

Transcriptome-wide quantitative mapping of 5-methylcytosine in Arabidopsis via RNA bisulfite sequencing reveals crucial links between this modification and cell division and plant stress pathways.
Abstract
Posttranscriptional methylation of RNA cytosine residues to 5-methylcytosine (m5C) is an important modification with diverse roles, such as regulating stress responses, stem cell proliferation, and RNA metabolism. Here, we used RNA bisulfite sequencing for transcriptome-wide quantitative mapping of m5C in the model plant Arabidopsis thaliana. We discovered more than a thousand m5C sites in Arabidopsis mRNAs, long noncoding RNAs, and other noncoding RNAs across three tissue types (siliques, seedling shoots, and roots) and validated a number of these sites. Quantitative differences in methylated sites between these three tissues suggest tissue-specific regulation of m5C. Perturbing the RNA m5C methyltransferase TRM4B resulted in the loss of m5C sites on mRNAs and noncoding RNAs and reduced the stability of tRNAAsp(GTC). We also demonstrate the importance of m5C in plant development, as trm4b mutants have shorter primary roots than the wild type due to reduced cell division in the root apical meristem. In addition, trm4b mutants show increased sensitivity to oxidative stress. Finally, we provide insights into the targeting mechanism of TRM4B by demonstrating that a 50-nucleotide sequence flanking m5C C3349 in MAIGO5 mRNA is sufficient to confer methylation of a transgene reporter in Nicotiana benthamiana.
INTRODUCTION
DNA 5-methylcytosine (m5C) is a well-characterized epigenetic modification with many functional roles in eukaryotes, such as transcriptional silencing and genomic imprinting (Suzuki and Bird, 2008). While DNA appears to have only a small number of other modifications, for example 5-hydroxymethylcytosine and N6-methyladenosine (Korlach and Turner, 2012), RNA has over 100 different modifications that have been identified in organisms across all three domains of life (Motorin et al., 2010; Edelheit et al., 2013; Jackman and Alfonzo, 2013; Machnicka et al., 2013).
The development of high-throughput techniques to identify RNA modification sites transcriptome wide has enabled deeper investigations into the function of m5C in diverse classes of RNA (Hussain et al., 2013a). Although m5C has been found to occur on tRNAs and rRNAs in many organisms (Edelheit et al., 2013; Burgess et al., 2015), it has only more recently been identified transcriptome wide on mRNA and long noncoding RNAs (lncRNAs) from humans (Squires et al., 2012; Hussain et al., 2013b; Khoddami and Cairns, 2013). tRNAs are heavily modified with modifications that affect their aminoacylation and codon identification and stabilize their secondary structure (Alexandrov et al., 2006; Helm, 2006; Agris, 2008; Motorin and Helm, 2010). Of these modifications, m5C sites in tRNAs commonly occur in the variable region and anticodon loop. m5C sites in tRNAs are required for tRNA stability and efficient translation (Schaefer et al., 2010; Tuorto et al., 2012, 2015) and are induced under oxidative stress conditions (Chan et al., 2010, 2012). Similarly, m5C sites play important roles in rRNA processing, structure, and translation (Hong et al., 1997; Sharma et al., 2013; Gigova et al., 2014). Additional roles for rRNA m5C sites in organismal lifespan have also been demonstrated (Schosserer et al., 2015). Several functions have been investigated for m5C in other noncoding RNAs (ncRNAs), such as the vault ncRNA in humans and lncRNAs. Loss of cytosine-5 methylation in vault RNAs causes aberrant processing into Argonaute-associated small RNA fragments that can function as microRNAs (Hussain et al., 2013b). While m5C is required for the stability of vault ncRNAs (Hussain et al., 2013b), m5C sites on the lncRNA, X-inactive specific transcript, prevent binding of the Polycomb repressive complex 2 complex in vitro (Amort et al., 2013).
In eukaryotes, there are two classes of RNA methyltransferases (RMTases), which catalyze m5C of both mRNAs and other types of ncRNAs. The first class of eukaryotic RMTase, TRNA aspartic acid methyltransferase 1 (TRDMT1), also known as DNA methyltransferase 2 (DNMT2), has been shown to methylate tRNAs in animals, plants, and fission yeast (Goll et al., 2006; Jurkowski et al., 2008; Becker et al., 2012; Burgess et al., 2015). The second class of RMTase is known as tRNA-specific methyltransferase 4 (TRM4) or NOP2/Sun domain protein 2 (NSUN2) in yeast and animals, respectively (Motorin and Grosjean, 1999; Auxilien et al., 2012; Tuorto et al., 2012). TRM4/NSUN2 depletion in mice results in male infertility, reduced growth, and epidermal differentiation defects, revealing a role for NSUN2 in stem cell self-renewal and differentiation (Blanco et al., 2011; Hussain et al., 2013c). In addition, NSUN2 mutations in humans are linked to inherited intellectual disability and reduced growth (Abbasi-Moheb et al., 2012; Khan et al., 2012; Martinez et al., 2012; Fahiminiya et al., 2014). Similar to humans, Drosophila melanogaster nsun2 mutants also display short-term memory deficits (Abbasi-Moheb et al., 2012). The underlying cause of these neurological symptoms is thought to be mediated by increased cleavage of nonmethylated tRNAs by the ribonuclease angiogenin during oxidative stress conditions (Blanco et al., 2014). Conserved and plant-specific functions for TRM4 homologs in plants require further investigation. The Arabidopsis thaliana genome encodes two putative TRM4/NSUN2 paralogs, TRM4A and TRM4B (Pavlopoulou and Kossida, 2009; Chen et al., 2010). Previously, we showed that trm4a mutants had a similar tRNA methylation pattern to that of wild-type Arabidopsis and suggested TR4M4A is catalytically inactive (Burgess et al., 2015). In contrast, trm4b mutants lost methylation at tRNA positions C48, C49, and C50 in Arabidopsis.
To further investigate the functions of m5C and to facilitate future studies of m5C in plants, we performed transcriptome-wide single nucleotide resolution of posttranscriptionally modified cytosine residues in Arabidopsis by combining RNA bisulfite conversion with second-generation Illumina sequencing (bsRNA-seq). We report over a thousand m5C sites in Arabidopsis transcribed mRNAs, lncRNAs, and other ncRNAs. Within mRNAs, the majority of m5C sites were identified in the coding sequence; however, when normalized for read coverage and sequence length, m5C sites were observed at higher frequencies than expected in 3′ untranslated regions (UTRs). We also show that the global methylation level of m5C sites varies among silique, shoot, and root tissues and that many m5C sites are dependent on TRM4B. To test the determinants for targeting of TRM4B, we performed a LOGO motif analysis but were unable to identify a consensus sequence that targets TRM4B for methylation. We subsequently identified a 50-nucleotide sequence that conferred TRM4B-dependent methylation of a transgene in vivo, suggesting that additional factors, such as RNA structure, may be involved. To determine the role of TRM4B in plant development, we investigated primary root growth. We found that trm4b mutants have a short-root phenotype as a consequence of reduced cell division compared with the wild type. Furthermore, trm4b mutants are more sensitive to oxidative stress and have reduced stability of nonmethylated tRNAs. These data provide a genome-wide, high-resolution view of m5C in plants and link this modification to biological function.
RESULTS
Transcriptome-Wide Detection of m5C Sites in Arabidopsis RNA
To identify transcriptome-wide m5C sites in Arabidopsis RNA at single-nucleotide resolution, we performed bisulfite conversion of ribosomal-depleted RNA from wild-type plants, combined with a nonmethylated in vitro transcribed Renilla Luciferase (R-Luc) mRNA bisulfite conversion control followed by stranded Illumina RNA-sequencing (bsRNA-seq). We obtained 255, 262, and 116 million paired-end (100 nucleotides) Illumina reads from Arabidopsis siliques, shoots, and roots, respectively, and aligned the sequences to an in silico bisulfite converted Arabidopsis transcriptome and R-Luc mRNA control (see Methods; Supplemental Data Set 1). Analysis of our R-Luc controls demonstrated almost complete C-to-T conversion for all samples. For example, the library derived from wild-type seedling shoots showed >99.8% conversion of the R-Luc control (Supplemental Figure 1A), and global endogenous cytosine abundance was <0.5% compared with ∼22% for nonbisulfite treated RNA-seq libraries (Supplemental Figure 1B). We were able to confirm previously identified m5C sites in rRNAs (mitochondrial 26S rRNA C1586) and tRNAs [tRNAGlu(TTC)], demonstrating the robustness of our bisulfite conversion protocol (Burgess et al., 2015) (Supplemental Data Sets 2 and 3). In addition, we PCR amplified, cloned, and Sanger sequenced the highly structured tRNAAsp(GTC), and all sequences demonstrated complete bisulfite conversion at known nonmethylated sites (Burgess et al., 2015) (Supplemental Figure 1C). Together, these data demonstrate that our bisulfite conversion was highly efficient.
