

Abstract
The HIV-1 accessory protein Vpr is required for efficient viral infection of macrophages and promotion of viral replication in T cells. Vpr’s biological activities are closely linked to the interaction with human DCAF1, a cellular substrate receptor of the Cullin4–RING E3 ubiquitin ligase (CRL4) of the host ubiquitin–proteasome-mediated protein degradation pathway. The molecular details of how Vpr usurps the protein degradation pathway have not been delineated. Here we present the crystal structure of the DDB1–DCAF1–HIV-1–Vpr–uracil-DNA glycosylase (UNG2) complex. The structure reveals how Vpr engages with DCAF1, creating a binding interface for UNG2 recruitment, in a manner distinct from the recruitment of SAMHD1 by Vpx protein for degradation by Vpx proteins. Vpr and Vpx use similar N-terminal and helical regions to bind the substrate receptor, whereas different regions target the specific cellular substrates. Furthermore, Vpr uses molecular mimicry of DNA by a variable loop for specific recruitment of the UNG2 substrate.
Introduction
Lentiviral genomes encode Gag, Pol, and Env proteins along with six accessory proteins1, which facilitate virus replication in vivo2,3. The accessory Viral protein R (Vpr), a 14 kDa protein, is highly conserved in HIV and simian immunodeficiency virus (SIV)4 and is involved in many processes that impact the pathophysiology of AIDS5.
Vpr is incorporated into budding virions via interaction with the C-terminus of Gag, and during virus maturation is recruited by viral RNA into the conical capsid core of the virus particle6. Since the core is released into the cytoplasm of a newly infected cell, Vpr is poised to play important roles in immediate post-entry events.
The most dramatic and widely acknowledged phenotype of Vpr expression in cycling cells is induction of cell cycle arrest in G2/M phase7,8. The determinants of Vpr’s activity in cell cycle arrest are not fully understood, but there is compelling evidence that Vpr executes its function by engaging the substrate recognition subunit, DDB1- and CUL4A-associated factor 1 (DCAF1), of the cullin4A-RING E3 ubiquitin ligase (CRL4), comprising cullin4A (CUL4A), RING H2 finger protein 1 (RBX1) and DNA damage-binding protein 1 (DDB1), and recruits cellular targets to the ligase for proteasome-mediated degradation9–15. Vpr mutants that abolish the interaction with DCAF1 or silencing of DCAF1 eliminate cell cycle arrest9–12,15.
Subversion of the host ubiquitin proteasome pathway is commonly exploited by lentiviral accessory proteins. Viral infectivity factor (Vif) and Viral protein U (Vpu) target Apolipoprotein B mRNA-editing, enzyme-catalytic, polypeptide-like 3G (APOBEC3G) and Bone marrow stromal cell antigen 2 (BST2) or Teterin, respectively, by co-opting specific CRL E3 ubiquitin ligases and altering their substrate specificities16–18.
HIV-1 Vpr directs the CRL4-DCAF1 E3 ubiquitin ligase complex towards several cellular substrates: for example, levels of MUS81, a component of the holiday junction resolvase SLX4 complex, are down-regulated in a Vpr- and DCAF1-dependent manner19, and degradation of minichromosome maintenance complex component 10 (MCM10), a known target of CRL4-DCAF1, is enhanced by Vpr20.
Viral protein X (Vpx) of HIV-2 and SIV, which evolved from a common ancestor of Vpr, binds to the same CRL4-DCAF1 E3 ubiquitin ligase and targets the sterile alpha motif and histidine/aspartate (HD) containing protein 1 (SAMHD1) for degradation21–23. SAMHD1 is an HIV restriction factor that acts by depleting the intracellular pool of deoxynucleoside triphosphates to levels below those required for the synthesis of the viral DNA by reverse transcriptase24. The structural basis of clade-specific SAMHD1 recruitment to the DCAF1-Vpx complex was recently elucidated25–27. However, no structural data on the interaction between DDB1 and DCAF1 or DCAF-1 and Vpr is available, and very little is known about how the closely related Vpr and Vpx proteins impart different substrate specificity to the same CRL4-DCAF1 E3 ligase for the selective recruitment of cellular targets.
The first identified target of HIV-1 Vpr-mediated redirection of the CRL4-DCAF1 E3 ubiquitin ligase was the nuclear isoform of uracil DNA glycosylase (UNG2)28,29. This enzyme recognizes and removes uracil bases from DNA, thereby catalyzing the first step in the base excision repair pathway (BER)30. The resulting abasic sites are fully repaired during subsequent steps. HIV-1 DNA acquires uracil through a variety of mechanisms, including APOBEC3-mediated deamination of cytidine residues in viral reverse transcripts and uracil missincorporation by viral reverse transcriptase31–33. Therefore, HIV-1 cDNA is a potential substrate for UNG2 mediated processing.
Substantially, UNG2 is antagonized by HIV-1 Vpr. HIV-1 Vpr loads UNG2 onto CRL4-DCAF1 E3 for polyubiquitynation and subsequent proteasome-mediated degradation29. As a result, UNG2 levels are depleted and uracil BER compromised in HIV-1 infected primary target cells34,35. Whereas the above findings are strong evidence for a role of UNG2 in HIV-1 infection, deciphering UNG2’s detailed effects so far have remained elusive. Evidence in support of a positive role36,37, a negative role38, or no role39 in HIV-1 replication has been reported, suggesting a more nuanced, possibly cell type specific role of UNG2 in HIV-1 infection.
To better understand the molecular mechanism underlying HIV-1 Vpr-mediated UNG2 downregulation through the CRL4 E3 ubiquitin ligase machinery, we determined the crystal structure of HIV-1 Vpr in complex with human UNG2 and the CRL4 acceptor–receptor proteins DDB1–DCAF1. We found that Vpr binds DCAF1 in a similar manner to Vpx, primarily through the N-terminal tail and α-helix α3, despite limited sequence similarity in these regions of Vpr and Vpx. Interestingly, for the UNG2 interaction, Vpr uses structural mimicry of DNA and engages the DNA-binding region of UNG2 in a cleft between helices α1 and α2. Together, our findings reveal a dual mechanism used by HIV-1 Vpr to antagonize UNG2: one involves degradation by the proteasome, and the other entails inhibition through molecular mimicry. The latter mechanism is also used by the bacteriophage gene product Ugi to inhibit a prokaryotic uracil-DNA glycosylase40.
