, Pitt HA. Periampullary adenocarcinoma: analysis of 5-year survivors. Ann Surg. 1998;227:821–831. doi: 10.1097/00000658-199806000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lillemoe KD, Cameron JL, Hardacre JM, Sohn TA, Sauter PK, Coleman J, Pitt HA, Yeo CJ. Is prophylactic gastrojejunostomy indicated for unresectable periampullary cancer? A prospective randomized trial. Ann Surg. 1999;230:322–328. doi: 10.1097/00000658-199909000-00005. discussion 328-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heby M, Elebro J, Nodin B, Jirstrom K, Eberhard J. Prognostic and predictive significance of podocalyxin-like protein expression in pancreatic and periampullary adenocarcinoma. BMC Clin Pathol. 2015;15:10. doi: 10.1186/s12907-015-0009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holzheimer RG, Mannick JA, editors. Surgical treatment: evidence-based and problem-oriented. Munich: 2001. [PubMed] [Google Scholar]
- 12.He J, Ahuja N, Makary MA, Cameron JL, Eckhauser FE, Choti MA, Hruban RH, Pawlik TM, Wolfgang CL. 2564 resected periampullary adenocarcinomas at a single institution: trends over three decades. HPB (Oxford) 2014;16:83–90. doi: 10.1111/hpb.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol. 2013;43:849–855. doi: 10.1093/jjco/hyt101. [DOI] [PubMed] [Google Scholar]
- 14.Tang L, Nogales E, Ciferri C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol. 2010;102:122–128. doi: 10.1016/j.pbiomolbio.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol. 2000;20:3137–3146. doi: 10.1128/mcb.20.9.3137-3146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15:655–664. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, Sun YH, Yang PY, Liu F. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35:867–876. doi: 10.1093/carcin/bgt398. [DOI] [PubMed] [Google Scholar]
- 20.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, Chow C, Fee J, Zayed A, Prentice L, Melnyk N, Turashvili G, Delaney AD, Madore J, Yip S, McPherson AW, Ha G, Bell L, Fereday S, Tam A, Galletta L, Tonin PN, Provencher D, Miller D, Jones SJ, Moore RA, Morin GB, Oloumi A, Boyd N, Aparicio SA, Shih Ie M, Mes-Masson AM, Bowtell DD, Hirst M, Gilks B, Marra MA, Huntsman DG. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, Lee SP, Ho SL, Chan AK, Cheng GH, Roberts PC, Rejto PA, Gibson NW, Pocalyko DJ, Mao M, Xu J, Leung SY. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 22.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Zhang Y, Yang Y, Niu M, Sun S, Ji H, Ma Y, Yao G, Jiang Y, Shan M, Zhang G, Pang D. Frequent low expression of chromatin remodeling gene ARID1A in breast cancer and its clinical significance. Cancer Epidemiol. 2012;36:288–293. doi: 10.1016/j.canep.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Cornen S, Adelaide J, Bertucci F, Finetti P, Guille A, Birnbaum DJ, Birnbaum D, Chaffanet M. Mutations and deletions of ARID1A in breast tumors. Oncogene. 2012;31:4255–4256. doi: 10.1038/onc.2011.598. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Nagl NG Jr, Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112:636. doi: 10.1002/ijc.20450. [DOI] [PubMed] [Google Scholar]
- 28.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, Fei QL, Chen H, Cai B, Zhou B, Xiao HS, Qin LX, Han ZG. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 30.Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D, Hruban RH, Eshleman JR, Iacobuzio-Donahue CA, Goggins M, Maitra A, Malek SN, Powell S, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33:100–103. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, Maitra A, Pollack JR. Convergent structural alterations define switch/sucrose nonfermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109:E252–259. doi: 10.1073/pnas.1114817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gingras MC, Covington KR, Chang DK, Donehower LA, Gill AJ, Ittmann MM, Creighton CJ, Johns AL, Shinbrot E, Dewal N, Fisher WE Australian Pancreatic Cancer Genome Initiative. Pilarsky C, Grutzmann R, Overman MJ, Jamieson NB, Van Buren G 2nd, Drummond J, Walker K, Hampton OA, Xi L, Muzny DM, Doddapaneni H, Lee SL, Bellair M, Hu J, Han Y, Dinh HH, Dahdouli M, Samra JS, Bailey P, Waddell N, Pearson JV, Harliwong I, Wang H, Aust D, Oien KA, Hruban RH, Hodges SE, McElhany A, Saengboonmee C, Duthie FR, Grimmond SM, Biankin AV, Wheeler DA, Gibbs RA. Ampullary cancers harbor ELF3 tumor suppressor gene mutations and exhibit frequent WNT dysregulation. Cell Rep. 2016;14:907–919. doi: 10.1016/j.celrep.