

Abstract
Understanding mechanisms of cancer metastasis is crucial for reduction of cancer mortality. Acyl-CoA medium-chain synthetase 3 (ACSM3) is an acyl-CoA synthetase which takes part in the first step of fatty acid metabolism. However, the expression, clinical significance and biological function of ACSM3 remain unknown in hepatocellular carcinoma (HCC). In this study, the expression and prognostic relevance of ACSM3 were investigated by tissue microarray and HCC clinical samples. Migration and invasion assays were carried out for functional analysis in vitro and a xenograft model was used to analyze the effects of ACSM3 on cancer metastasis in vivo. Furthermore, human phospho-kinase array assays were performed to explore molecular mechanisms of ACSM3 in HCC. The results showed ACSM3 was downregulated in HCC tissues. HCC patients with low expression of ACSM3 exhibited poor prognosis. Overexpression of ACSM3 attenuated migration and invasion of HCC cells in vitro and in vivo and downregulated the phosphorylation of WNK1 and AKT. Our findings indicate ACSM3 is a novel prognostic marker and a potential therapeutic target for HCC.
Keywords: ACSM3, hepatocellular carcinoma, prognosis, metastasis
Introduction
Hepatocellular carcinoma (HCC) is one of the most common worldwide visceral neoplasms [1]. Although liver surgical resection, chemotherapy, radiotherapy and transplantation have become curative therapies in the early stages of the disease, the prognosis of HCC patients still remains poor due to the high rate of recurrence and metastasis [2]. Therefore, it is critical to identify novel genes related to metastasis.
ACSM3 is one member of acyl-CoA medium-chain (C4-C14) synthetase (ACSM) family, which at least comprise 5 proteins; O-ACSM, SAH and ACSM1~3. O-ACSM is specifically expressed in the olfactory epithelium and may play a role in processing odorants in a zone-specific manner [3]. The other four members have a putative mitochondrial targeting signal at the N terminus of their primary amino acid sequence, and are localized in the mitochondrial matrix [4]. The linkage disequilibrium with genetic polymorphism of them may contribute to multiple risk factors [4]. Especially, SAH and ACSM1, which are mainly detected in the liver and kidney, are associated with plasma high-density lipoprotein cholesterol levels [5]. ACSM2 Leu513Ser polymorphism is also reported to have an association with risk factors of the metabolic syndrome [6]. However, biological functions of ACSM3 remain unclear in human cancer.
In this study, we investigated the expression, explored clinicopathologic significance, and addressed biological functions of ACSM3 in HCC. Our data indicated ACSM3 was downregulated in HCC, and could be served as a promising biomarker for HCC prognosis. ACSM3 inhibited metastasis of HCC in vitro and in vivo, and we speculated the inhibitory effects may be induced by downregulating phosphorylation of WNK1 and AKT.
Materials and methods
Clinical samples and tissue microarray
Primary HCC tissues and their adjacent histologic normal liver tissues were obtained between 2004 and 2005, 8 pairs of which were chosen to perform western blot to detect ACSM3 protein level, and 70 pairs of which were chosen to perform quantitative real-time PCR to detect mRNA level of ACSM3. All samples were frozen at -80°C until RNAs and proteins were extracted.
Tissue microarrays (TMAs) were purchased from Eastern Hepatobiliary Surgery Hospital (Shanghai, PR China), the HCC specimens included in the TAMs were obtained from patients who underwent hepatic resection between 1996 and 2001, and followed until October 2010. No patients in this study received either radiotherapy or chemotherapy before the surgery, and exhibited any other cancers co-occurrence. Written informed consent was obtained from all patients, and this study was performed on the basis of the protocol approved by the Declaration of Helsinki. The Edmondson grading system was used to classify tumor stage [7] and the AJCC/UICC classification system was applied to grade clinical staging. The overall survival (OS) was defined as the interval between the dates of surgery and death. The time to recurrence (TTR) was calculated from the date of tumor resection until the detection of tumor recurrence, death or the last observation.
Protein extraction and western blot
Cell lysates were harvested by T-PER tissue protein extraction reagent (Thermo Scientific) with a cocktail of protease inhibitors, phosphatase inhibitors, and PMSF. Similar amounts of proteins were separated by 10% SDS-PAGE and transferred to NC membranes. After blocking with 5% skimmed milk dissolved in PBST (1% Tween in PBS), the membranes were incubated with primary antibodies at 4°C overnight. Subsequently, the membranes were washed three times in PBST, and probed with HRP-conjugated secondary antibodies for 1 h at room temperature. After washing three times in PBST again, target protein bands were visualized by a Bio-rad ChemiDoc XRS system (USA).
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen) and reversely transcribed using PrimeScript™ RT Reagent Kit (TaKaRa Biotechnology). Quantitative real-time PCR was subsequently performed by an ABI 7500 instrument (Applied Biosystems Inc). The PCR conditions were as follows: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. The relative expression levels were determined by 2-ΔΔCt method. One set of primers used for ACSM3 was as follows: Forward: 5’-GGACAGACTGAAACGGTGCT-3’; Reverse: 5’-GCCAAATGGTCGGTTGGGTA-3’.