Discovery of Novel m5C Sites in Arabidopsis Siliques, Shoots, and Roots
To determine tissue-specific methylation profiles, we next examined all reads aligning to the Arabidopsis transcriptome to identify hundreds of novel m5C sites in three different tissue types. In Arabidopsis siliques, seedling shoots, and roots, 128, 201, and 859 m5C sites were identified, respectively, with methylation levels ranging from 2 to 92% (false discovery rate [FDR] ≤ 0.3) (Figure 1A; Supplemental Data Sets 2 and 3). The majority of these sites were tissue specific, and only 15 sites were commonly methylated between all three tissue types. The two tissues showing the greatest number of conserved methylated sites were siliques and shoots, sharing 48 common m5C sites. When global methylation levels of m5C sites within siliques, shoots, and roots were compared, roots had lower average methylation of m5C sites compared with siliques or shoots (P value ≤ 0. 0001) (Supplemental Figure 2).
Figure 1.

Transcriptome-Wide Mapping of m5C Using bsRNA-Seq in Arabidopsis.
(A) Venn diagram showing the number of m5C sites in Arabidopsis wild-type (Col-0) siliques at 1 d after pollination, seedling shoots at 10 d after germination (DAG), and seedling roots at 6 DAG using bsRNA-seq/RBS-seq (n = 2–3). Methylated sites are called as significant at FDR ≤ 0.3 and ≥1% methylation.
(B) Gene schematic and genome browser view of a differentially methylated gene, MAG5, in three different tissue types. In the MAG5 gene schematic, the methylated cytosine C3349 is indicated. Boxes represent exons; lines represent introns. In the genome browser view, black arrows indicate m5C position C3349 (chr5: 19, 262, 126). Top: m5C methylation percentage (proportion of blue in column represents methylation percentage or numbers of nonconverted cytosines). Bottom: a subset of bsRNA-seq reads mapped to the MAG5 locus (chr5: 19, 262, 122-19, 262, 130) from the separate tissue data sets: siliques, shoots, and roots. Each row represents one sequence read and each column a nucleotide. Gray boxes represent nucleotides matching with the MAG5 reference sequence, and red boxes indicate mismatching nucleotides and/or nonmethylated, bisulfite converted cytosines. Sequencing gaps are shown in white.
(C) Distribution of m5C sites within different RNA types from siliques, shoots, and roots. The numbers in parentheses are percentage of total.
(D) Scatterplot showing that increased methylation correlates with lower mRNA abundance in Arabidopsis roots (rs = −0.32, *P value ≤ 0.0001, Spearman’s correlation).
(E) Histogram showing relative enrichment of observed m5C sites versus expected number of m5C sites in the 3′UTR compared with the CDS and 5′UTR across all three tissue types (*P value ≤ 0.0001, binomial test).
(F) Metagene profile of m5C site abundance along 5′UTR, CDS, and 3′UTRs, normalized for transcript length (relative distance) across all three tissue types (FDR ≤ 0.3 and ≥2% methylation).
(G) Methylation of candidate m5C sites using bsRNA-amp-seq/RBS-amp-seq was analyzed to independently confirm bsRNA-seq results. Heat map depicts log arcsine transformed methylation percentages of five m5C sites in Arabidopsis wild-type siliques, shoots, and roots using bsRNA-seq and bsRNA-amp-seq. The numbers in parentheses are genome coordinates, and + or – indicates the strand.
We then asked whether the percentage of methylation at specific sites in mRNA transcripts expressed in shoots, siliques, and roots varied and might indicate functional significance. We compared the m5C level at common sites and discovered that the level of methylation at specific m5C sites varied in shoot, silique, and root tissues (Supplemental Data Sets 2 and 3). For example, C3349 in MAIGO5 (MAG5) had 55% methylation in shoots, 33% in shoots, and 26% in roots (P < 0.05, Student’s t test; Figure 1B).
Annotation of m5C-containing transcripts from the three tissue types revealed many diverse RNA types, including mRNAs, pseudogenes, lncRNAs, natural antisense transcripts, small nucleolar RNAs (snoRNAs), small nuclear RNAs (snRNAs), and other ncRNAs (Figure 1C). While methylation of specific tRNAs and other types of ncRNAs is associated with increased stability and abundance (Chen et al., 1993; Tuorto et al., 2012; Hussain et al., 2013b), no link between mRNA abundance and m5C levels in mammals has been reported to date (Tuorto et al., 2012; Hussain et al., 2013b, 2013c). To examine if a correlation between m5C methylation and gene expression exists in Arabidopsis, we plotted the percentage of m5C methylation (≥2%) identified in the bsRNA-seq root tissue data against RNA-seq gene expression data generated from the same tissue type (Figure 1D). We observed a small negative correlation of mRNA abundance with higher levels of methylation in Arabidopsis roots (rs = −0.32, P value ≤ 0.0001), indicating m5C deposition in these transcripts may affect its stability or turnover rate within the cell.
To further investigate the role of m5C in mRNAs, we searched for any bias in m5C site distribution within mRNAs. This revealed a significant enrichment of m5C sites within 3′UTRs (P value ≤ 0.0001), when normalized for read coverage and sequence length (Figure 1E). When examining the abundance of m5C sites that occur along the features of mRNAs, the greatest number of m5C sites was identified within the coding sequence (Figure 1F). While m5C sites were evenly distributed within 5′UTRs and coding sequences, m5C sites were most abundant in the first quarter of 3′UTRs.
Confirmation and Validation of m5C Candidate Sites by bsRNA Amplicon Sequencing
To confirm the results of global analysis of m5C sites in silique, shoot, and root transcriptomes and to test the range of sensitivity of our bsRNA-seq approach, we independently tested five candidate m5C sites using RNA bisulfite amplicon sequencing (bsRNA-amp-seq). BSRNA-amp-seq involves next-generation sequencing of targeted PCR amplicons derived from bisulfite-treated total RNA (see Methods). All five selected m5C sites had similar, reproducible methylation levels in both the transcriptome-wide and targeted amplicon sequencing experiments, ranging from ∼3 to 55% methylation (Figure 1G; Supplemental Data Set 4). Methylation percentages as low as 3.2% ± 0.6% for mRNA obtained from the methionine synthase gene ATMS1 (AT5G17920) using bsRNA-seq in wild-type seedling shoots were able to be reproduced and independently validated using bsRNA-amp-seq (3.3% ± 0.8%) (Figure 1G; Supplemental Data Set 4). Similarly, our targeted amplicon sequencing validated sites that showed differential methylation levels among the tissues using bsRNA-seq, such as MAG5 C3349, which shows higher methylation levels in siliques than in seedling shoot and roots (Supplemental Data Set 4). Together, these data demonstrate that a majority of the m5C sites identified in our transcriptome-wide bsRNA-seq data are robust.
To validate our transcriptome-wide bsRNA-seq results, we performed m5C RNA immunoprecipitations (IPs) on RNA alongside an IgG control. Using quantitative RT-PCR (qRT-PCR) on fractions of RNAs IP with either the antibody specific to m5C or an IgG control, we measured the abundance of four mRNAs and one rRNA that bsRNA-seq identified to contain m5C and five mRNAs that were found to be devoid of m5C (Figure 2). We normalized our qRT-PCR measurements in the m5C IP fraction to the IgG fraction. We found that all five transcripts tested were significantly (P value < 0.01, Student’s t test) enriched in the m5C fractions compared with the nonspecific antibody control (Figure 2). This provided evidence for the robustness of the transcriptome-wide bsRNA-seq identification of m5C sites. For the five putative negative control loci (those with no m5C sites), we found that all of these RNAs had a similar level (P value < 0.01, Student’s t test) in the m5C IP fractions compared with the IgG control (Figure 2). Together, our data demonstrate that the m5C sites identified by bsRNA-seq data are independently verifiable.
Figure 2.

Immunoprecipitation of Transcripts Found to Contain m5C Modifications by bsRNA-Seq.
qRT-PCR analysis of five transcripts (At3g63450, At5g17929, At5g40395, At5g47480, and 25S rRNA) found to contain m5C by bsRNA-seq and five transcripts (At1g50010, At1g65480, At3g18780, At3g22480, and At5g39420) found to lack m5C. The qRT-PCR data for all transcripts were normalized to the nonspecific IgG control. Fold enrichment over an IgG nonspecific antibody control (y axis) is plotted for each transcript. qRT-PCRs were performed on two biological and three technical replicates. Error bars indicate ±se of the mean. P values were calculated with a Student’s t test. Asterisk denotes P value < 0.05.
TRM4B Is Required for Methylation of m5C Sites in Shoot, Silique, and Root Transcriptomes
To gain insight into the enzymes mediating m5C methylation, we performed transcriptome-wide bsRNA-seq on rRNA-depleted RNA from wild-type and trm4b mutant silique, seedling shoot, and root tissues and from trdmt1 mutant seedling shoots (Figure 3; Supplemental Data Set 3). In siliques and seedling root tissues, 40 and 17 sites had no or reduced methylation in trm4b mutants, respectively, compared with the wild type (FDR ≤ 0.3). Similarly, for seedling shoots of trm4b, 69 sites had completely absent or significantly reduced methylation compared with the wild type, whereas no m5C sites were differentially methylated in trdmt1 mutants (FDR ≤ 0.3) (Figure 3A; Supplemental Data Sets 3 and 5). Furthermore, we used bsRNA-amp-seq to investigate the requirement for the proposed catalytically inactive TRM4A at 15 m5C sites and found no change in methylation levels in trm4a mutants compared with the wild type (Supplemental Figure 3 and Supplemental Data Set 6). We looked more closely at TRM4B-dependent sites and discovered that TRM4B mediates many m5C sites on diverse classes of RNAs, such as mRNAs, tRNAs, snoRNAs, snRNAs, lncRNAs, and natural antisense transcripts across all three tissue types examined (Figure 3B). Next, we asked if sites dependent on TRM4B were located within specific regions of mRNA transcripts. Most TRM4B-dependent sites were located in the coding sequence, which is consistent with the global distribution of m5C sites within mRNAs (Figure 1F; Supplemental Data Set 3). One such TRM4B-dependent m5C site, which is methylated in all three tissues types, occurs at cytosine position C3349 in the coding sequence of MAG5 (Figure 3C). While 33% of MAG5 transcripts are methylated at C3349 in wild-type seedling shoots, methylation is reduced to 0% in trm4b.