Results
The overall structure of the DDB1-DCAF1-Vpr-UNG2 complex
Crystals of the DDB1-DCAF1-Vpr-UNG2 complex, belonging to space group P1, were used for structure determination (Table 1). The structure was solved by molecular replacement in Molrep (www.ccp4.ac.uk/html/molrep.html), using each protein component in turn as search models (DDB1, PDB 3E0C41, DCAF1, PDB 4Z8L27, Vpr, PDB 4Z8L27 and UNG2, PDB 2OYT42. Initial Rcryst/Rfree (%) values of 38.3/39.5 were obtained after rigid body and B-group refinement with Phenix (https://www.phenix-online.org/). Given that data diffracted to 3.5 Å, the initial model was subjected to several rounds of manual model building, with B-factor sharpening in Coot (http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/) and refinement with Buster (https://www.globalphasing.com/buster/). The use of B-factor sharpening permitted visualization of most amino acid side chains (Figure 1A) allowing residue placement and model refinement to a final Rcryst/Rfree (%) of 17.6/20.6. The final (Buster-refined) 2Fobs-Fcalc map of the complex is illustrated in Figure 1B and Supplementary Figure 1A to 1D. The structure includes approximately 1800 residues per monomer in the asymmetric unit (ASU) and includes full length human DDB1, the C-terminal region of DCAF1, including WD40 domain (residues 1046–1396), full length HIV-1 Vpr, and the catalytic domain of human UNG2 (residues 83–304) (Figure 1C and Supplementary Figure 1F). Two copies of the hetero-tetramer complex were present per ASU and pack against each other (Supplementary Figure 1E), with UNG2 forming extensive contacts with DDB1, DCAF1 and Vpr from the symmetry related molecule, suggesting that the presence of UNG2 was critical for crystallization (Supplementary Figure 1G). These contacts, however, are solely crystallographic, since size exclusion chromatography and multi-angle light scattering experiments revealed that only a monomeric complex is present in solution (Supplementary Figure 1H).
Table 1.
Data collection and refinement statistics
|
DDB1-DCAF-UNG2-Vpr (PDB: 5JK7) |
DDB1-DCAF-UNG2-Vpr (N/A)c |
|
|---|---|---|
| Data collection | ||
| Space group | P1 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 119.90, 128.20, 129.20 | 119.80, 127.00, 127.00 |
| α, β, γ (°) | 75.11, 89.44, 65.37 | 75.10, 89.50, 65.50 |
| Resolution (Å) | 40-3.49 | 40-3.99 |
| Rmerge | 14.9 (262) | 8.7 (83.2) |
| Rpim | 6.4 (115) | 6.1 (55.6) |
| I/σ (I) | 7.8 (0.7) | 7.5 (1.4) |
| CC1/2 | 99.3 (24.8) | 99.8 (60.1) |
| Completeness (%) | 90.6 (40.3) | 88.2 (85.8) |
| Redundancy | 6.6 (6.2) | 2.2 (2.2) |
| Refinement | ||
| Resolution (Å) | 40-3.49 | |
| No. reflections | 77,315 | |
| Rwork / Rfree | 17.6 / 20.6 | |
| No. atoms | ||
| Protein | 27,701 | |
| B factors | ||
| Protein | 193.89 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.01 | |
| Bond angles (°) | 1.34 |
Numbers in parentheses correspond to the highest resolution shell (3.68–3.49 Å and 4.09–3.99 Å, respectively).
Resolution limits were extended to include weak intensity data59.
This data set was only used for obtaining the initial molecular replacement model.
Figure 1. Overall structure of DDB1-DCAF1-Vpr-UNG2 complex.
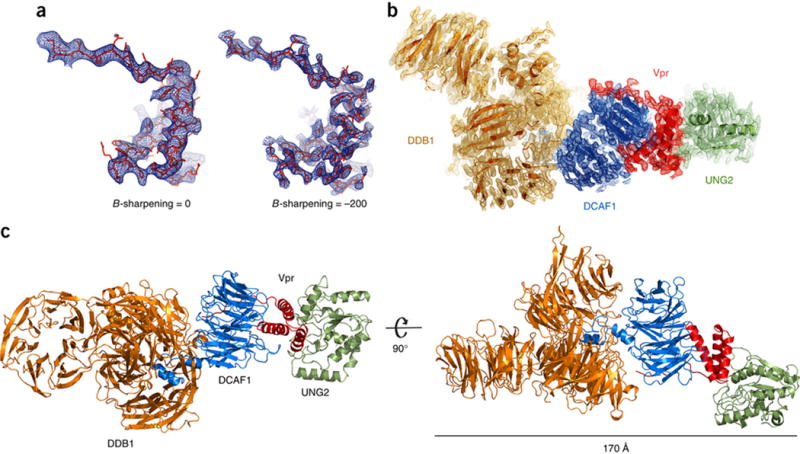
(A) B-factor sharpening applied to the Fobs-Fcalc electron density map of Vpr in the complex, contoured at 2.5 σ. Use of negative B-factors, in this case −200, resulted in enhanced electron density features, which allowed placement of side-chains58 and refinement of the full structure. The atomic model is shown in red stick representation and the electron density in blue.
(B) The final refined 2 Fobs-Fcalc electron density map of the DDB1-DCAF1-Vpr-UNG2 complex contoured at 1 σ. The individual proteins in the complex are colored: DDB1, orange; DCAF1, blue; Vpr, red; UNG2, green; this color scheme is used throughout the manuscript.
(C) Two views of the overall DDB1-DCAF1-Vpr-UNG2 complex in ribbon representation.
The four components in the complex are arranged in series, with DDB1 binding to DCAF1, which interacts with Vpr, which in turn binds to UNG2 (Figure 1C). This arrangement, resulting in an overall length of the complex of 170 Å, may have important functional implications for HIV-1 biology (see below).
Vpr mimics UNG2 binding to DNA
Vpr forms a 3-helical bundle in the complex, and the helical arrangement is very similar to the NMR solution structure solved at low pH43. Four structural motifs that are important in Vpr’s interaction with partners can be discerned: a random coil N-terminal tail, a hydrophobic cleft between helices α1, α2 and the first turn of α3, the loop connecting α2 and α3 (insert loop), and the C-terminal region of helix α3 (Supplementary Figure 2A). Vpr interacts with DCAF1 using its N-terminus and helix α3 (see below) and with UNG2 via the insert loop and residues in the hydrophobic cleft, burying a surface area of 1650 Å2 and 940 Å2, respectively (Supplementary Figure 2B).
Intriguingly, the binding site of UNG2 that is contacted by Vpr is essentially identical to the one used in the UNG2-DNA complex (Figure 2A, B and Supplementary Figure 2C). In particular, the loop (residues 266–283) of UNG2 that interacts with DNA, uses Leu272 to insert into the minor groove42 and in the DDB1-DCAF1-Vpr-UNG2 complex, Leu272 inserts deeply into the cleft of Vpr (Figure 2C).
Figure 2. Vpr employs structural mimicry and engages UNG2 residues that are also used in DNA binding.
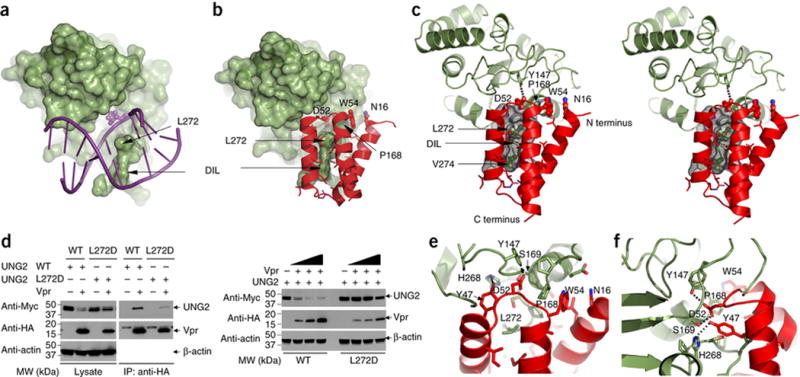
(A) Structure of the UNG2 (green) - DNA (purple) complex (PDB:id 2OYT,)42). UNG2 is depicted in surface representation and the double stranded DNA in stick representation. The flipped-out abasic sugar inside UNG2’s active site pocket is shown in space filling representation. The Leu272 residue and the DNA intercalation loop (DIL) are labeled by arrows.