Cell culture and transfection
Human normal liver cell lines L02 and SMMC7721 were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. SK-Hep1, HepG2, and Hep3B were purchased from the American Type Culture Collection (ATCC, VA, USA). MHCC97H, MHCC97L, and HCCLM3 cell lines were provided by the Liver Cancer Institute of Zhongshan Hospital of Fudan University (Shanghai, China). Huh7 cells were established from Riken Cell Bank (Tsukuba, Japan). All the cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, CA, USA) supplemented with 10% FBS (Gibco) at 37°C in an atmosphere of 5% CO2.
The ACSM3 ORF sequence (NM_005622.3) was amplified by PCR using specific primers and cloned into the lentiviral expression vector pWPXL (Addgene) to develop a pWPXL-ACSM3 recombinant plasmid. Virus packaging was performed by the co-transfection of pWPXL-ACSM3, packaging plasmid psPAX2 (Addgene) and envelope plasmid pMD2.G (Addgene) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in HEK 293T cells. Viruses were harvested 48 h after transfection, and viral titers were determined. HepG2 and SMMC7721 were infected with either recombinant lentivirus constitutively expressing ACSM3, or control empty vector.
Immunohistochemistry
Immunohistochemistry analysis was performed as described previously [8,9]. The sections were incubated with primary rabbit anti-ACSM3 monoclonal antibody (1:100, Santa Cruz Biotechnology) at 4°C overnight, and then carried out according to the protocol of GTVisionTM III Detection System/Mo & Rb IHC Kit (Gene Tech Company limited). Average sum of integrated optical density (IOD) of each sample was calculated using Image J software.
Cell migration and invasion assays
Cell migration and invasion assays were performed using Transwell filter chambers (8μm pore size, BD Falcon, CA, USA) and 24-well transwell plates (BD Biosciences, CA, USA). For migration, 5×104 SMMC7721 or HepG2 cells in 200 μl serum-free DMEM were seeded in the upper chamber of a transwell and 800 μl medium supplemented with 15% FBS was added to the lower chamber. For invasion, transwell filter chambers were coated with Matrigel (BD Biosciences) according to manufacturer’s instructions, and followed the same protocol as for migration. After incubated at 37°C for 24 h (migration), or 48 h (invasion), cells were fixed and stained with Crystal violet (1% in methanol) for 15 min. The stained cells were imaged through an inverted microscope (Olympus), and five random microscopic fields were counted per well for each group. The experiment was performed in triplicates and repeated three times independently.
Cell proliferation
Proliferation ability of HCC cells was measured by Cell Counting Kit-8 reagent (CCK8, Dojin Laboratories, Kumamoto, Japan). Approxi-mately, 1×103 cells were seeded per well in 96-well flat-bottom plates, after a respective period of time (0, 12, 24, 48, 72 h), 10 μl CCK-8 reagent was added to each well and plate was incubated for 2 h at 37°C. Absorbance was read at 450 nm, using a microplate reader (BIO-TEK, USA).
Statistical analysis
Kaplan-Meier analysis was used to assess survival analysis and log-rank test was chosen to compare patients’ survival between subgroups. Experiments were performed in triplicates and data were presented as mean ± SEM. Two-tailed student’s tests were used to analyze differences (P<0.05 was considered significant). Statistical analysis was performed using GraphPad Prism 5.01 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
ACSM3 is downregulated in HCC tissues
Oncomine data-mining analysis showed ACSM3 expression was mainly downregulated in HCC (Figure 1A). Therefore, we detected ACSM3 expression in both mRNA (Figure 1B-D) and protein (Figure 1E) levels. As the results shown in Figure 1B-D, ACSM3 expression in tumorous (T) tissue was obviously downregulated compared to adjacent non-tumorous (N) liver tissue of the same HCC patient. To validate these results, we next performed IHC analysis for ACSM3 using a tissue microarray containing 77 paired samples of HCC tumorous (T) tissue and adjacent non-tumorous (N) liver tissue. The IHC results showed the staining density of ACSM3 in non-tumorous (N) tissue group was obviously stronger than that in HCC tumorous (T) tissue group (Figure 1F, 1G).
Figure 1.