Figure 3.

TRM4B-Dependent m5C Sites in mRNAs and ncRNAs of Siliques, Shoots, and Roots in Arabidopsis.
(A) Heat map showing log arcsine transformed methylation percentages of TRM4B-dependent m5C sites in Arabidopsis wild-type (Col-0) and trm4b-1 siliques at 1 d after pollination, seedling shoots at 10 DAG, and seedling roots at 6 DAG using bsRNA-seq (n = 2–3). Differentially methylated sites are called as significant at (FDR ≤ 0.3 and ≥1% methylation). Coordinate for m5C site is shown in the format chromosome number_genomic position_Watson or Crick strand is in parentheses.
(B) Distribution of TRM4B-dependent m5C sites in different RNA classes across all three tissue types. The numbers in parentheses are percentage of total.
(C) MAG5 (AT5G47480) requires TRM4B for methylation at cytosine position chr5: 19, 262, 126 (black arrows). Top, wild type; bottom, trm4b-1 shoots 10 DAG. Integrative Genomics Viewer browser snapshots of percentage m5C (proportion of blue in column represents methylation percentage or numbers of nonconverted cytosines) and mapped reads, a subset of the bsRNA-seq reads mapped to the MAG5 locus (chr5: 19, 262, 122-19, 262, 130). Each row represents one sequence read and each column a nucleotide. Gray boxes represent nucleotides matching with the MAG5 reference sequence, and red boxes indicate mismatching nucleotides and/or nonmethylated, bisulfite converted cytosines. Sequencing gaps are shown in white.
(D) Methylation of candidate m5C target sites of TRM4B in the wild type and trm4b-1 mutants using bsRNA-amp-seq was analyzed to independently confirm bsRNA-seq results. Heat map depicts log arcsine transformed methylation percentages of five m5C sites in Arabidopsis wild-type and trm4b-1 siliques, shoots, and roots using bsRNA-seq (n = 2–3) and bsRNA-amp-seq (n = 3).
(E) Methylation of TRM4B-dependent m5C sites in 3-week-old leaves of wild-type and transgenic plants overexpressing TRM48 (TRM48-OX) using bsRNA-amp-seq. Heat map depicts log arcsine transformed methylation percentages of ten m5C sites, all of which show increased methylation in TRM48-OX (wild type, n = 3; TRM4B-OX, n = 1).
To independently confirm the TRM4B-dependent sites, including MAG5 C3349, that were identified using our bioinformatics pipeline and statistical cutoff of FDR ≤ 0.3 and ≥1% methylation, we performed bsRNA-amp-seq on wild-type and trm4b mutant silique, seedling shoot, and root tissue (Figure 3D; Supplemental Data Set 7). All five m5C sites tested showed reproducible methylation in the wild type and loss or severely reduced methylation in trm4b mutants across all three tissue types.
TRM4B Overexpression Specifically Increases m5C Methylation
To investigate whether TRM4B expression contributes to altered methylation rates between different tissues, we compared the methylation percentages of TRM4B-dependent sites in plants overexpressing TRM4B mRNA versus the wild type (Figure 3E; Supplemental Data Set 8). We overexpressed TRM4B (Supplemental Figure 4A) and performed bsRNA-amp-seq on 10 TRM4B-dependent m5C sites. At all 10 sites, methylation levels were higher in TRM4B overexpression plants than in wild-type plants. We observed between 3- and 124-fold increases in methylation percentages, specifically at TRM4B-dependent m5C sites. No additional, spurious cytosines were methylated when TRM4B expression was increased, suggesting the occurrence of controlled, specific targeting of TRM4B (Supplemental Figure 4B).
After identifying and validating TRM4B-dependent m5C sites transcriptome-wide, we investigated how TRM4B is targeted to these specific cytosines in RNA. To test the sequence determinants for targeting of TRM4B, we performed a LOGO motif analysis using a 50-nucleotide region flanking both sides of TRM4B-dependent m5C sites identified from siliques, seedling shoots, and roots (Figure 3A). Using this approach, we were unable to identify a consensus sequence for targeting TRM4B methylation (Supplemental Figure 5). This is similar to findings in human cancer cell lines (Squires et al., 2012; Hussain et al., 2013b) and led us to further investigate the sequence requirements for targeting of TRM4B in plants.
TRM4B Flanking Sequence Context for Effective Methylation
As the LOGO motif analysis did not reveal any consensus sequences, we next investigated the sequence requirements for TRM4B to confer methylation in mRNA. We developed a rapid, transient expression assay in Nicotiana benthamiana to express m5C sensor constructs, which contained sequences of varying lengths flanking the m5C site (C3349) in the MAG5 transcript. The MAG5 m5C site was chosen because it is highly methylated across a range of tissues, the methylation percentage varies across tissues, and methylation is dependent on TRM4B (Figures 1B, 3A, and 3D). Three sensor fragments (51, 101, and 189 nucleotides) containing the MAG5 C3349 m5C site and a negative control sensor fragment containing a region of MAG5 that is not methylated endogenously in Arabidopsis were cloned downstream of a GFP reporter gene. Transgene expression was confirmed by GFP fluorescence in the infiltrated N. benthamiana leaves (Figure 4A), and RNA was purified for bsRNA-amp-seq. For each construct, three replicate leaves were infiltrated, and bsRNA-amp-seq was performed separately. The results show differential methylation of MAG5 C3349 site for each of the four sensor constructs tested (Figure 4B). As expected, efficient bisulfite conversion was observed for all cytosines flanking the methylated site, as no methylation was detected using a 0.9% threshold for nonconversion and no methylation was detected in the negative control sensor construct (Figure 4B). Sensors 2 and 3 (S2 and S3), which contained 101- and 189-nucleotide MAG5 inserts, respectively, had the highest methylation level of 53.2% ± 1% at C3349. In contrast, the smallest sensor (S1), with 51 nucleotides, was the least methylated at 4.6% ± 0.4%. This indicates that shorter sequences of up to 25 nucleotides flanking the m5C site may not carry all the substrate information required for effective methylation by TRM4B. While the level of methylation of N. benthamiana MAG5 is not known, the cytosine methylation level of 53% observed in sensors 2 and 3 is similar to that observed in Arabidopsis silique tissue (55%). Together, these results demonstrate that longer sequences of 50 and 94 nucleotides flanking the methylated C3349 site are able to confer moderate methylation, while the 25-nucleotide flanking sequence severely decreased methylation, suggesting that additional factors such as RNA structure may be involved.
Figure 4.

In Vivo TRM4B-Dependent Substrate Recognition.
(A) Schematic representation of m5C methylation sensor constructs. Three different sized sequences flanking the MAG5 C3349 methylated site were cloned in between the GFP coding region and the beginning of the 3′UTR sequence and are referred to as sensor 1 (S1; 51 nucleotides), sensor 2 (S2; 101 nucleotides), and sensor 3 (S3; 189 nucleotides). The C3349 methylated site is located in the center of all three sensor fragments. A fourth region, derived from MAG5 exon one, was used as the “negative” control in addition to a “no insert” empty vector control. Representative images of N. benthamiana leaves transiently expressing GFP reporter for each of the sensor constructs are shown. Bar = 100 µm.
(B) Cytosine methylation in sensor fragments identified by bsRNA-amp-seq. Top: Average MAG5 C3349 methylation percentage in sensors 1 to 3 from three biological replicates. Each biological replicate was constructed by purifying RNA from three infiltrated leaves of N. benthamiana, and the RNA was then bisulfite treated, cDNA synthesis performed, and sensor regions were PCR amplified and Illumina sequenced. No nonconverted cytosines were identified in the negative control construct. Error bars indicate ± se of the mean (****P < 0.001, one-way ANOVA; n = 3). Bottom: Heat maps showing the cytosine methylation status within sensor fragments. No nonconverted cytosines were identified in the flanking sequences.