(B) Structure of the UNG2 (green) - Vpr (red) portion of the DDB1-DCAF1-Vpr-UNG2 complex. UNG2 is depicted in surface representation and Vpr in ribbon representation. Several side chains of Vpr that are interacting with UNG2 are shown in ball and stick representation and labeled with residue name and number.
(C) Stereo-view of the contact region between UNG2 (green) and Vpr (red), illustrating details of the interaction. Leu272 and Val274 of UNG2 are buried inside Vpr’s hydrophobic cleft (rendered in gray surface representation). Asp52 of Vpr (ball and stick representation) is located inside UNG2’s active site pocket, forming a hydrogen bond with Tyr147 of UNG2. Trp54 of Vpr (ball and stick representation) is in van der Waals contact with Pro168 of UNG2 and Asn16 of Vpr is stacked onto the Trp54 side chain.
(D) Co-immunoprecipitation experiments to assess the interaction between UNG2 and Vpr (left panels). UNG2 WT or Leu272Asp was transfected into HEK293 cells with increasing amounts of Vpr (right panels).
(E–F) Two expanded views of Vpr’s insert loop contacts with the active site pocket of UNG2.
The importance of Leu272 of UNG2 for Vpr binding was verified by mutagenesis (Figure 2D). In co-immunoprecipitation experiments, the interaction between Vpr and UNG2 was substantially affected by the Leu272Asp mutation (Figure 2D, left panels). In addition, the Leu272Asp mutant was resistant to the Vpr-mediated down-regulation, whereas a substantial reduction of UNG2 levels was observed after transient co-transfection of Vpr and WT UNG2, (Figure 2D, right panels).
The insert loop of Vpr engages UNG2 via a proline rich motif (165-PPPPS-169), creating a second interface. Stacking interactions between Asp52 and Trp54 of Vpr and Pro168 of UNG2 are present (Figure 2C and E), as well as hydrogen bonding between Tyr47 of Vpr and His268 of UNG2, and Asp52 of Vpr and Tyr147 of UNG2 (Figure 2E and F). In agreement with this observation, the Vpr mutants W54R and W54G do not bind UNG244 and lose the ability to down-regulate UNG2 in cells22,35,36. Interestingly, the Vpr insert loop mimics the phosphate backbone trace around the abasic sugar in the DNA (Supplementary Figure 2 D–F). A similar mechanism of molecular mimicry was observed in the crystal structure of human UNG2 bound to B. subtilis bacteriophage uracil-DNA glycosylase inhibitor (Ugi), a 9.5 kDa protein40. However, the two structures are very different in architecture. Ugi contains a five-strand antiparallel β-sheet, flanked by two α helices and Vpr consists of a three α-helical bundle. Glu20 of Ugi and Asp52 of Vpr interact with UNG2’s DNA binding site (Supplementary Figure 2G), and UNG2’s Leu272 (Supplementary Figure 2H) inserts into their respective pockets. To test whether binding of Vpr to UNG2 could effectively inhibit the enzyme’s activity we performed base excision and inhibition assays with a single-stranded uracilated DNA substrate36. As predicted based on our structural findings, in the presence of the DDB1-DCAF1-Vpr complex, but not DDB1-DCAF1 alone, substrate cleavage was inhibited, confirming our hypothesis that Vpr mimics DNA and thereby interferes with substrate binding (Supplementary Figure 2I).
Vpr binding to the DCAF1 WD40 domain
The DCAF1 construct in the present complex includes a helix-loop-helix motif (Phe1050-Arg1079) at the N-terminus and the canonical seven-blade WD40 β-propeller domain (Arg1081-Glu1388; Supplementary Figure 3A). The surface of the β-propeller is not flat; loops connecting the β-strands on one hemi-solenoid of the propeller create a large cleft: this is where Vpr binds (Figure 3A and Supplementary Figure 3 B–D). The cleft is lined by acidic residues at one end (Supplementary Figure 3B) and hydrophobic residues at the center (Figure 3A). Residues Arg62, Gln65, and Arg73 on α3 of Vpr form hydrogen bonds with Glu1088, Ser1136, and Thr1139 of DCAF1, respectively (Figure 3B, left panel). In addition, Phe69 of Vpr is buried inside a small hydrophobic pocket, formed by residues Ala1137, Phe1330 and Phe1355 of DCAF1 (Figure 3B, right panel and Supplementary Figure 3D). On Vpr, residues Glu25, Leu26 and Glu29 on α1, together with residues Gln65 and Leu68 on α3, form a small pocket into which DCAF1 residue Trp1156 binds (Figure 3B, right panel and Supplementary Figure 3D). Furthermore, the N-terminal tail of Vpr wraps around one side of the propeller, following a small groove between two blades (Figures 3C).
Figure 3. Interface between DCAF1 (blue and gold) - Vpr (red) in the DDB1-DCAF1-Vpr-UNG2 complex.
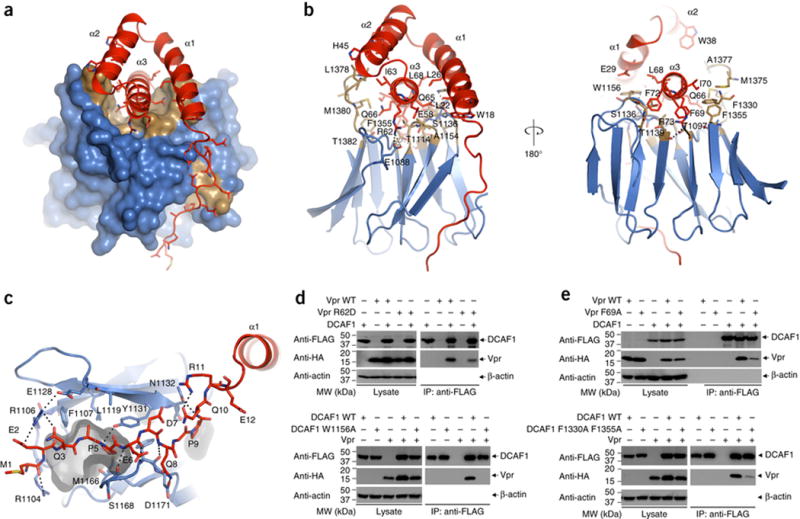
(A) Surface (DCAF1) and ribbon (Vpr) representations of the DCAF1-Vpr portion of the DDB1-DCAF1-Vpr-UNG2 complex structure. The hydrophobic cleft in the rim of the DCAF1 WD40 propeller, which accommodates the a3 helix of Vpr is shown in gold.
(B) Two views of DCAF1-Vpr interactions, illustrating details of contact residues. Pertinent side chains are shown in stick representation. Vpr is shown in red, and DCAF1 is in gold (hydrophobic) and blue (polar).