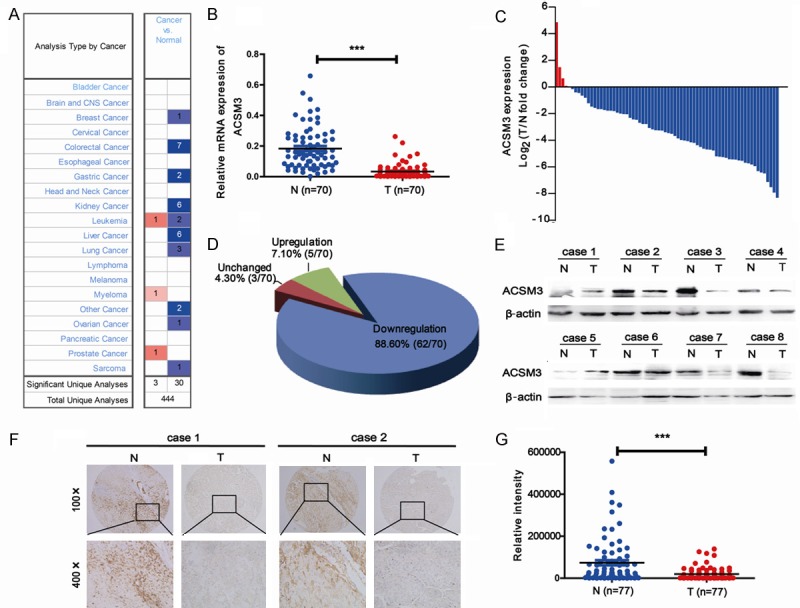
Downregulation of ACSM3 in HCC tissues. A: Downregulation of ACSM3 was shown in Oncomine database. B-D: mRNA levels of ACSM3 were detected in 70 paired samples of tumorous (T) tissues and corresponding adjacent nontumorous (N) liver tissues from HCC patients by quantitative real-time PCR. E: Protein levels of ACSM3 were detected in 8 paired samples of tumorous (T) tissues and corresponding adjacent nontumorous (N) liver tissues from HCC patients by western blot. F, G: IHC staining of ACSM3 in a microarray includes 77 paired samples of tumorous (T) tissues and matched adjacent nontumorous (N) liver tissues. Integrated optical density (IOD) for ACSM3 was obtained for analysis. ***; P<0.001.
Clinical significance of ACSM3 in HCC patients
In order to determine the value of ACSM3 for the prognosis of postsurgical HCC patients, tissue microarray analysis of HCC tissues from 235 patients underwent liver resection was performed. According to the density of staining (Figure 2A), levels of ACSM3 protein in tumor tissues were classified as high expression (score ++, moderate; score +++, strong) in 55 cases (55/235, 23.4%), and low expression (score +, weak) or not stained (score -, negative) in 180 cases (180/235, 76.6%). Clinical and pathological characteristics of these patients were listed in Table 1. We found ACSM3 had a correlation with serum AFP (P=0.042), but no association with other clinicopathological parameters.
Figure 2.
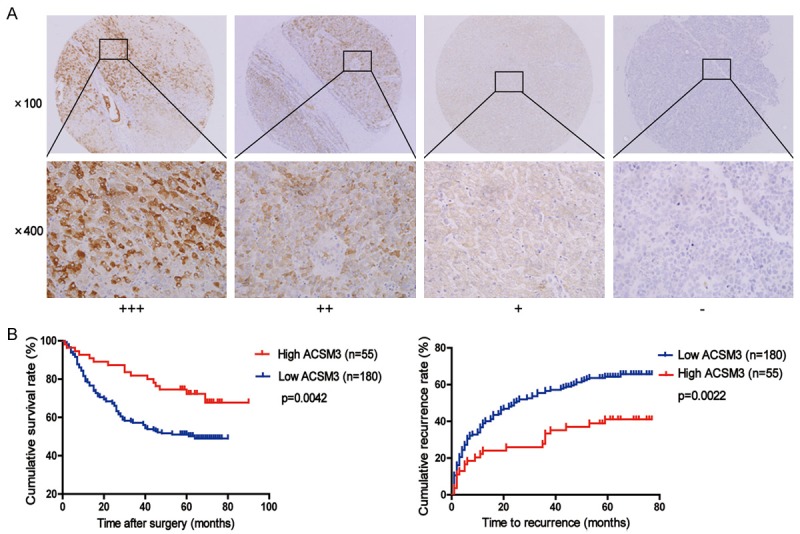
Downregulation of ACSM3 is correlated with poor prognosis in HCC patients. A: Representative photomicrographs showed strong (+++), moderate (++), weak (+), or negative (-) staining of ACSM3 in HCC specimens (magnification, ×100, ×400). B: Kaplan-Meier analysis of overall survival and time to recurrence in 235 HCC patients based on ACSM3 expression.
Table 1.
Correlation between ACSM3 expression and clinicopathological features in 235 HCC patients
| ACSM3 | |||
|---|---|---|---|
|
| |||
| Variable | Low (n=180) | High (n=55) | p-value |
| Sex | 0.486 | ||
| Male | 162 | 49 | |
| Female | 18 | 6 | |
| Age | 0.397 | ||
| ≤50 | 87 | 23 | |
| >50 | 93 | 32 | |
| HBsAg | 0.307 | ||
| Negative | 30 | 6 | |
| Positive | 148 | 48 | |
| Serum AFP | 0.042* | ||
| ≤20 ng/ml | 53 | 24 | |
| >20 ng/ml | 126 | 30 | |
| Liver cirrhosis | 0.459 | ||
| No | 59 | 21 | |
| Yes | 121 | 34 | |
| TNM | 0.799 | ||
| I | 57 | 18 | |
| II | 97 | 31 | |
| III-IV | 26 | 6 | |
| Child-pugh class | 0.328 | ||
| A | 165 | 48 | |
| B | 15 | 7 | |
| Tumor size | 0.323 | ||
| ≤3 cm | 78 | 28 | |
| >3 cm | 102 | 27 | |
| Tumor number | 0.239 | ||
| Single | 137 | 46 | |
| Multiple | 43 | 9 | |
| Tumor differentiation | 0.307 | ||
| Well | 12 | 7 | |
| Moderate | 167 | 48 | |
| Poor | 1 | 0 | |
| Vascular invasion | 0.400 | ||
| No | 64 | 23 | |
| Yes | 116 | 32 | |
Abbreviations: HCC, hepatocellular carcinoma; HBsAg, hepatitis B surface antigen; AFP, α-fetoprotein; TNM, tumor-nodes-metastases.