TRM4B Is Required for Cell Proliferation in the Arabidopsis Root Apical Meristem
In yeast and animals, TRM4/NSUN2 has broad functional roles in mediating stem cell self-renewal and oxidative stress tolerance (Blanco et al. 2011, 2014; Chan et al., 2012; Tuorto et al., 2012; Hussain et al., 2013c). Loss of NSUN2 in mice results in male infertility and reduced size, which is thought to be due to reduced proliferative capacity in stem cells (Tuorto et al., 2012; Hussain et al., 2013c). In plants, stem cell niches are located in the shoot and root apical meristems and are responsible for postembryonic growth of shoot and root systems (Beemster and Baskin, 1998; Stahl and Simon, 2010). While shoot and inflorescence growth appeared unaffected in Arabidopsis trm4b mutants (Burgess et al., 2015; Supplemental Figure 6), primary roots were significantly shorter compared with the wild type in both trm4b T-DNA mutant alleles analyzed (Figure 5). Monitoring of primary root length over a 7-d period revealed decreased elongation rates in the trm4b mutant, indicating that TRM4B positively regulates root growth at early stages of seedling growth (Figure 5B). Furthermore, complementation of the trm4b-1 mutant with a TRM4B genomic DNA construct confirmed that the loss of TRM4B causes the observed short-root phenotype (Supplemental Figure 7). Root growth in trdmt1, the second RMTase mutant analyzed in this study, was indistinguishable from that of wild-type plants. Meanwhile, trm4b-1 trdmt1 double mutants showed short roots similar to those of trm4b-1 single mutants, indicating the phenotype is solely linked to TRM4B loss of function (Supplemental Figure 8).
Figure 5.

TRM4B Is Required for Primary Root Growth Elongation in Young Seedlings.
(A) Root elongation of 7 DAG wild type (Col-0) and two TRM4B T-DNA mutants, trm4b-1 and trm4b-2, grown on 0.5× MS medium with 1% sucrose. Bar = 1 cm.
(B) Graph of primary root growth measurements over time in the wild type and trm4b-1 and trm4b-2 mutants.
(C) Confocal images of 7 DAG wild-type and trm4b-1 root tips stained with propidium iodide. White arrowheads indicate the position of the quiescent center, and yellow arrowheads indicate the first rapidly elongated cell in the cortical layer marking the end of the meristem. Insets (50-µm length) are enlarged images of the boxed areas showing cortical cells in the meristematic zone marked with asterisks. Bar = 100 µm.
(D) to (F) Root apical meristem size (D), cell number (E), and cell length (F) of wild-type and trm4b-1 mutant plants.
(G) Activity of CYCB1;1:GUS reporter in the root meristems of wild-type and trm4b-1 plants, detected by GUS staining. Bar = 100 µm. Error bars in (D) to (G) indicate the se of the mean (*P < 0.05, Student’s t test; n ≥ 8 seedlings).
At the cellular level, primary root growth is determined by cell production rates in the meristematic zone and by the final cell lengths achieved in the differentiation zone (Beemster and Baskin, 1998). To further understand the role of TRM4B in coordinating root growth, we examined the cellular anatomy in the different root growth zones in trm4b mutants and wild-type plants. The cellular organization within the root apical meristem (RAM) and its overall size were unaffected in the trm4b-1 mutant (Figure 5C). The RAM size, defined as the length from the quiescent center to the first significantly elongated cell, was measured in the epidermal (outer) and cortical (inner) cell files. Interestingly, quantification of meristematic cell numbers revealed trm4b-1 to have significantly fewer epidermal and cortical cells (reduced by 21 and 17%, respectively) compared with the wild type. The reduced cell numbers and unchanged meristem size observed in the trm4b-1 mutant correlated with an increased average cell length (Figures 5D to 5F). However, the final cell lengths achieved in the differentiation zone were similar in the mutant and the wild type, indicating that the larger meristem cells in trm4b-1 represent a possible compensatory mechanism to maintain overall meristem size (Supplemental Figure 9). Thus, the short-root phenotype observed for trm4b-1 is most likely linked to the reduced capacity for the cells to divide in the meristem. Supporting evidence was also obtained from a CYCB1;1 (CYCLIN B1;1)-GUS reporter line crossed into the wild type and the trm4b-1 mutant background. CYCB1;1 is expressed during the G2-M phase of the cell cycle, allowing mitotic activity to be visualized (Doerner et al., 1996). Based on the intensity of GUS staining in the RAM, a marked reduction in mitotic activity was evident in the trm4b-1 roots compared with the wild type (Figure 5G). Together, this suggests that TRM4B positively regulates root growth by controlling cell production numbers in the meristem.
trm4b Mutants Display Increased Sensitivity to Oxidative Stress
Loss of TRM4/NSUN2 in yeast and mice results in heightened sensitivity to oxidative stress (Chan et al., 2012; Blanco et al., 2014). We next asked if TRM4B in Arabidopsis is also required for the oxidative stress response by challenging the mutants with the oxidative stress-inducing compound paraquat and H2O2 in a root growth sensitivity assay. The trm4b mutant displayed increased sensitivity to oxidative stress damage compared with wild-type seedlings across a range of paraquat and H2O2 concentrations (Figure 6). In contrast, trm4b mutant plants showed a similar level of sensitivity to that of the wild type when grown on salt (Figure 6A), indicating that TRM4B is unlikely to be part of the general stress pathway and that it specifically acts in mediating oxidative stress responses in Arabidopsis.
Figure 6.

Loss of TRM4B Leads to Oxidative Stress Sensitivity and Decreased tRNA Stability.
(A) Relative root lengths of seedlings on paraquat, H2O2, and NaCl compared with untreated controls. Seedlings of the wild type, trmb4-1, and trmb4-2 were grown on 0.5× MS medium for 4 d before being transferred to control medium or medium containing paraquat, H2O2, or NaCl at the indicated concentrations. Root growth was measured for three subsequent days after transfer. Error bars represent se of the mean (*P < 0.05, one-way ANOVA comparison to the wild type; n ≥ 15–30 seedlings).
(B) Left: Representative RNA gel blot analysis of nuclear tRNAsAsp(GTC) and chloroplast tRNALeu(CAA) for the wild type and trm4b-1. The loading control miR156 was used for analysis. Normalized intensities are given beneath each lane. Right: Quantitative analysis of TRM4B substrate tRNAAsp(GTC) and controls: tRNALeu(CAA) and tRNAGln(TTG) from two to three independent RNA gel blots, with two replicate RNA samples run for each blot. The tRNA signal intensities were normalized to the miR156 loading control and then compared with the wild type. Error bars represent se of the mean (*P < 0.05, Student’s t test; n = 3–4 replicate RNA samples).
In humans and mouse, methylation by TRM4/NSUN2 protects tRNAs from oxidative stress-induced cleavage (Blanco et al., 2014), and in yeast, TRM4-dependent m5C sites in tRNAs are dynamically modulated in response to oxidative stress (Chan et al., 2010, 2012). As we recently showed that TRM4B methylates a number of nuclear tRNAs in Arabidopsis (Burgess et al., 2015), we investigated the steady state levels of tRNAAsp(GTC) as a representative TRM4B substrate. Three m5C sites (C48, C49, and C50) in the variable region of tRNAAsp(GTC) were identified as being TRM4B dependent (Burgess et al., 2015). RNA gel blot analysis revealed a 50% reduction in tRNAAsp(GTC) abundance in the trm4b-1 mutant compared with the wild type, indicating that TRM4B-mediated methylation is required to stabilize tRNAAsp(GTC) substrate (Figure 6B). In contrast, both chloroplast tRNALeu(CAA) and nuclear tRNAGln(TTG), which are not dependent on TRM4B methylation, display similar abundance levels in the mutant and wild type (Figure 6B). We also observed a modest reduction in steady state levels for tRNAHis(GTG), which contains one TRM4B-dependent m5C site, indicating that additional m5C sites like that observed for tRNAAsp(GTC) may increase tRNA stability or resistance to cleavage (Supplemental Figure 10). These findings, together with earlier reports of the functions of TRM4 and TRDMT1 in other species, suggest reduced tRNA stability as a possible cause of the hypersensitivity to oxidative stress exhibited by trm4b plants (Schaefer et al., 2010; Tuorto et al., 2012; Blanco et al., 2014).
Genes in the Oxidative Response Pathway Are Constitutively Activated in trm4b
To further investigate the molecular basis of the short-root phenotype and hypersensitivity to oxidative stress of trm4b, we performed RNA sequencing of the mutant and wild type under paraquat-stressed and nonstressed control conditions using whole root tissue (Supplemental Data Set 9). As expected, a large number of genes in the oxidative response pathway were differentially expressed between wild-type control and wild-type paraquat-treated seedlings (Supplemental Figure 11). Gene Ontology (GO) term analysis of differentially expressed genes showed enrichment of oxidative stress-related biological processes (P values ≤ 0.001). Surprisingly, we also found oxidative response-related genes to be differentially expressed in the trm4b mutant compared with the wild type under nonstressed conditions (P values ≤ 0.001) (Supplemental Figure 11B). Consistent with this observation, GO terms related to oxidative responses were also enriched in the subset of differentially expressed genes common to both paraquat-treated wild-type seedlings compared with untreated wild-type seedlings and in the trm4b mutant compared with the wild type (Figure 7). Closer examination of this common subset of differentially expressed genes revealed that 77% of upregulated genes and 78% of downregulated genes in the trm4b mutant compared with the wild type were also differentially expressed in the paraquat-treated wild-type plants compared with control conditions (Figure 7B). Furthermore, we investigated genes that showed a significant interaction between loss of TRM4B and oxidative stress. Of note, 229 genes showed significant interaction effects based on the RNA-seq data (FDR ≤ 0.05) (see “R_interaction_term” tab in Supplemental Data Set 9). Together, these results support the role of TR4MB in regulating oxidative stress responses and suggest that the short-root phenotype of trm4b may be mediated by partial activation of oxidative stress pathways.
Figure 7.