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yachida S, Wood LD, Suzuki M, Takai E, Totoki Y, Kato M, Luchini C, Arai Y, Nakamura H, Hama N, Elzawahry A, Hosoda F, Shirota T, Morimoto N, Hori K, Funazaki J, Tanaka H, Morizane C, Okusaka T, Nara S, Shimada K, Hiraoka N, Taniguchi H, Higuchi R, Oshima M, Okano K, Hirono S, Mizuma M, Arihiro K, Yamamoto M, Unno M, Yamaue H, Weiss MJ, Wolfgang CL, Furukawa T, Nakagama H, Vogelstein B, Kiyono T, Hruban RH, Shibata T. Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell. 2016;29:229–240. doi: 10.1016/j.ccell.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ku JL, Yoon KA, Kim IJ, Kim WH, Jang JY, Suh KS, Kim SW, Park YH, Hwang JH, Yoon YB, Park JG. Establishment and characterisation of six human biliary tract cancer cell lines. Br J Cancer. 2002;87:187–193. doi: 10.1038/sj.bjc.6600440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ku JL, Park JG. Biology of SNU cell lines. Cancer Res Treat. 2005;37:1–19. doi: 10.4143/crt.2005.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, Yu W, Wu Y, Lee M, Wu J, Poh D, Wan WK, Rha SY, So J, Salto-Tellez M, Yeoh KG, Wong WK, Zhu YJ, Futreal PA, Pang B, Ruan Y, Hillmer AM, Bertrand D, Nagarajan N, Rozen S, Teh BT, Tan P. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 39.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, O’Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA Jr, Von Hoff DD, Johansen JS, Drebin JA, Velculescu VE. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686. doi: 10.1038/ncomms8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, Edgren H, Kallioniemi O, Aleynikova O, Przybytkowski E, Malcolm K, Mousses S, Tonin PN, Basik M. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene. 2012;31:2090–2100. doi: 10.1038/onc.2011.386. [DOI] [PubMed] [Google Scholar]
- 41.Nikolaev SI, Sotiriou SK, Pateras IS, Santoni F, Sougioultzis S, Edgren H, Almusa H, Robyr D, Guipponi M, Saarela J, Gorgoulis VG, Antonarakis SE, Halazonetis TD. A single-nucleotide substitution mutator phenotype revealed by exome sequencing of human colon adenomas. Cancer Res. 2012;72:6279–6289. doi: 10.1158/0008-5472.CAN-12-3869. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25:615–624. doi: 10.1038/modpathol.2011.189. [DOI] [PubMed] [Google Scholar]
- 43.Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, Ooi L, Chung A, Chow P, Cheow PC, Lee SY, Choo SP, Tan IB, Duda D, Nastase A, Myint SS, Wong BH, Gan A, Rajasegaran V, Ng CC, Nagarajan S, Jusakul A, Zhang S, Vohra P, Yu W, Huang D, Sithithaworn P, Yongvanit P, Wongkham S, Khuntikeo N, Bhudhisawasdi V, Popescu I, Rozen SG, Tan P, Teh BT. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45:1474–1478. doi: 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- 44.Hechtman JF, Liu W, Sadowska J, Zhen L, Borsu L, Arcila ME, Won HH, Shah RH, Berger MF, Vakiani E, Shia J, Klimstra DS. Sequencing of 279 cancer genes in ampullary carcinoma reveals trends relating to histologic subtypes and frequent amplification and overexpression of ERBB2 (HER2) Mod Pathol. 2015;28:1123–1129. doi: 10.1038/modpathol.2015.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, Feng J, Zhang Y, Gao H, Liu DX, Lu J, Huang B. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012;72:4597–4608. doi: 10.1158/0008-5472.CAN-12-1045. [DOI] [PubMed] [Google Scholar]
- 46.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schubeler D, van Nimwegen E, Christofori G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23:768–783. doi: 10.1016/j.ccr.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 47.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M, Conejo-Garcia JR, Speicher DW, Zhang R. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samartzis EP, Gutsche K, Dedes KJ, Fink D, Stucki M, Imesch P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget. 2014;5:5295–5303. doi: 10.18632/oncotarget.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]