Furthermore, Kaplan-Meier analysis and Cox’s proportional hazards model were used to calculate the effects of ACSM3 expression on OS and TTR time. Kaplan-Meier analysis showed patients with low expression of ACSM3 exhibited worse overall survival (OS, P=0.0042) and shorter time to recurrence (TTR, P=0.0022) than patients with high expression of ACSM3 (Figure 2B). In addition, ACSM3 expression was an independent prognostic factor for OS (P=0.024) and TTR time (P=0.034) in HCC (Table 2) according to Cox’s multivariate proportional hazards model.
Table 2.
Univariate and multivariate analysis of factors associated with survival and recurrence in 235 HCC patients
| Variable | OS | TTR | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Multivariate | Multivariate | |||||||
|
|
|
|||||||
| Univariate p | HR | 95% CI | p-value | Univariate p | HR | 95% CI | p-value | |
| Sex: male vs female | 0.782 | NA | 0.775 | NA | ||||
| Age: ≤50 vs >50 | 0.318 | NA | 0.162 | NA | ||||
| HBsAg: negative vs positive | 0.450 | NA | 0.269 | NA | ||||
| Serum AFP: ≤20 ng/ml vs >20 ng/ml | 0.000 | 2.074 | 1.279-3.365 | 0.003 | 0.000 | 1.567 | 1.048-2.344 | 0.029 |
| Liver cirrhosis: no vs yes | 0.097 | NA | 0.001 | 1.809 | 1.209-2.708 | 0.004 | ||
| TNM: I vs II vs III-IV | 0.000 | NS | 0.000 | NS | ||||
| Child-pugh class: A vs B | 0.346 | NA | 0.511 | NA | ||||
| Tumor size: ≤3 cm vs >3 cm | 0.000 | 2.678 | 1.726-4.153 | 0.000 | 0.000 | 2.097 | 1.463-3.005 | 0.000 |
| Tumor number: single vs multiple | 0.000 | 2.160 | 1.435-3.250 | 0.000 | 0.000 | 2.043 | 1.406-2.969 | 0.000 |
| Tumor differentiation: I-II vs III-IV | 0.186 | NA | 0.055 | NA | ||||
| Vascular invasion: no vs yes | 0.043 | NS | 0.035 | NS | ||||
| ACSM3: low vs high | 0.004 | 0.539 | 0.315-0.923 | 0.024 | 0.002 | 0.606 | 0.381-0.964 | 0.034 |
Abbreviations: HCC, hepatocellular carcinoma; HBsAg, hepatitis B surface antigen; AFP, α-fetoprotein; TNM, tumor-nodes-metastases; HR, hazard ratio; 95% CI, 95% confidential interval; NA, not applicable; NS, not significant.
Inhibitory effects of ACSM3 in cell migration and invasion
To further understand the functions of ACSM3 in HCC progression, we detected the expression of ACSM3 in one normal liver cell line and a panel of HCC cell lines by quantitative real-time PCR. We found the expression of ACSM3 was very low in HCC cell lines (Figure 3A). Then, ACSM3 was overexpressed in HepG2 and SMMC7721 cells through lentiviral infection, and the protein levels of ACSM3 were effectively upregulated (Figure 3B). The transwell migration and invasion assay were utilized to determine the effects of ACSM3 on cell metastasis. The results showed ACSM3 overexpression significantly inhibited migration and invasion ability of HepG2 and SMMC7721 cells (Figure 3C). The effect of ACSM3 on cell proliferation was determined by CCK8 assay. Results showed that ACSM3 overexpression had no significant effect on proliferation of HCC cells (Figure 3D), and excluded the possibility that the suppression in migratory and invasive ability was caused by different growth rates. Next, we established tail vein injected models to further verify the roles of ACSM3 in HCC metastasis. The mice were handled and housed according to protocols approved by the Shanghai Medical Experimental Animal Care Commission. Compared to the mice injected with control cells, the mice bearing HepG2-ACSM3 cells exhibited decreased numbers of lung metastatic nodules (Figure 3E).
Figure 3.