(C) Detailed view of the interaction between the N-terminal tail of Vpr and residues of DCAF1 at the edges of two propeller blades. A small hydrophobic pocket, accommodating Pro5 of Vpr is rendered in gray.
(D) – (E) Co-immunoprecipitation experiments to examine the effect of selected mutants on Vpr-DCAF1 binding.
Verification of several contacts by immunoprecipitation is provided in Figure 3D and 3E. For the Arg62Asp mutation of Vpr, DCAF1 binding is substantially reduced (Figure 3D, upper panels), and the critical interaction between residues on α1 of Vpr and Trp1156 of DCAF1 is abolished in the Trp1156Ala mutant of DCAF1, for which no stable binding is observed (Figure 3D, lower panels). The disruption of hydrophobic interactions between Phe69 of Vpr and Phe1330/Phe1335 of DCAF1 by Alanine substitution for any of these residues abolished complex formation (Figure 3E).
DCAF1 is anchored into DDB1 by a helix-loop-helix motif
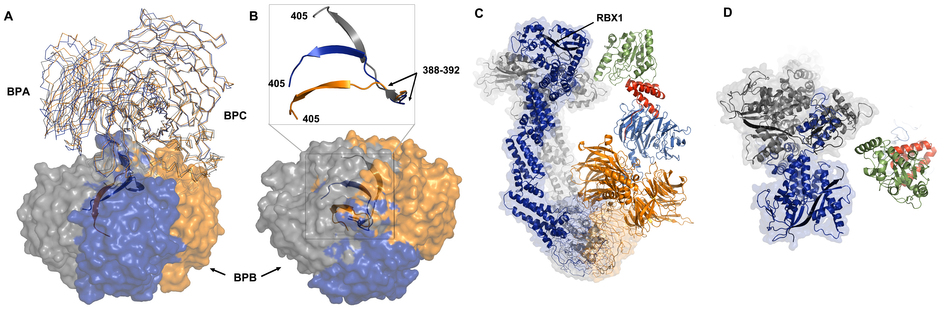
The N-terminal helix-loop-helix of DCAF1 anchors DCAF1 into DDB1 (Supplementary Figure 3A). DDB1 is a large multi-domain protein, comprising three WD40 β-propeller domains (BPA, BPB, and BPC) and a C-terminal helical domain (CTD)45,46. BPA and BPC pack against each other at an approximately 65° angle, forming a large opening (approximately 28 Å in diameter) at the interface of the two propellers (Supplementary Figure 3e–g). Helix a1 and the loop bind inside this opening, while helix a2 lines the entrance of the opening. The orientation of the helices in the helix-loop-helix region is stabilized by hydrogen bonds between the Arg1053 side chain and the backbone carboxyl group of Ala1046 and the side chain of Asp1072 (Supplementary Figure 3H). Intermolecular contacts comprise, for example, a hydrogen bond between Arg1057 of DCAF-1 and the backbone carboxyl of Met954 of DDB1 (Supplementary Figure 3H). Additional DDB1-DCAF1 interactions include contacts between DCAF1 loops with BPA and BPC residues on the opposite site of the Vpr binding site. The importance of specific residues in this interaction was probed by mutagenesis: replacement of amino acids in the helix-loop-helix motif of DCAF1, such as Arg1053Glu/Arg1057Glu, abolishes binding to DDB1 (Supplementary Figure 3I).
A helix-loop-helix motif is commonly found in substrate receptor proteins that bind to DDB1 of the CRL4 E3 ubiquitin ligase46–49. Comparison of the current DDB1-DCAF1 arrangement with the one present in the DDB1-DNA-repair protein DNA damage binding protein 2 (DDB2) complex reveals that they interact in very similar fashion47,48,50. Structural alignment between DCAF1 and DDB2 (PDB 4A0L)47 yields an RMSD of 3.02 Å for Cα atoms and a Z-score of 7.5, despite only 8% amino acid sequence identify. Consequently, different amino acids are involved in the interaction. Most interestingly, however, structural superposition of Vpr-bound DCAF1 and DNA-bound DDB2 indicate that Vpr and damaged DNA interact with very similar regions on the surface of DCAF1 and DDB2, respectively, suggesting that Vpr engages in structural mimicry (Supplementary Figure 3J and K).
Vpr recruits substrates differently from Vpx
Analysis and comparison of the Vpr structure with Vpx from SIV reveal canonical, yet distinctive modes of binding to DCAF1 and their specific cellular substrates. Two structures of Vpx complexes have been determined, one containing Vpx isolated from Mandrill SIV (VpxMND, PDB 5AJA and 4Z8L)26,27 and the other with Vpx from Sooty Mangabey SIV (VpxSM, PDB 4CC9)25. In both structures, Vpx forms a very similar three-helix bundle, with a RMSD value (Cα atoms) of 1.21 Å. The Vpr architecture in the current structure is similar to both, VpxMND and VpxSM, with RMSD values of 1.57 Å and 1.58 Å, respectively, for the helical part (Vpr residues 15–74). Despite this similarity in overall structure, functionally important differences for specific substrate recruitment can be discerned: in particular, the cleft in the Vpr structure, which is used by UNG2 (Figure 4A and Supplementary Figure 4A), is absent in Vpx due to the presence of bulky side chains, such as Trp45 and Tyr67 in VpxMND, and Trp49 and Tyr71 in VpxSM (Supplementary Figure 4B & 4C). In addition, Vpx contains a much longer insert loop than Vpr, and the amino acid identity in the loop is not conserved between Vpr and Vpx. This region of the Vpr structure is intimately involved in the UNG2 interaction and it is unlikely that a long insert loop could be accommodated (Figure 4B). Thus length and residue type of the insert loop of Vpr and Vpx are important determinants for binding specificity.
Figure 4. Comparison between Vpr and Vpx structures and their interactions with binding partners.
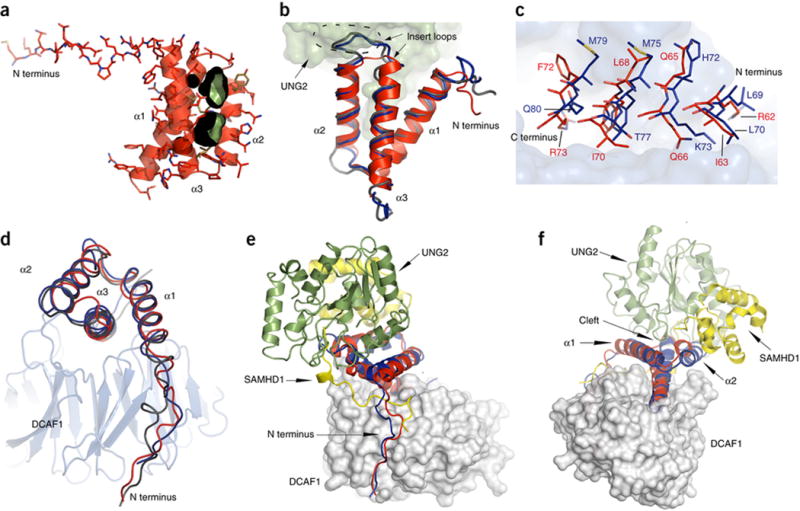
(A) Ribbon and stick representation of Vpr illustrating the cleft between helices α1 and α2 (rendered in black and green).