Oxidative Stress-Responsive Pathways Are Constitutively Activated in trm4b Mutants.
Whole roots at 6 DAG were harvested from wild-type, trm4b-1, and paraquat treated wild-type seedlings, and RNA-seq analysis was performed (n = 3). Most genes differentially expressed in trm4b-1 mutants compared with the wild type are also differentially expressed in paraquat-treated wild-type plants compared with control conditions.
(A) Biological process GO terms involved in the oxidative stress response are significantly enriched (FDR < 0.05) in trm4b-1 mutants compared with the wild type and in paraquat-treated wild-type plants compared with untreated wild-type plants. GO terms significantly enriched in both comparisons are depicted, with black stars indicating GO terms relating to oxidative stress responses. Heat map shows the significance level using the negative log of the P value, where red = very significant and yellow = significant (GO term FDR < 0.05).
(B) Venn diagrams show the overlap of differentially upregulated (left) and downregulated (right) genes (FDR < 0.05).
DISCUSSION
The discovery of m5C in cellular RNAs of a wide range of eukaryotic and prokaryotic organisms underscores its importance as a key regulator of RNA metabolism (Squires et al., 2012; Edelheit et al., 2013; Hussain et al., 2013b; Khoddami and Cairns, 2013; Burgess et al., 2015). While previous studies have shown m5C to be required for tRNA stability (Chen et al., 1993; Schaefer et al., 2010; Tuorto et al., 2012; Blanco et al., 2014) and rRNA processing (Hong et al., 1997), its function in other RNA classes, such as mRNAs and the enzymes mediating m5C methylation, remain largely unexplored. As an important step in determining their biological function, we identified m5C sites in diverse classes of coding and noncoding RNAs in Arabidopsis shoots, roots, and siliques using bsRNA-seq. To confirm the accuracy and to test the range of sensitivity of our transcriptome-wide bsRNA-seq approach, we independently tested candidate m5C sites and previously known methylated sites using bsRNA-amp-seq as well as conventional Sanger sequencing. We found that the methylation rates of m5C sites were in close agreement with our bsRNA-seq data and were highly reproducible across a wide methylation range (Figures 1G and 2D; Supplemental Data Sets 4 and 7). Together, these data demonstrate that we performed robust and sensitive detection of m5C sites using our approach. As several modifications other than m5C may inhibit the conversion of C to T during bisulfite treatment (Schaefer et al., 2009; Squires et al., 2012), we confirmed that many of these modified sites are indeed m5C sites, as they are dependent on the RNA methyltransferase TRM4B and could be immunoprecipitated with an m5C antibody.
We detected 201, 859, and 128 m5C sites in Arabidopsis shoots, roots, and siliques, respectively (FDR ≤ 0.3, ≥1% methylation) (Figure 1A; Supplemental Data Sets 2 and 3). Only a small proportion of these sites were commonly methylated across all three tissue types, suggesting possible tissue-specific functions. Methylated sites were identified predominately in mRNAs and were also found in diverse noncoding types of RNA, such as snRNA, snoRNAs, lncRNAs, and natural antisense transcripts (Figure 1C). The presence of m5C in many different types of RNA is a conserved feature of RNA methylation across mammals and plants (Squires et al., 2012; Hussain et al., 2013b; Khoddami and Cairns, 2013). Within Arabidopsis mRNAs, m5C sites most commonly occur within the coding sequence (CDS), and fewer sites were observed in UTRs (Figure 1F). When normalized for sequence length and read coverage, this revealed enrichment of m5C sites within Arabidopsis 3′UTRs (Figure 1E), which is similar to the results obtained for human cancer cells (Squires et al., 2012). To investigate the functions of m5C in mRNAs, we asked if methylation levels affected mRNA abundance in Arabidopsis roots. We observed a small negative correlation of mRNA abundance with higher levels of methylation, suggesting that, in contrast to tRNAs (Tuorto et al., 2012) and other ncRNAs (Hussain et al., 2013b), m5C in mRNA does not generally increase RNA stability, and the effects of m5C in mRNA are likely context dependent (Figure 1D). In addition, no correlations were observed between mRNA abundance and m5C levels in mammals, as no global changes in mRNA abundance were observed in mouse or human nsun2 mutants (Tuorto et al., 2012; Hussain et al., 2013b, 2013c), suggesting that m5C may play alternate roles in specific mRNAs, such as affecting the translation of transcripts.
The bsRNA-seq approach provides both single base-pair resolution and quantitative measurements of methylation percentages of candidate m5C sites. We used this information to compare m5C methylation in three tissue types in Arabidopsis: seedling shoots, roots, and siliques. All three tissue types displayed a distinct methylation profile, indicating that m5C deposition is dynamically controlled and may contribute to tissue-specific functions and regulation (Figures 1A and 1B; Supplemental Figure 2 and Supplemental Data Sets 2 and 3). In our previous study, we observed m5C levels at specific cytosines in Arabidopsis tRNAs and found little or no variation in methylation levels between floral and seedling shoot tissues (Burgess et al., 2015). In comparison, greater variation in methylation levels of specific m5C sites in mRNAs between different tissue types was observed (Figure 1B; Supplemental Figure 2 and Supplemental Data Sets 2 and 3). These differences support the proposed function for m5C in tRNAs in maintaining stability across all tissues equally, while mRNAs may be differentially methylated in a tissue-specific manner to perform tissue-specific functions.
We previously showed that TRM4B methylated 39 nuclear-derived tRNA isodecoders (tRNAs with the same anticodon but different body sequences) in Arabidopsis (Burgess et al., 2015). Here, we demonstrate an expanded role for TRM4B, i.e., to mediate methylation of over 100 m5C sites in mRNAs and snoRNAs across three different plant organs. In the trm4b mutant, methylation at these m5C sites was completely lost or reduced to background levels, whereas overexpression of TRM4B resulted in increased methylation of these sites, in some cases over 124-fold, compared with wild-type levels (Figure 3E). In comparison, no TRM4A- or TRDMT1-dependent sites were identified in our data set, suggesting that the remaining TRM4B-independent m5C sites are mediated by another RNA methyltransferase, such as Arabidopsis RCMT9. Together, these data indicate that TRM4B plays a major role in mediating transcriptome-wide methylation in Arabidopsis. A similar role was demonstrated for NSUN2 in humans, which was shown to have broad substrate range, including mRNAs, tRNAs, and other noncoding RNAs such as vault ncRNAs, suggesting that it shares conserved functions and targeting mechanisms with Arabidopsis TRM4B (Squires et al., 2012; Hussain et al., 2013b; Khoddami and Cairns, 2013). Furthermore, as with NSUN2, no consensus target sequence was identified in our bsRNA-seq data set for TRM4B-dependent m5C sites.
To gain further insights into substrate specificity, we used a transgene-based approach to test TRM4B’s requirements for m5C site recognition in N. benthamiana (Figure 4). Using the internal MAG5 mRNA m5C site as a candidate sensor, we found that TRM4B requires longer sequences of 50 or 100 nucleotides flanking the m5C site for effective methylation (27.7% ± 2% to 53.2% ± 1%), while shorter flanking sequences of 25 nucleotides reduce methylation efficiency (4.6% ± 0.4%.). It remains to be studied whether properties related to sequence or structure are required to confer cytosine methylation. Future experiments involving site-directed mutagenesis of nucleotides within the sensor fragments will provide clues about the nature of TRM4B’s targeting mechanism. Intriguingly, we did not identify a MAG5 ortholog or sequences that shared significant similarity surrounding the MAG5 m5C site in the N. benthamiana genome. This raises the question of how the endogenous N. benthamiana TRM4B is able to effectively methylate m5C sites in sensor 2 and sensor 3 in our transient expression system and suggests local RNA structure may influence TRM4B specificity. The importance of RNA structure in targeting of m5C sites by RNA methyltransferases was clearly observed for TRDMT1 (Goll et al., 2006; Jurkowski et al., 2008; Shanmugam et al., 2014).
Importantly, our results demonstrate a link between the components required for m5C methylation and cell proliferation capacity in plants. We show that the trm4b mutant exhibits a short-root phenotype and displays fewer meristematic cells in the primary RAM than the wild type (Figure 5). The cells in the RAM are characterized by their high proliferative capacity, suggesting TRM4B controls the extent of cell cycle progression in the root meristem, ultimately affecting root growth. Consistent with this hypothesis, we observed reduced expression of the cell cycle marker CYCB1;1-GUS (Doerner et al., 1996) in the trm4b mutant (Figure 5G). Indeed, NSUN2 has also been shown to be regulated by the cell cycle, and its intracellular localization varies depending on cell cycle stage in human cell lines (Sakita-Suto et al., 2007). In addition, the loss of NSUN2 in mice disrupts the balance between stem cell self-renewal and differentiation by delaying cell cycle progression (Blanco et al., 2011; Hussain et al., 2013c), further mirroring TRM4B’s functions in plants. Future experiments that combine trm4b with mutants in the cell cycle pathway as well as identification of protein cofactors will help elucidate the role of TRM4B in plant cell division.