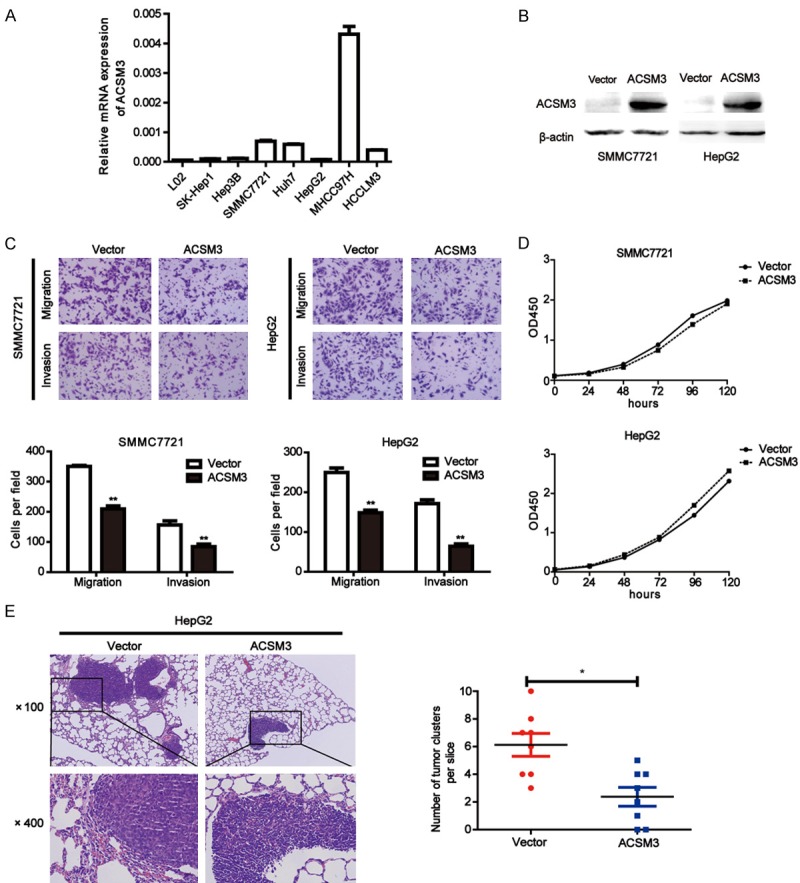
Overexpression of ACSM3 suppresses migration and invasion of HCC cells. A: ACSM3 expression was measured by quantitative real-time PCR in one human normal liver cell line (L02) and a panel of HCC cell lines. B: Overexpression of ACSM3 in SMMC7721 and HepG2 cells was confirmed by western blot. C: Representative images and quantitative results of migration and invasion in SMMC7721 and HepG2 cells with ACSM3 overexpression and control (original magnification, ×400). D: Cell proliferation of SMMC7721 and HepG2 cells with ACSM3 overexpression and control was observed by CCK8 assay. E: Representative images for lung metastatic nodules in HepG2-Vector or HepG2-ACSM3 group (magnification, ×100, ×400). Number of lung metastatic nodules was quantified on serial sections of H&E staining. **; P<0.01, *; P<0.05.
WNK1-AKT signaling may be responsible for ACSM3-inhibited cell metastasis
In order to explore the molecular mechanisms of ACSM3 in inhibition of HCC metastasis, human phospho-kinase array assays (Proteome Profiler; R&D Systems, Minneapolis, MN, USA) were performed in HepG2 cells stably transfected with ACSM3 or control. As shown in Figure 4A, 4B, the phosphorylation levels of WNK1 (Thr60) and AKT (Thr308) were decreased in HepG2-ACSM3 group compared with HepG2-Vector group. The results were also confirmed in SMMC7721-ACSM3 cell lines by western blot (Figure 4C). In order to confirm ACSM3 inhibition of HCC metastasis is mediated by suppressing WNK1-AKT signaling, we transfected vectors expressing constitutively activated AKT (HA-AKT) into HepG2-ACSM3 and 7721-ACSM3 cells, the HA-AKT plasmid was provided by Zhongzhou Yang (Model Animal Research Center of Nanjing University). We found the cancer cell migration and invasion ability, and WNK1 phosphorylation level were restored by HA-AKT (Figure 4D, 4E). These results suggest the possibility that ACSM3 inhibits metastasis of HCC by suppressing WNK1-AKT signaling.
Figure 4.
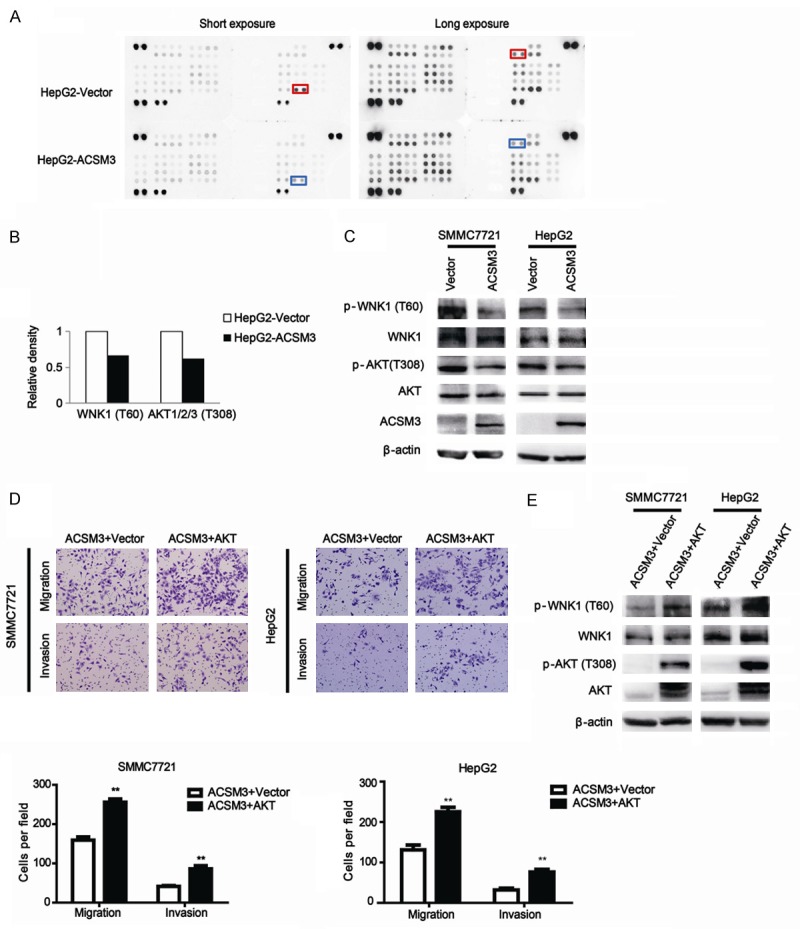
Overexpression of ACSM3 inhibits HCC metastasis via the phosphorylation of WNK1 and AKT. A, B: Results of phospho-kinase array suggested that activities of WNK1 and AKT were inhibited in HepG2-ACSM3 cells. C: Impacts of ACSM3 overexpression on the activity of WNK1 and AKT were detected by western blot assay. D, E: Migration and invasion assays, and western blot analysis of p-WNK1 were done for SMMC7721 and HepG2 cells transiently expressing constitutively activated AKT. **; P<0.01.