(B) Superposition of VprHIV-1 (red, ribbon) in the present complex with UNG2 (green surface representation), VpxMnd (blue worm representation) and VpxSM (silver worm representation) Vpx structures. The insert loops in the Vpx structures are enclosed by a dashed oval.
(C) Close up view of helix α3 residues (VprHIV-1, red; VpxMND, blue) in the DCAF1 binding site. Vpr and Vpx residues are depicted in stick representation and the DCAF1 binding site is shown in gray surface representation.
(D) Best fit superposition of Vpr (red), Vpx (Mnd, blue; SM, silver) structures (worm representation) in complexes with DCAF1 (gray ribbon representation).
(E) – (F) Two views of the superposition of the DCAF1 (gray)-VprHIV-1 (red)-UNG2 (green) and DCAF1 (gray)-VpxMND (blue)-SAMHD1 (yellow) complexes. DCAF1 is show in surface representation and Vpr, Vpx, UNG and SAMHD1 are shown in ribbon representation.
Despite low amino acid sequence identity between Vpr and Vpx (Supplemental Figure 4G), in the interaction with DCAF1 the same structural elements are used, namely the N-terminal tail and helix α3. However, at each contact point, different amino acids are engaged, although the length and shape of the residues tend to be similar (Figure 4C). Moreover, in contrast to Vpx, no Vpr residues establish hydrogen bond interactions with the acidic loop of DCAF1 (Supplementary Figure 4D)25,26. On the other hand, for the N-terminal tail of Vpr and Vpx, a different path along the DCAF1 binding surface is seen (Figure 4D). This is, in part, caused by the presence of the N-terminal tail of SAMHD1 in the Vpx complex, which intertwines between DCAF1 and Vpx (Figure 4E and Supplementary Figure 4E), illustrating the remarkable plasticity that is used by Vpr and Vpx proteins in engaging their interaction partners.
Binding of Vpr to UNG2 involves the cleft between helices α1 and α2 on Vpr while binding of Vpx to SAMHD1 involves helix α2 (Figure 4d–f), the N-terminal tail and the insert loop (Supplementary Figure 4d−f). Thus, it appears that the Vpr and Vpx families of proteins use related, but distinct structural regions to bind and recruit cellular targets to the E3 ligase complex, which will ultimately be targeted for degradation by the proteasome.
Discussion
By promoting physical interactions with cellular proteins and modulating the output of cellular signaling events, retroviral accessory proteins manipulate these signaling pathways to enhance viral replication. This is clearly seen for HIV-1 Vpr which acts as a molecular bridge, using non-overlapping surfaces to link DCAF1 and UNG2. The binding interface with DCAF1 involves predominantly hydrophobic residues on helix α3 and Vpr’s N-terminal disordered tail. The interface with UNG2 comprises Vpr’s insert loop and cleft, and the DCAF1-Vpr interaction enhances DCAF1 binding to DDB151.
Although, both, Vpr and Vpx are packaged into virions and delivered into host cells upon viral infection, their presence results in completely different phenotypic outcomes: Vpx strongly enhances lentiviral infection in monocytes and monocyte-derived dendritic cells, while HIV-1 Vpr promotes virus replication in macrophages52, possibly by overcoming macrophage-specific restriction of Env expression53, and triggers cell cycle arrest at G2 in cycling cells7,8,54,55. These different outcomes are mediated by Vpr and Vpx usurping the same CRL4-DCAF1 E3 ubiquitin ligase (see below), but targeting and loading different cellular substrates9–15,54,55. Our structure elegantly explains how this is achieved, namely by employing different regions for substrate binding (Figure 4F & 4G). Importantly, Vpr uses the binding cleft between the α1 and α2 helices to engage UNG2 (Figure 4A) while Vpx uses its N-terminal tail and helix α2 to bind SAMHD1 (Figure 4G).
The crystal structures of Vpr and Ugi bound to UNG2 illustrate how these two proteins engage in molecular mimicry of DNA-binding. Both block the active site of UNG2 and this steric obstruction impairs the enzymatic activity of UNG2 (Supplementary Figure 2I) and thus the first step in uracilated DNA repair. Although Vpr and Ugi exhibit strikingly different protein folds, both bind the UNG2 residue Leu272, which is used in DNA binding, and similar surface areas are buried upon complex formation (940 Å2 for Vpr versus 1200 Å2 for Ugi). In addition Vpr and Ugi use the insert loop and a β-strand, respectively, for contacting UNG2. This striking observation explains why the phage protein Ugi can bind with high affinity to UNG240 and inactivate the human enzyme.
Analyses of multiple DDB1 structures in complex with different binding partners (including peptides of different DCAF proteins, a series of pathogenic viral proteins and substrate receptor proteins, such as DDB2 and the Cockayne syndrome A (CSA)) have revealed that a canonical α-helix within the helix-loop-helix motif anchors substrate receptor proteins to DDB145–48,50. In the present structure, DCAF1 also interacts with DDB1 using the helix-loop-helix motif (Supplementary Figure 3A), although the lengths of the helices and their orientation differ from the DDB1-DBB2 structure (Supplementary Figure 3F), It appears that the helix-loop-helix motif is not the only region interfacing with DDB1, and the WD40 propeller domain potentially play an additional role. This may most likely apply to the majority of DCAFs, since 52 of 60 DCAFs possess a WD40 propeller in addition to the helix-loop-helix motif56. Indeed, within the WD40 propeller of DCAF1, a conserved WDXR motif has been identified as important in the interaction with DDB157. In the current structure Arg1283 of DCAF1 interacts with Glu201 in the BPA domain of DDB1. Thus, DCAF1 interacts with DDB1 similarly to other substrate receptors of the CRL4 E3 ubiquitin ligase, using the common helix-loop-helix and WDXR motifs.
The hinge connecting the BPB β-propeller to the clam-shaped BPA-BPC double-propeller (Supplementary Figure 3E) allows for large conformational changes and enables positioning of the substrates for accepting the ubiquitin mark47. To evaluate how HIV-1 Vpr may function in host factor recruitment to the CRL4-DCAF1 E3 ubiquitin ligase machinery and in particular how human UNG2 is positioned for ubiquitination, we built a molecular model of the entire CRL4-DCAF1-Vpr-UNG2 assembly by superimposing the BPA-BPC portion of DDB1 of the current structure onto the BPA-BPC portion of DDB1 in the DDB1-CUL4A-RBX1 structure (Figure 5A, PDB 2HYE)57. Our model shows a 30° degree rotation of the BPB β-propeller with respect to the BPB propeller in PDB 2HYE57 (Supplementary Figure 5 A–D). In this orientation Vpr bridges DCAF1 to UNG2 such that UNG2 is placed within 10 Å of RBX1 (Figure 5B and 5C). Positioning of UNG2 next to RBX1 in the overall complex should allow for polyubiquitination of UNG2, necessary for proteasomal degradation.