Previous studies have demonstrated that m5C methyltransferases are important regulators of oxidative stress responses in several species (Chan et al., 2010, 2012; Schaefer et al., 2010; Blanco et al., 2014; Schosserer et al., 2015). The lack of TRDMT1/DNMT2 (Schaefer et al., 2010) and TRM4-mediated (Chan et al., 2010, 2012) methylation confers hypersensitivity to oxidative stress in Drosophila and yeast, respectively. In each of these cases, the loss of tRNA m5C methylation and reduced tRNA stability have been suggested to contribute to the increased oxidative stress sensitivity. We previously reported that TRM4B is required to methylate m5C sites in a number of nuclear-derived tRNAs in Arabidopsis (Burgess et al., 2015). Here, we demonstrate trm4b mutant plants are more sensitive to paraquat and hydrogen peroxide than the wild type and show reduced stability of tRNAAsp(GTC) targeted by TRM4B (Figure 6). We propose that the loss of TRM4B-mediated tRNA methylation reduces tRNA stability, thereby reducing the pool of available tRNAs and possibly sensitizing the cell to oxidative stress. Indeed, emerging evidence indicates that tRNAs are important mediators of oxidative stress responses in several species (Thompson et al., 2008; Chan et al., 2012; Blanco et al., 2014). Notably, in NSUN2-deficient mice, the loss of tRNA methylation leads to the accumulation of 5′ tRNA-derived fragments, which is sufficient to activate cellular stress responses (Blanco et al., 2014). Whether the loss of TRM4B-specific methylation induces similar downstream responses in Arabidopsis remains to be investigated. Interestingly, however, we found that genes in the oxidative stress pathway are constitutively activated in the trm4b mutant even in the absence of stress, perhaps reflecting a similar tRNA-activated stress pathway that may be conserved in Arabidopsis (Figure 7). Given the broad substrate range demonstrated for TRM4B, it is difficult to tease apart the contributions of tRNA and mRNA methylation to the phenotypes observed in this study. Further analysis of the regulatory functions of m5C in different RNA contexts will help clarify the biological significance of these modifications in plant development and stress responses.
The dynamic regulation of m5C in Arabidopsis and its impact on gene regulation is just beginning to be elucidated. Here, we described the distribution of m5C in the plant transcriptome and identified crucial links between this modification and cell division and stress pathways. The parallel roles of TRM4B and NSUN2 in m5C deposition patterns, as well the phenotypic similarities of the corresponding mutants, provide a fascinating insight into this conserved posttranscriptional regulatory mechanism in plants and animals.
METHODS
Plant Material and Root Growth Experiments
Arabidopsis thaliana (Columbia ecotype) plants were grown in Phoenix Biosystems controlled environment rooms at 21°C under metal halide lights that provided a level of PAR of 110 μmol of photos/m2/s. For the plate experiments, Arabidopsis seeds were surface sterilized in a solution containing one part 10% sodium hypochlorite and nine parts 100% ethanol and plated on 0.5× Murashige and Skoog (MS) medium supplemented with 1% sucrose. The plates were sealed with porous tape (3M) and placed in the dark for 3 d at 4°C to allow the seeds to imbibe before being transferred to the growth chamber. All plants were grown under long-day photoperiod conditions of 16 h light and 8 h darkness. To monitor root growth, the seedlings were grown on plates oriented vertically, with primary root tips marked daily until 8 d postgermination. For the oxidative stress experiments, 4-d-old seedlings growing vertically on 0.5× MS medium supplemented with 1% sucrose were transferred to control medium or medium supplemented with the specified concentrations of paraquat, hydrogen peroxide, or NaCl. Primary root growth was recorded for 3 d after transfer. The seedlings were scanned (Epson flat-bed scanner) at 600 pixels inch−1 and primary root length measured in millimeters using ImageJ software.
Characterization of the mutant alleles trdmt1 (SALK_136635), trm4a (SALK_121111), trm4b-1 (SAIL_318_G04), and trm4b-2 (SAIL_667_D03) and the derived double mutant trdmt1 trm4b are as described previously (Alonso et al., 2003; Goll et al., 2006; Burgess et al., 2015).
Plasmid Construction and Generation of Transgenic Plants
For the TRM4B overexpression construct, the full-length genomic region of TRM4B including the 5′UTR and 3′UTR was amplified using Col-0 genomic DNA template with primers provided in Supplemental Data Set 10 and cloned into Gateway entry vector PCR8 TOPO-TA (Invitrogen). The insert was sequenced and then cloned into the destination vector pMDC32, using the Gateway cloning system (Curtis and Grossniklaus, 2003) following the manufacturer’s protocol (Invitrogen), resulting in the Pro35S:TRM4B construct. The construct was transformed into Arabidopsis wild type Col-0 plants by the Agrobacterium tumefaciens-mediated floral dip method (Davis et al., 2009). Transgenic plants were selected on 0.5× MS medium supplemented with 15 μg mL−1 Hygromycin B. TRM4B transcript abundance was assessed in at least five independent T1 plants using qRT-PCR, and two lines showing the highest TRM4B transcript levels were carried through to the homozygous T3 generation for phenotypic analysis. cDNA synthesis for qRT-PCR was performed using an Invitrogen SuperScript III kit as per the manufacturer’s recommendations from 2 μg of total RNA and oligo(dT) primed cDNA synthesis. qRT-PCR detection of TRM4B and the housekeeping gene PDF2A mRNA was performed using a LightCycler480 (Roche) and SYBR green (Roche). Primers are provided in Supplemental Data Set 10.
For the m5C sensor constructs, synthesized oligos corresponding to the three sensor fragments flanking MAG5 C3349 were annealed and cloned into pGreen (Hellens et al., 2000) using the restriction enzyme sites ApaI and SpeI. The ApaI site was inserted into the pGreen multiple cloning site using an oligo adapter. Synthesized oligos are provided in Supplemental Data Set 10. The constructs were transiently expressed in Nicotiana benthamiana using Agrobacterium infiltration into leaves as described previously (Sparkes et al., 2006).
For Sanger sequencing of tRNAAsp(GTC) clones, tRNAAsp(GTC) was PCR amplified from cDNA from bisulfite-treated RNA and cloned into the pGEM-T Easy Vector system (Promega), and individual clones were sequenced to determine bisulfite conversion efficiency.
Propidium Iodide Staining, GUS Staining, and Imaging
Eight-day-old Arabidopsis seedlings were transferred to 10 μM propidium iodide solution (Sigma-Aldrich), incubated for 2 to 3 min, rinsed, and mounted in MilliQ water. Longitudinal optical sections of the different tissues were visualized using Leica confocal microscopy (Leica SP5 spectral scanning confocal microscope) with the following settings: excitation wavelength, 488 nm; emission, 550 to 800 nm; beam splitter, 488/543/633-nm triple dichroic; objective, HC PL APO CS 20.0x0.70 IMM/COR. For GUS staining, 6-d-old Arabidopsis seedlings were stained for 16 h at 37°C with staining solution (50 mM NaPO4, pH 7, 2 mM potassium-ferrocyanide, 2 mM potassium-ferricyanide, 0.2% Triton X-100, and 2 mM X-Gluc). Roots were then cleared with serial ethanol and methanol washes and rehydrated in water. Roots were visualized using bright-field microscopy. Images were processed using ImageJ.
RNA Gel Blot Analyses
Total RNA was extracted from 10-d-old seedlings, and 20 to 25 μg per sample was loaded onto each lane of a 12.5% polyacrylamide-urea gel, blotted onto a Hybond-N+ membrane, detected using [γ-32P]ATP-labeled oligonucleotide probes, and scanned using a phosphor imager (Typhoon 3410). Blot signal intensity was quantified using ImageJ. tRNA and loading control miR156 probe sequences are provided in Supplemental Data Set 10.
RNA Isolation and Bisulfite Conversion of RNA for bsRNA-Seq
Total RNA was isolated from wild-type and trm4b-1 10-d-old Arabidopsis seedling shoots, 6-d-old seedling roots, 1-d postfertilization siliques, and trdmt1 10-d-old seedling shoots using the Spectrum Plant total RNA kit (Sigma-Aldrich), and contaminating DNA was removed using DNase I (Sigma-Aldrich). To enrich for the mRNA fraction, 5 μg of purified RNA was subjected to rRNA depletion using the Ribo-Zero kit (Illumina MRZPL116), and successful depletion was monitored using a Bioanalyzer 2001 (Agilent Technologies). For bisulfite conversion, 150 to 200 ng of ribosomal-depleted RNA was converted with sodium metabisulfite (Sigma-Aldrich) as previously described (Schaefer et al., 2009; Squires et al., 2012; Burgess et al., 2015). As a control, 200 pg of in vitro-transcribed Renilla luciferase (Sigma-Aldrich) was added to each RNA sample prior to conversion with sodium metabisulfite. Bisulfite-converted RNA was used as a template for strand-specific RNA-seq library preparation using the NEB Ultra directional RNA library kit. As bisulfite-treated RNA is sheared, the fragmentation step of the library preparation was omitted and samples were quickly processed for first-strand cDNA synthesis after the addition of the fragmentation buffer. The remaining steps of library construction were performed as per the manufacturer’s instructions. Bisulfite-treated RNA library samples were sequenced on the Illumina HiSeq 2500 (2 × 100 nucleotide paired end) platform at ACRF, Adelaide. For each tissue type, a minimum of two biological replicate library samples were prepared, and detailed biological replicate and sequencing information is shown in Supplemental Data Set 1.