ACSM3 has no effect on fatty acid metabolism
ACSM3 and its family have acyl-CoA synthetase activity. They can produce acyl-CoA by interacting with medium-chain fatty acids which is the first step of fatty acid metabolism [13]. We used Oil Red O staining to determine whether ACSM3 could affect the fatty acid metabolism in HCC. Representative images of Oil Red O staining were shown and quantitative analysis was performed (Figure 5A, 5B). Results showed there was no difference between SMMC7721-ACSM3 and SMMC7721-Vector groups, or between HepG2-ACSM3 and HepG2-Vector groups. We further performed expression analysis of targeted genes by quantitative real-time PCR for key lipogenic enzymes including fatty acid synthase (FASN) and stearoyl-CoA desaturase (SCD). We also analyzed the expression of key molecules (carnitine palmitoyltransferase 1A, CPT1A and acyl-CoA oxidase 1, ACOX1) in fatty acid oxidation (Figure 5C). None of them showed any difference between ACSM3 overexpression cells and control cells. Our results indicate ACSM3 may have no important impacts on fatty acid metabolism in HCC.
Figure 5.
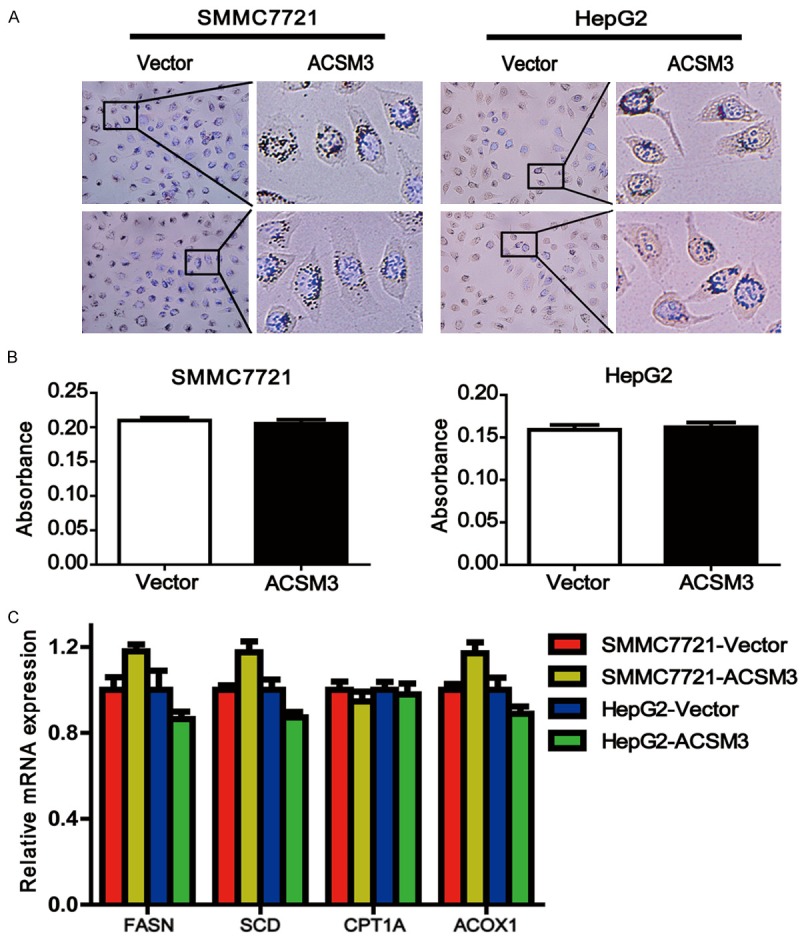
ACSM3 has no effect on fatty acid metabolism. A: Oil Red O staining showed the neutral lipid content of cells. Nucleus was counterstained with DAPI. B: Quantitative analysis of Oil Red O staining by measuring the absorbance (OD at 490 nm) of extracted dye. C: Quantitative real-time PCR analysis for expression of FASN, SCD, CPT1A and ACOX1 in SMMC7721 and HepG2 cells with ACSM3 overexpression or control.