Figure 5. Model of the CRL4-DCAF1 E3 ubiquitin ligase complex. Vpr modifies the surface of the common DCAF1 substrate receptor such, that UNG2 or other cellular components can now interact and become a substrate for ubiquitination.
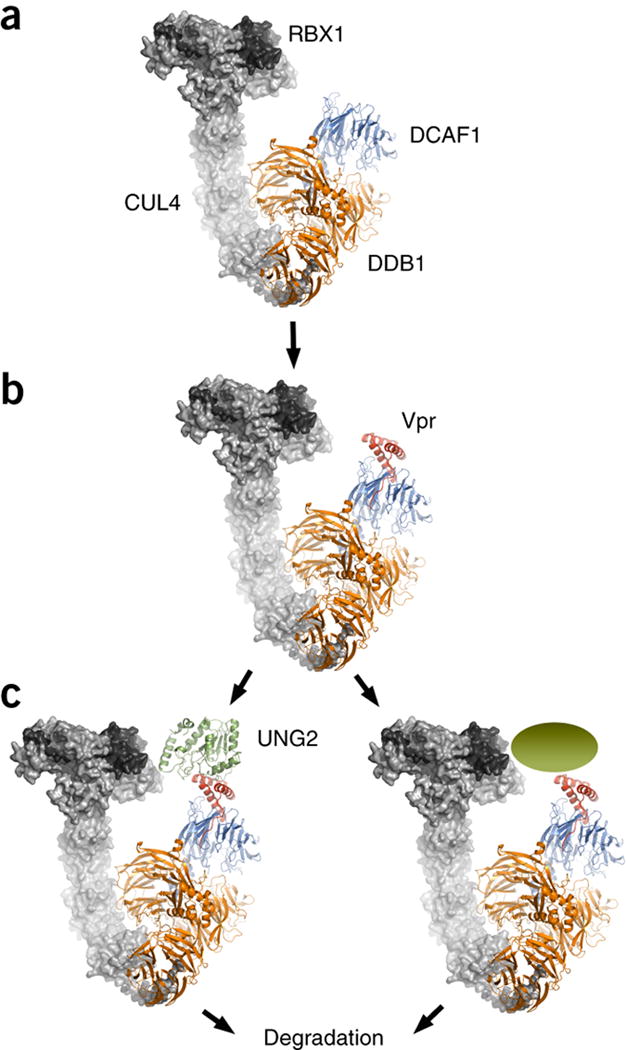
(A) RBX1 and CUL4 in complex with DDB1 (gray surface representation, PDB 2HYE)57 was superimposed on the DDB1 portion of the current DDB1-DCAF1-Vpr-UNG2 complex structure (ribbon representation).
(B) Vpr alters the substrate-binding surface of DCAF1, thereby allowing
(C) Recruitment of UNG2 or other cellular substrates to the E3 ligase for ubiquitination and proteasomal degradation.
Retroviral accessory proteins frequently antagonize antiviral proteins, thereby promoting virus replication in host cells16–18. They frequently achieve their function by co-opting host cellular E3 ubiquitin ligases and loading restriction factors onto the ligases for ubiquitination and proteasome-dependent degradation. Our present findings demonstrate that Vpr uses a powerful dual mechanism to antagonize UNG2. One step exploits molecular mimicry to block substrate recognition by UNG2. The other involves proteasome-dependent degradation of UNG2, with the UNG2–Vpr complex being loaded onto CRL4–DCAF1 E3. Importantly, inspection of HIV-1 Vpr protein sequences deposited in the LosAlamos database (http://www.hiv.lanl.gov/content/index/) revealed that the Vpr interaction surface for UNG2 is well conserved among HIV-1 and SIVcpz Vpr proteins, as is Vpr’s ability to antagonize UNG234. This strong conservation of Vpr’s UNG2 antagonism in HIV-1 and SIVcpz lineage viruses is consistent with the possibility that UNG2 exerts a negative effect on HIV-1 replication. Further virologic studies are required to resolve these issues.
Our structural data support a general mechanism of Vpr-mediated host-factor ubiquitination, which is accomplished by the viral protein modifying the substrate-binding surface of the host adaptor protein DCAF1. In addition, our biochemical data show that Vpr binding to UNG2 results in inhibition of UNG2’s enzymatic activity, which in turn might also contribute to Vpr-mediated infectivity. Thus, Vpr delivers a two-step ‘punch’, inhibiting UNG2 enzymatic activity and steering it toward destruction via the host degradation machinery. Having established the structural and molecular basis of the assembly of the viral modified E3 ubiquitin ligase machinery, our studies should provide a foundation for improved understanding of Vpr’s functions in viral replication and in pathogenesis.
Online Methodes
Cloning and Plasmid constructions
The Vpr with H78C mutation from HIV-1 (NL4-3 clone) was cloned into the pET43 (EMDBioscience) vector with a C-terminal His6-tag, modified to include a TEV protease site between the N-terminal soluble fusion NusA and the Vpr, as described previously29. The WT Vpr was also cloned into pCDNA3 vector (Life Technologies) with an N-terminal HA tag. The cDNA encoding full-length UNG2 was cloned into the pCDNA3 vector with an N-terminal Myc tag, and the catalytic core domain (residues 83–304) was cloned into the pE-SUMO vector (LifeSensors). The N-terminally Myc-tagged DDB1 was cloned in to the pCDNA3 vector. The C-terminal region of DCAF1 (residues 1021–1400) was cloned into the pCDNA5/FRT (Life Technologies) with an N-terminal 3XFLAG-tag. Site-specific mutants of Vpr, UNG2 and DCAF1 were prepared using the Quickchange mutagenesis kit (Agilent Technologies). All other clones were described previously29.
Protein Expression and Purification
NusA-Vpr and UNG2 were expressed in E. coli Rosetta 2 (DE3), and DCAF1 and DDB1 were expressed in insect cells sf21 (Life Technologies). Proteins were purified as described previously23,29. For preparation of multi-protein complexes, DDB1-DCAF1 and NusA-Vpr were mixed at a molar ratio of 1: 3, digested with TEV protease, and purified over an 5 mL MONO Q column (GE Lifesceinces) at pH 7.5 using NaCl gradient 0 – 1 M.
Mammalian Cell Lines, Transfection, and Western blotting
Human embryonic kidney cell lines (HEK293T purchased from ATCC with Catalog CRL-3216, not test for mycoplasma contamination) were grown in advanced DMEM, supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) 100× Glutamine at 37 °C, 5% CO2. Typically, 3 × 106 cells were plated on 100 mm dish and transfected next day with a mixture of pCDNA plasmids encoding specific cDNAs using Lipofectamine 3000 (Life Technologies) according to manufacturer’s protocol. Cells were harvested 48 h later, washed with PBS and lysed with sonication in lysis buffer (50 mM Tris, 150 mM NaCl, 0.5% NP-40, pH 7.5) with 1 tablet of Complete EDTA-free protease inhibitor mixture (Sigma) per 100 ml solution. After centrifugation at 10,000 × g for 10 min, anti-HA affinity matrix (Sigma) or anti-FLAG M2 Magnetic beads (Sigma) were added to the cell lysates supernatant and the mixture were incubated with shaking at 4°C for 5 h. Then, the samples were washed three times with the lysis buffer. Bound proteins were eluted with appropriate peptides, subjected to SDS-PAGE and Western blotting analysis with appropriate antibodies. The antibodies used in this study are anti-HA (COVANCE, Catalog MMS-101P), anti-Myc (COVANCE, Catalog MMS-150P), anti-Flag (Abnova, Catalog PAB0900), anti-Actin (Sigma, Catalog A5316), anti-UNG2 (Abnova, Catalog MAB1099), anti-Rabbit IgG (Sigma, Catalog A8275), anti-Mouse IgG (Sigma, Catalog A2304). Validation is provided on the manufacturers’ websites.