RNA Isolation and Library Construction for RNA-Seq
Total RNA was isolated from wild-type and trm4b-1 6-d-old Arabidopsis seedling roots grown on 0.5× MS medium containing no paraquat, or supplemented with 0.01 µM paraquat, using the Spectrum Plant total RNA kit (Sigma-Aldrich). DNA was removed using DNase I (Sigma-Aldrich). rRNA was removed using the Ribo-Zero kit (Illumina MRZPL116), and successful depletion was monitored using a Bioanalyzer 2001 (Agilent Technologies). The resulting mRNA-enriched RNA was used as a template for strand-specific RNA-seq library preparation using the NEB Ultra directional RNA library kit. RNA-seq library construction was performed as per the manufacturer’s instructions. RNA-seq library samples were sequenced on the Illumina HiSeq 2000 (2 × 100 nucleotide paired end) platform at ACRF. Each biological replicate was derived from one plate of pooled seedlings. Three biological replicates were prepared and sequenced for each condition. Detailed sequencing information is shown in Supplemental Data Set 1.
Bioinformatic Analysis of bsRNA-Seq and RNA-Seq
Global Mapping
Reads were first adapter trimmed using Trimmomatic in palindromic mode, allowing single-base precision, with stringent 3′ quality filtering (if any four-base window in the read had a Phred quality score below 20, then the remaining 3′ region of the read was trimmed) (Bolger et al., 2014). Reads were then globally mapped to in silico bisulfite-converted Arabidopsis reference genomes (TAIR10) (Lamesch et al., 2012) based on the method and implementation of B-Solana (Kreck et al., 2012), but adapted to RNA (“B-Solana RNA” in the sequel). In particular, TopHat (Trapnell et al., 2009) was used rather than Bowtie (Langmead et al., 2009) to first map junction reads to a refseq transcriptome (to reliably detect known transcript isoforms) and then to the reference genome. To ensure maximal specificity of predicted sites, only uniquely mapped reads were retained (“uniquely mapped” was defined by a Bowtie2 mapping quality [mapq] score of ≥ 20).
Differential and overall methylation calls were based on a statistical model and method (B. Parker, T. Sibbritt, and T. Preiss, unpublished data)
Proportion Statistic
The primary statistic used for detection of methylation was a proportion statistic P = (C+Ψ)/(T+C), where Ψ is added pseudo counts (1/8 counts), which was transformed to an approximate Gaussian distribution by an arcsine of square root transformation (Snedecor and Cochran, 1980), followed by a log transformation, as empirically the distribution is weighted toward lower methylation levels, with transformed proportion p′ defined by the following equation:
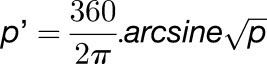 |
Transformed silique data were additionally normalized between samples by applying cyclic loess normalization (Law et al., 2014)
Nonspecific Filtering
Nonspecific filtering (i.e., blinded to experimental/control labels) to initially exclude low expressed sites and clearly nonmethylated C bases was performed by requiring ≥5 reads across all samples in experimental and control samples. In addition, for the differential methylation models, we required half the samples to show a significantly (P value < 0.05; binomial test) >5% methylation fraction. For overall methylation calls, we required all samples to have >0% methylation fraction.
Statistical Model
A weighted moderated linear model based on transformed proportion data, p', was used for differential methylation estimation using Limma (Smyth, 2005). The moderation utilizes global variance estimates across methylation sites to improve the variance estimates of the individual methylation sites.
The weights corrected for mean-variance trend were estimated by quadratic fit of variance of the transformed data versus total read counts over each site using the Voom method (Law et al., 2014). Contrasts appropriate to the particular analyses discussed in Results were used. For differential methylation site calling, balanced contrasts between methyltransferase mutant and wild-type controls were used. For overall methylation calls, wild-type samples were compared against a fixed value of 0% methylation.
RNA-Seq Data
Root RNA-seq data were adaptor-trimmed using Trimmomatic (Bolger et al., 2014) and mapped to the Arabidopsis TAIR10 reference genome (Lamesch et al., 2012) using TopHat (Trapnell et al., 2009). Data were normalized between samples using TMM normalization (Robinson and Oshlack, 2010). Differentially expressed genes were detected using Limma (Smyth, 2005), with interaction terms included for the root factorial design. GO analysis was performed using the GOrilla package (Eden et al., 2009) and REVIGO (Supek et al., 2011) to create nonredundant GO term sets. The background set of genes used for GO term analysis was all Arabidopsis genes listed in TAIR10.
FDR was estimated by the Storey method (Storey, 2002).
Post-Filtering
Called methylation sites were required to be homogeneous (C + T ≥ 95% of reads) to exclude calls in bases of high read error rate or unannotated SNPs. A minimum mean methylation fraction of 1% across wild-type samples was required to exclude very low methylation calls that may be enriched for nonconversion artifacts due to RNA structure or other systematic biases.
Distribution Analyses
Distribution of candidate m5C sites across the 5′UTR, CDS, and 3′UTR was performed by determining the total number of candidate m5C sites detected in each region relative to the proportion of total potential m5C sites (for cytosines ≥5 reads) within each region. Statistical significance was assessed by binomial test.
LOGO Motif Analysis
Sequence LOGOs were generated over flanking regions centered around the genomic coordinates of candidate m5C sites dependent on TRM4B using the seqLogo R package.
Sequencing and Analysis of bsRNA-Amp-Seq
Total RNA was isolated from wild-type and trm4b-1 10-d-old Arabidopsis seedling shoots, 6-d-old seedling roots, 1-d postfertilization siliques, and trm4a 10-d-old seedling shoots using the Spectrum Plant total RNA kit (Sigma-Aldrich), and contaminating DNA was removed using DNase I (Sigma-Aldrich). For bisulfite conversion, 2 to 5 µg of total RNA was converted with sodium metabisulfite (Sigma-Aldrich) as described above. cDNA synthesis for bsRNA-amp-seq was performed using an Invitrogen SuperScript III kit as per the manufacturer’s instructions using 1 to 2 μg of bisulfite-treated total RNA and bulked gene-specific reverse transcription primers (Supplemental Data Set 10). Amplicons were PCR amplified manually or on an Access Array (Fluidigm) using the primers listed (Supplemental Data Set 10) as per the manufacturer’s instructions. PCR amplicons derived from the same biological replicate were bulked and indexed as recommended by the manufacturer. Illumina sequencing was performed on a MiSeq platform (2 × 160 nucleotide paired end) at ACRF. Three biological replicates were prepared and sequenced for each condition.
Analysis of bsRNA-amp-seq libraries was performed using CLC Genomics Workbench (Qiagen). Briefly, sequences were trimmed for adapters and filtered for low-quality reads. BSRNA-amp-seq reads were then mapped to in silico-converted reference sequences corresponding to the expected PCR amplicons. To identify methylated cytosines, nonconversion of a cytosine in aligned read sequences was taken to indicate the presence of m5C. Percentage of methylation at specific positions was calculated as the number of mapped cytosines divided by the combined total number of mapped cytosines and mapped thymines.
RNA Immunoprecipitation
Total RNA was immunoprecipitated with 12 μg of an undiluted IgG nonspecific control antibody (Cell Signaling) or an anti-5-methylcytosine (m5C) antibody (Epigentek) at a 1:2500 dilution. Fifty microliters of Dynabeads Protein A (Thermo Fisher Scientific) was washed with 1× Dulbecco’s PBS (DPBS; Thermo Fisher Scientific) and coupled to the 12 μg of antibody in DPBS by rocking at room temperature for 1 h. The beads were washed again twice with DPBS solution. Five micrograms of RNA was denatured at 70°C for 5 min, placed on ice for 3 min, and incubated with the bead-linked antibodies in IP buffer (140 mM NaCl, 0.05% [v/v] Triton X-100, and 10 mM Tris, all from Ultrapure, RNase-free stocks dissolved in DEPC-treated water and filter sterilized at 0.2 μM). Bead/RNA mix was rocked at 4°C for 4 h. Bound RNA was washed three times in IP buffer and then eluted in Trizol (Thermo Fisher Scientific), precipitated, and washed.
Accession Numbers
Nucleotide sequence data for the following genes can be found in the TAIR database under the following accession numbers: TRDMT1 (At5g25480), TRM4A (At4g40000), TRM4B (At2g22400), and MAIGO5 (MAG5; At5g47480). Sequence data from this article can be found in the GenBank/EMBL libraries under accession number GSE80054.
Supplemental Data
Supplemental Figure 1. Efficient bisulfite conversion of nonmethylated cytosine residues.
Supplemental Figure 2. Differences in global methylation levels of m5C sites in Arabidopsis siliques, shoots, and roots.
Supplemental Figure 3. Methylation of selected m5C sites analyzed using bsRNA-amp-seq is not perturbed in trm4a mutants.
Supplemental Figure 4. qRT-PCR expression of TRM4B-OX line and bisulfite conversion efficiency of bsRNA-amp-seq.
Supplemental Figure 5. LOGO motif analysis of TRM4B-dependent m5C sites.
Supplemental Figure 6. Vegetative and flowering time traits of the wild type and trm4b mutants.
Supplemental Figure 7. Complementation of the short-root phenotype in trm4b.
Supplemental Figure 8. Primary root length of trdmt1 mutants is similar to that of wild-type plants.
Supplemental Figure 9. Mature cortical cell length of wild-type and trm4b-1 mutant roots.