Discussion
Recurrence and metastasis are the most primary causes contributing to the high mortality of HCC. In the past years, great efforts have been made to clarify molecular mechanisms of HCC recurrence and metastasis, but the prognosis of HCC patients still remains dismal. Therefore, identifying biomarkers of HCC diagnosis and understanding molecular mechanisms responsible for metastasis and postsurgical recurrence will help extend survival rate of HCC patients.
In this study, we first uncovered ACSM3 was downregulated in HCC tissues compared with their corresponding adjacent non-tumorous tissues (Figure 1) and downregulation of ACSM3 had influences on HCC prognosis (Figure 2). In addition, our data indicated that ACSM3 might act as a suppressor against HCC metastasis, but had no effects on proliferation (Figure 3), as well as cell cycle and apoptosis (data not shown). ACSM3 and its family have acyl-CoA synthetase activity, they can produce acyl-CoA by interacting with medium-chain fatty acids on the outer membrane of mitochondria [10]. And then, Acyl-CoA is transported into the matrix for β-oxidation of acyl-group. However, our results showed ACSM3 as a metabolic enzyme had no effect on the fatty acid metabolism in HCC (Figure 5).
AKT and WNK1 were identified by human phospho-kinase array assays in this study. AKT, also referred to as PKB or Rac, plays a critical role in controlling survival, apoptosis [11-13], cell cycle [14] and glycogen synthesis [15,16]. PI3K-AKT-mTOR signaling is one of the most frequently studied pathways in HCC development and metastasis [17-19]. Previous studies have demonstrated many acyl-CoA synthetase enzymes (ACSs) such as ACSL4 [20,21] and ACSVL3 [22] exert biological functions via multiple downstream signaling pathways among which AKT activation (i.e. phosphorylation) plays a prominent role.
WNK1 (with-no-lysine) is serine-threonine protein kinases with an unusual position of the catalytic lysine in subdomain I instead of subdomain II [23]. WNK1 mutations in humans cause pseudohypoaldosteronism type 2 (PHA2), an autosomal-dominant disease which is characterized by hypertension and hyperkalemia [24]. WNK1 has also been reported to regulate renal Na+ and K+ transporters, whose dysregulation contributes to hypertension and hyperkalemia in PHA2 [25]. Loss of expression or inactivating WNK1 mutations could promote epithelial-mesenchymal transition (EMT) of epithelial tumour cells [26], and EMT is a typical biological behavior associated with cancer metastasis. Moreover, a recent report links WNK1 to Rho GTPases, which control the dynamics of the actin cytoskeleton, and are important for cell migration and invasion [27,28]. Besides, it has also been reported that Akt kinase phosphorylates WNK1 [29,30], and downregulation of WNK1 phosphorylation reduces cell proliferation, migration and differentiation in the C17.2 mouse neural progenitor cell line [31]. Therefore, it is possible that ACSM3 suppresses HCC metastasis by inhibition of WNK1 and AKT phosphorylation. However, detailed molecular mechanisms underlying tumor suppressive effects of ACSM3 need further investigation.
Acknowledgements
This work was supported by grants from National Key Basic Research Program of China (973 Program: 2015CB553905), National Natural Science Foundation of China (81301818, 81402278, 81572311, 81421001), Project of Special Research Fund for Healthy (201402003), Shanghai Natural Science Foundation of China (16ZR1434700), Shanghai Jiao Tong University School of Medicine (YG2015QN34, YG2014MS44), and State Key Laboratory of Oncogenes and Related Genes (SB16-04).