Crystallization and data collection
The DDB1-DCAF1-Vpr is cocrystalized with UNG2 in a solution containing 20 mM Tris-HCl, pH 7.8, 200 mM NaCl, 5 mM MgCl2, 0.02% azide and 2 mM DTT, 3% Glycerol, 0.1X TEZ (40 mM Tris, 1 mM EDTA, 10 mM ZnCl2) buffer and incubated at 4°C for 30 min before crystallization. Crystals were grown at 16°C with the hanging drop vapor diffusion method by mixture of 0.5 uL protein (5 mg/ml) with 0.5 uL crystallization buffer (100 mM Na Citrate, pH 5.6, 11% PEG 20000) and were improved by streak seeding. Crystals were cryoprotected by the addition of 25% (v/v) MPD. Diffraction data were collected at the Advanced Photon Source beamline 23-IDD and 12-2 SSRL. (wavelength 0.979 Å and temperature 100 K). The data statistics are summarized in Table 1.
Structure determination and refinement
Diffraction data were processed, integrated and scaled using XDS (http://xds.mpimf-heidelberg.mpg.de). The structures were solved by molecular replacement using coordinates of DDB1 (PDB 3E0C)41, DCAF1 and VpxMND in the ternary complex of DCAF1-VpxMND-SAMHD1 (PDB 4Z8L)27, and UNG2 (PDB 2OYT)42 as a search model. The initial molecular replacement model was obtained using a data set diffracting to 4.0 Å collected at SSRL. We assumed that the crystal included DDB1, DCAF-1, Vpr and UNG2 (based on biochemical data) and since DDB1 comprises half of the residues in the complex, an initial search using the DDB1 structure (PDB 3E0C)41 in Molrep was performed. This search yielded a unique solution. A second search was performed with the DCAF-1-Vpx complex (PDB 4Z8L)27 as search model, and the solution for DDB1 as fixed model. The solution of the second search showed that the helix-loop-helix domain of DCAF-1 was buried inside the large cleft formed by two DDB1 WD40 propellers, reminiscent of the interactions between the DDB1 and DDB250. No clashes with symmetry related molecules were observed. A third search was started by using UNG2 (PDB 2OYT)42 as the search model, and the solution was found in the second step, which used DDB1–DCAF-1–Vpx as the fixed model. A unique solution was obtained, with UNG2 interacting with Vpx. The solution comprising the four components (heterotetramer) was loaded in Coot (http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/), and after calculation of the symmetry-related molecules, it was evident that a second heterotetramer had to be present in the asymmetric unit. The search was successful and the solutions exhibited no steric clashes between the two heterotetramers or with symmetry related molecules. The initial Molrep model of the DDB1-DCAF-1-Vpr-UNG2 showed that the insert loop (IL) of Vpx search model clashed with UNG2. Calculating the difference map (Fo-Fc) using Buster60 revealed a region of positive density close to the position of the insert loop and reconstituting and repositioning the loop of Vpr during refinement alleviated all steric clashes with UNG2.
The initial refinement of the structure was performed at 4.0 Å; however, we obtained data from a crystal diffracting to 3.5 Å at the APS. These data permitted further refinement using exclusively the 3.5-Å data set. Manual model building in Coot (with B-sharpening) for the DDB1–DCAF-1– UNG2 part of the structure (omitting the model for Vpr) combined with model refinement using Buster improved the electron density maps substantially. At this stage, a difference map (Fo − Fc) of the refined complex showed clear electron density for the Vpr part of the structure (Fig. 1a). Bulky residues such as Trp18, Trp38, Trp54 and the two histidines (His33 and His71) that form part of the zinc-finger domain allowed residue placement with the correct register. The final molecular replacement models were refined iteratively in Buster, interleaved with several cycles of manual building with B-factor sharpening in Coot. All figures were rendered with PyMOL (http://www.pymol.org/).
Base excision and inhibition assay
The glycosylase activity of UNG2 (1 pmol) toward a 29-mer single-stranded DNA substrate with the following sequence TTTTTTTTTTTTTTUTTTTTTTTTTTTTT (100 pmol) was analyzed in the absence or presence of the DDB1–DCAF1 or DDB1–DCAF1–Vpr complexes in 10-μl reaction mixtures containing 50 mM Tris-HCl, pH 7.8, 0.5 mM EDTA, 10 μM ZnCl2 and 5% glycerol36. The reaction mixtures were incubated at 37 °C for 5 min. Reactions were stopped by the addition of 10 μl of quench buffer containing 0.5 M NaOH and 30 mM EDTA, and were heated to 95 °C for 5 min to cleave the phosphate chain at the abasic site. Samples were analyzed on 20% polyacrylamide and 8 M urea TBE gels and stained with SYBR Gold (Invitrogen). The control reaction was treated in the same manner, except that no UNG2 was added.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like thank J. Skowronski for carefully reading the manuscript and providing us with valuable critical comments and suggestions. We would also like to acknowledge M. Becker and C. Ogata at GM/CA (Argonne National Laboratory), and I. Mathews, C. Smith, and A. Gonzalez at Stanford Synchrotron Radiation Lightsource (SSRL) for their magnificent support during data collection. We thank D. Lee for home source X-ray technical support. This work was supported by NIH grant P50GM082251 (to A.M.G.). G.C. acknowledges support from BioXFEL-STC1231306. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
A.M.G., J.A. and G.C conceived the study. Y.W., X.Z., A.M.G., J.A and G.C designed the experiments and analysed the data. Y.W. and G. C. performed crystallization, data collection, structure refinement and analysis. X.Z, M.D. and J.A. performed mutagenesis and structure validation analysis. C.O.B. and A.C. performed X-ray data process. M.D. and J.A. prepared recombinant protein complexes. Y.W., X.Z., A.M.G., J.A. and G.C. wrote the manuscript. All authors discussed the results, commented and approved the manuscript.
Accession codes.
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code PDB 5JK7.