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844–855. doi: 10.1136/gutjnl-2013-306627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oka Y, Kobayakawa K, Nishizumi H, Miyamichi K, Hirose S, Tsuboi A, Sakano H. O-MACS, a novel member of the medium-chain acyl-CoA synthetase family, specifically expressed in the olfactory epithelium in a zone-specific manner. Eur J Biochem. 2003;270:1995–2004. doi: 10.1046/j.1432-1033.2003.03571.x. [DOI] [PubMed] [Google Scholar]
- 4.Iwai N, Mannami T, Tomoike H, Ono K, Iwanaga Y. An acyl-CoA synthetase gene family in chromosome 16p12 may contribute to multiple risk factors. Hypertension. 2003;41:1041–1046. doi: 10.1161/01.HYP.0000064944.60569.87. [DOI] [PubMed] [Google Scholar]
- 5.Haketa A, Soma M, Nakayama T, Sato M, Kosuge K, Aoi N, Matsumoto K. Two medium-chain acyl-coenzyme A synthetase genes, SAH and MACS1, are associated with plasma high-density lipoprotein cholesterol levels, but they are not associated with essential hypertension. J Hypertens. 2004;22:1903–1907. doi: 10.1097/00004872-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Lindner I, Rubin D, Helwig U, Nitz I, Hampe J, Schreiber S, Schrezenmeir J, Doring F. The L513S polymorphism in medium-chain acyl-CoA synthetase 2 (MACS2) is associated with risk factors of the metabolic syndrome in a Caucasian study population. Mol Nutr Food Res. 2006;50:270–274. doi: 10.1002/mnfr.200500241. [DOI] [PubMed] [Google Scholar]
- 7.Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, Zhou J, Li BZ, Shi YH, Xiao YS, Xu Y, Fan J. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971–979. doi: 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- 8.Jin H, Zhang Y, You H, Tao X, Wang C, Jin G, Wang N, Ruan H, Gu D, Huo X, Cong W, Qin W. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration, and invasion of human hepatocellular carcinoma. Sci Rep. 2015;5:10466. doi: 10.1038/srep10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruan H, Yang C, Jin G, Gu D, Deng X, Wang C, Qin W, Jin H. Co-expression of LASS2 and TGF-β1 predicts poor prognosis in hepatocellular carcinoma. Sci Rep. 2016;6:32421. doi: 10.1038/srep32421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujino T, Kang MJ, Suzuki H, Iijima H, Yamamoto T. Molecular characterization and expression of rat acyl-CoA synthetase 3. J Biol Chem. 1996;271:16748–16752. doi: 10.1074/jbc.271.28.16748. [DOI] [PubMed] [Google Scholar]
- 11.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 12.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 14.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 15.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 16.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344 Pt 2:427–431. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Dai Y, Chen J, Zeng T, Li Y, Chen L, Zhu YH, Li J, Li Y, Ma S, Xie D, Yuan YF, Guan XY. Maelstrom promotes hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition by way of Akt/GSK-3beta/Snail signaling. Hepatology. 2014;59:531–543. doi: 10.1002/hep.26677. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, Song C, Cheung YS, Lai PB, Cheng SH, Ng MH, Huang A, Ko BC. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3beta/beta-catenin signaling. Hepatology. 2013;57:2287–2298. doi: 10.1002/hep.26278. [DOI] [PubMed] [Google Scholar]
- 19.Wen W, Ding J, Sun W, Fu J, Chen Y, Wu K, Ning B, Han T, Huang L, Chen C, Xie D, Li Z, Feng G, Wu M, Xie W, Wang H. Cyclin G1-mediated epithelial-mesenchymal transition via phosphoinositide 3-kinase/Akt signaling facilitates liver cancer progression. Hepatology. 2012;55:1787–1798. doi: 10.1002/hep.25596. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Deng F, Li Y, Daniels G, Du X, Ren Q, Wang J, Wang LH, Yang Y, Zhang V, Zhang D, Ye F, Melamed J, Monaco ME, Lee P. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget. 2015;6:44849–44863. doi: 10.18632/oncotarget.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyares RL, Stein C, Renisch B, Anderson JL, Hammerschmidt M, Farber SA. Long-chain Acyl-CoA synthetase 4A regulates Smad activity and dorsoventral patterning in the zebrafish embryo. Dev Cell. 2013;27:635–647. doi: 10.1016/j.devcel.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei Z, Sun P, Huang P, Lal B, Laterra J, Watkins PA. Acyl-CoA synthetase VL3 knockdown inhibits human glioma cell proliferation and tumorigenicity. Cancer Res. 2009;69:9175–9182. doi: 10.1158/0008-5472.CAN-08-4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 24.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 25.Cheng CJ, Huang CL. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am Soc Nephrol. 2011;22:460–471. doi: 10.1681/ASN.2010060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee BH, Chen W, Stippec S, Cobb MH. Biological cross-talk between WNK1 and the transforming growth factor beta-Smad signaling pathway. J Biol Chem. 2007;282:17985–17996. doi: 10.1074/jbc.M702664200. [DOI] [PubMed] [Google Scholar]
- 27.Moniz S, Jordan P. Emerging roles for WNK kinases in cancer. Cell Mol Life Sci. 2010;67:1265–1276. doi: 10.1007/s00018-010-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Xu X, Zhang Y, Zhou J, Yu Z, He C. LINGO-1 interacts with WNK1 to regulate nogo-induced inhibition of neurite extension. J Biol Chem. 2009;284:15717–15728. doi: 10.1074/jbc.M808751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitari AC, Deak M, Collins BJ, Morrice N, Prescott AR, Phelan A, Humphreys S, Alessi DR. WNK1, the kinase mutated in an inherited high-blood-pressure syndrome, is a novel PKB (protein kinase B)/Akt substrate. Biochem J. 2004;378:257–268. doi: 10.1042/BJ20031692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai JG, Tsai SM, Tu HC, Chen WC, Kou FJ, Lu JW, Wang HD, Huang CL, Yuh CH. Zebrafish WNK lysine deficient protein kinase 1 (wnk1) affects angiogenesis associated with VEGF signaling. PLoS One. 2014;9:e106129. doi: 10.1371/journal.pone.0106129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun X, Gao L, Yu RK, Zeng G. Down-regulation of WNK1 protein kinase in neural progenitor cells suppresses cell proliferation and migration. J Neurochem. 2006;99:1114–1121. doi: 10.1111/j.1471-4159.2006.04159.x. [DOI] [PubMed] [Google Scholar]