References
- 1.Subbramanian RA, Cohen EA. Molecular biology of the human immunodeficiency virus accessory proteins. J Virol. 1994;68:6831–5. doi: 10.1128/jvi.68.11.6831-6835.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malim MH, Emerman M. HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–98. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe. 2010;8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Tristem M, Purvis A, Quicke DL. Complex evolutionary history of primate lentiviral vpr genes. Virology. 1998;240:232–7. doi: 10.1006/viro.1997.8929. [DOI] [PubMed] [Google Scholar]
- 5.Sharifi HJ, Furuya AM, de Noronha CM. The role of HIV-1 Vpr in promoting the infection of nondividing cells and in cell cycle arrest. Curr Opin HIV AIDS. 2012;7:187–94. doi: 10.1097/COH.0b013e32835049e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Accola MA, Ohagen A, Gottlinger HG. Isolation of human immunodeficiency virus type 1 cores: retention of Vpr in the absence of p6(gag) J Virol. 2000;74:6198–202. doi: 10.1128/jvi.74.13.6198-6202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Re F, Braaten D, Franke EK, Luban J. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J Virol. 1995;69:6859–64. doi: 10.1128/jvi.69.11.6859-6864.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogel ME, Wu LI, Emerman M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J Virol. 1995;69:882–8. doi: 10.1128/jvi.69.2.882-888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belzile JP, et al. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007;3:e85. doi: 10.1371/journal.ppat.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeHart JL, et al. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol J. 2007;4:57. doi: 10.1186/1743-422X-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hrecka K, et al. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci U S A. 2007;104:11778–83. doi: 10.1073/pnas.0702102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Rouzic E, et al. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6:182–8. doi: 10.4161/cc.6.2.3732. [DOI] [PubMed] [Google Scholar]
- 13.Schrofelbauer B, Hakata Y, Landau NR. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc Natl Acad Sci U S A. 2007;104:4130–5. doi: 10.1073/pnas.0610167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan L, Ehrlich E, Yu XF. DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J Virol. 2007;81:10822–30. doi: 10.1128/JVI.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen X, Duus KM, Friedrich TD, de Noronha CM. The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J Biol Chem. 2007;282:27046–57. doi: 10.1074/jbc.M703955200. [DOI] [PubMed] [Google Scholar]
- 16.Blanchet FP, Mitchell JP, Piguet V. beta-TrCP dependency of HIV-1 Vpu-induced downregulation of CD4 and BST-2/tetherin. Curr HIV Res. 2012;10:307–14. doi: 10.2174/157016212800792441. [DOI] [PubMed] [Google Scholar]
- 17.Strebel K. HIV accessory proteins versus host restriction factors. Curr Opin Virol. 2013;3:692–9. doi: 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Baig TT, Love RP, Chelico L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front Microbiol. 2014;5:450. doi: 10.3389/fmicb.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laguette N, et al. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell. 2014;156:134–45. doi: 10.1016/j.cell.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Romani B, Shaykh Baygloo N, Aghasadeghi MR, Allahbakhshi E. HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J Biol Chem. 2015;290:17380–9. doi: 10.1074/jbc.M115.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hrecka K, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–61. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn J, et al. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem. 2012;287:12550–8. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLucia M, Mehrens J, Wu Y, Ahn J. HIV-2 and SIVmac accessory virulence factor Vpx down-regulates SAMHD1 catalysis prior to proteasome-dependent degradation. J Biol Chem. 2013;288:19116–19126. doi: 10.1074/jbc.M113.469007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstone DC, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–82. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 25.Schwefel D, et al. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature. 2014;505:234–8. doi: 10.1038/nature12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwefel D, et al. Molecular determinants for recognition of divergent SAMHD1 proteins by the lentiviral accessory protein Vpx. Cell Host Microbe. 2015;17:489–99. doi: 10.1016/j.chom.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, et al. Structural Basis of Clade-specific Engagement of SAMHD1 (Sterile alpha Motif and Histidine/Aspartate-containing Protein 1) Restriction Factors by Lentiviral Viral Protein X (Vpx) Virulence Factors. J Biol Chem. 2015;290:17935–45. doi: 10.1074/jbc.M115.665513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouhamdan M, et al. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J Virol. 1996;70:697–704. doi: 10.1128/jvi.70.2.697-704.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn J, et al. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J Biol Chem. 2010;285:37333–41. doi: 10.1074/jbc.M110.133181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy EM, Amie SM, Bambara RA, Kim B. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J Biol Chem. 2012;287:14280–8. doi: 10.1074/jbc.M112.348482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan N, O’Day E, Wheeler LA, Engelman A, Lieberman J. HIV DNA is heavily uracilated, which protects it from autointegration. Proc Natl Acad Sci U S A. 2011;108:9244–9. doi: 10.1073/pnas.1102943108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364:675–87. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hrecka K, et al. HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1605023113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eldin P, et al. Vpr expression abolishes the capacity of HIV-1 infected cells to repair uracilated DNA. Nucleic Acids Res. 2014;42:1698–710. doi: 10.1093/nar/gkt974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen R, Le Rouzic E, Kearney JA, Mansky LM, Benichou S. Vpr-mediated incorporation of UNG2 into HIV-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J Biol Chem. 2004;279:28419–25. doi: 10.1074/jbc.M403875200. [DOI] [PubMed] [Google Scholar]
- 37.Guenzel CA, et al. Recruitment of the nuclear form of uracil DNA glycosylase into virus particles participates in the full infectivity of HIV-1. J Virol. 2012;86:2533–44. doi: 10.1128/JVI.05163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weil AF, et al. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc Natl Acad Sci U S A. 2013;110:E448–57. doi: 10.1073/pnas.1219702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80:875–82. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mol CD, et al. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–8. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 41.Xu C, Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2:202–14. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker JB, et al. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–7. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morellet N, Bouaziz S, Petitjean P, Roques BP. NMR structure of the HIV-1 regulatory protein VPR. J Mol Biol. 2003;327:215–27. doi: 10.1016/s0022-2836(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 44.Selig L, et al. Uracil DNA glycosylase specifically interacts with Vpr of both human immunodeficiency virus type 1 and simian immunodeficiency virus of sooty mangabeys, but binding does not correlate with cell cycle arrest. J Virol. 1997;71:4842–6. doi: 10.1128/jvi.71.6.4842-4846.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–17. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 46.Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. A promiscuous alphahelical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol. 2010;17:105–11. doi: 10.1038/nsmb.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer ES, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–39. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 48.Yeh JI, et al. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proc Natl Acad Sci U S A. 2012;109:E2737–46. doi: 10.1073/pnas.1110067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scrima A, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–23. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerard FC, et al. Defining the interactions and role of DCAF1/VPRBP in the DDB1-cullin4A E3 ubiquitin ligase complex engaged by HIV-1 Vpr to induce a G2 cell cycle arrest. PLoS One. 2014;9:e89195. doi: 10.1371/journal.pone.0089195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–44. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 53.Mashiba M, Collins DR, Terry VH, Collins KL. Vpr overcomes macrophage-specific restriction of HIV-1 Env expression and virion production. Cell Host Microbe. 2014;16:722–35. doi: 10.1016/j.chom.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivastava S, et al. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008;4:e1000059. doi: 10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharova N, et al. Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008;4:e1000057. doi: 10.1371/journal.ppat.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell. 2007;26:775–80. doi: 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 57.Angers S, et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–3. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 58.DeLaBarre B, Brunger AT. Considerations for the refinement of low-resolution crystal structures. Acta Crystallogr D Biol Crystallogr. 2006;62:923–32. doi: 10.1107/S0907444906012650. [DOI] [PubMed] [Google Scholar]
- 59.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blanc E, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr. 2004;60:2210–21. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.