Supplemental Figure 10. RNA gel blot analysis of tRNAHis(GTG).
Supplemental Figure 11. Oxidative stress-responsive biological process GO terms are constitutively activated in trm4b mutants.
Supplemental Data Set 1. Read coverage of all libraries sequenced
Supplemental Data Set 2. Overall m5C site calls.
Supplemental Data Set 3. trm4b-dependent m5C sites.
Supplemental Data Set 4. Wild-type heat map raw values for Figure 1G.
Supplemental Data Set 5. trdmt1-dependent m5C sites.
Supplemental Data Set 6. trm4a heat map raw values for Supplemental Figure 3A.
Supplemental Data Set 7. trm4b heat map raw values for Figure 3D.
Supplemental Data Set 8. TRM4B-OX raw values used to generate the heat map in Figure 3E.
Supplemental Data Set 9. RNA-seq analysis of differentially expressed genes in wild-type and trm4b mutant roots under controlled and paraquat-stressed conditions.
Supplemental Data Set 10. Primer sequences used in the study.
Supplementary Material
Acknowledgments
We thank Wendy Parker and Joel Geoghegan for their expertise for the Fluidigm Access Array experiments and Ashley Jones for technical expertise for the initial bsRNA-seq library construction. This research was supported by ARC grants DP110103805 and FT130100525 awarded to I.R.S. and an APA and a GRDC PhD top-up scholarship awarded to A.B. T.P. acknowledges support by NHMRC Project Grant APP1061551.
AUTHOR CONTRIBUTIONS
R.D., A.B., T.S., T.P., and I.R.S. designed the experiments. R.D., A.B., and K.P. performed the experiments. B.P., R.D., A.B., K.P., and I.R.S. performed data analysis. R.D., A.B., and I.R.S. equally prepared and edited the manuscript. All authors read and approved the final manuscript.
Glossary
- lncRNA
long noncoding RNA
- ncRNA
noncoding RNAs
- RMTase
RNA methyltransferase
- bsRNA-seq
RNA bisulfite sequencing
- UTR
untranslated region
- FDR
false discovery rate
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- bsRNA-amp-seq
RNA bisulfite amplicon sequencing
- RAM
root apical meristem
- GO
Gene Ontology
- CDS
coding sequence
- MS
Murashige and Skoog
- DAG
days after germination
References
- Abbasi-Moheb L., et al. (2012). Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90: 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris P.F. (2008). Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 9: 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A., Chernyakov I., Gu W., Hiley S.L., Hughes T.R., Grayhack E.J., Phizicky E.M. (2006). Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21: 87–96. [DOI] [PubMed] [Google Scholar]
- Alonso J.M., et al. (2003). Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301: 653–657. [DOI] [PubMed] [Google Scholar]
- Amort T., Soulière M.F., Wille A., Jia X.Y., Fiegl H., Wörle H., Micura R., Lusser A. (2013). Long non-coding RNAs as targets for cytosine methylation. RNA Biol. 10: 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auxilien S., Guérineau V., Szweykowska-Kulińska Z., Golinelli-Pimpaneau B. (2012). The human tRNA m (5) C methyltransferase Misu is multisite-specific. RNA Biol. 9: 1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker M., Müller S., Nellen W., Jurkowski T.P., Jeltsch A., Ehrenhofer-Murray A.E. (2012). Pmt1, a Dnmt2 homolog in Schizosaccharomyces pombe, mediates tRNA methylation in response to nutrient signaling. Nucleic Acids Res. 40: 11648–11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beemster G.T., Baskin T.I. (1998). Analysis of cell division and elongation underlying the developmental acceleration of root growth in Arabidopsis thaliana. Plant Physiol. 116: 1515–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S., et al. (2014). Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 33: 2020–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S., Kurowski A., Nichols J., Watt F.M., Benitah S.A., Frye M. (2011). The RNA-methyltransferase Misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet. 7: e1002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A.L., David R., Searle I.R. (2015). Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae. BMC Plant Biol. 15: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.T., Dyavaiah M., DeMott M.S., Taghizadeh K., Dedon P.C., Begley T.J. (2010). A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6: e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.T.Y., Pang Y.L.J., Deng W., Babu I.R., Dyavaiah M., Begley T.J., Dedon P.C. (2012). Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3: 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Jäger G., Zheng B. (2010). Transfer RNA modifications and genes for modifying enzymes in Arabidopsis thaliana. BMC Plant Biol. 10: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Sierzputowska-Gracz H., Guenther R., Everett K., Agris P.F. (1993). 5-Methylcytidine is required for cooperative binding of Mg2+ and a conformational transition at the anticodon stem-loop of yeast phenylalanine tRNA. Biochemistry 32: 10249–10253. [DOI] [PubMed] [Google Scholar]
- Curtis M.D., Grossniklaus U. (2003). A Gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 133: 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A.M., Hall A., Millar A.J., Darrah C., Davis S.J. (2009). Protocol: Streamlined sub-protocols for floral-dip transformation and selection of transformants in Arabidopsis thaliana. Plant Methods 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerner P., Jørgensen J.E., You R., Steppuhn J., Lamb C. (1996). Control of root growth and development by cyclin expression. Nature 380: 520–523. [DOI] [PubMed] [Google Scholar]
- Edelheit S., Schwartz S., Mumbach M.R., Wurtzel O., Sorek R. (2013). Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 9: e1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahiminiya S., et al. (2014). Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin. Genet. 86: 134–141. [DOI] [PubMed] [Google Scholar]
- Gigova A., Duggimpudi S., Pollex T., Schaefer M., Koš M. (2014). A cluster of methylations in the domain IV of 25S rRNA is required for ribosome stability. RNA 20: 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll M.G., Kirpekar F., Maggert K.A., Yoder J.A., Hsieh C.L., Zhang X., Golic K.G., Jacobsen S.E., Bestor T.H. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–398. [DOI] [PubMed] [Google Scholar]
- Hellens R.P., Edwards E.A., Leyland N.R., Bean S., Mullineaux P.M. (2000). pGreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol. Biol. 42: 819–832. [DOI] [PubMed] [Google Scholar]
- Helm M. (2006). Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 34: 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B., Brockenbrough J.S., Wu P., Aris J.P. (1997). Nop2p is required for pre-rRNA processing and 60S ribosome subunit synthesis in yeast. Mol. Cell. Biol. 17: 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Aleksic J., Blanco S., Dietmann S., Frye M. (2013a). Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol. 14: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Sajini A.A., Blanco S., Dietmann S., Lombard P., Sugimoto Y., Paramor M., Gleeson J.G., Odom D.T., Ule J., Frye M. (2013b). NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Reports 4: 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S., Tuorto F., Menon S., Blanco S., Cox C., Flores J.V., Watt S., Kudo N.R., Lyko F., Frye M. (2013c). The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol. Cell. Biol. 33: 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman J.E., Alfonzo J.D. (2013). Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 4: 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowski T.P., Meusburger M., Phalke S., Helm M., Nellen W., Reuter G., Jeltsch A. (2008). Human DNMT2 methylates tRNA(Asp) molecules using a DNA methyltransferase-like catalytic mechanism. RNA 14: 1663–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.A., et al. (2012). Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90: 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoddami V., Cairns B.R. (2013). Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31: 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J., Turner S.W. (2012). Going beyond five bases in DNA sequencing. Curr. Opin. Struct. Biol. 22: 251–261. [DOI] [PubMed] [Google Scholar]
- Kreck B., Marnellos G., Richter J., Krueger F., Siebert R., Franke A. (2012). B-SOLANA: an approach for the analysis of two-base encoding bisulfite sequencing data. Bioinformatics 28: 428–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamesch P., et al. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40: D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka M.A., et al. (2013). MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F.J., Lee J.H., Lee J.E., Blanco S., Nickerson E., Gabriel S., Frye M., Al-Gazali L., Gleeson J.G. (2012). Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 49: 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y., Grosjean H. (1999). Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: identification of the gene and substrate specificity of the enzyme. RNA 5: 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y., Helm M. (2010). tRNA stabilization by modified nucleotides. Biochemistry 49: 4934–4944. [DOI] [PubMed] [Google Scholar]
- Motorin Y., Lyko F., Helm M. (2010). 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 38: 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulou A., Kossida S. (2009). Phylogenetic analysis of the eukaryotic RNA (cytosine-5)-methyltransferases. Genomics 93: 350–357. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., Oshlack A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakita-Suto S., Kanda A., Suzuki F., Sato S., Takata T., Tatsuka M. (2007). Aurora-B regulates RNA methyltransferase NSUN2. Mol. Biol. Cell 18: 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M., Pollex T., Hanna K., Lyko F. (2009). RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 37: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M., Pollex T., Hanna K., Tuorto F., Meusburger M., Helm M., Lyko F. (2010). RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 24: 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schosserer M., et al. (2015). Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 6: 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam R., et al. (2014). The Dnmt2 RNA methyltransferase homolog of Geobacter sulfurreducens specifically methylates tRNA-Glu. Nucleic Acids Res. 42: 6487–6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Yang J., Watzinger P., Kötter P., Entian K.D. (2013). Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 41: 9062–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. (2005). Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W., eds (New York: Springer; ), pp. 397–420. [Google Scholar]
- Snedecor G.W., Cochran W.G. (1980). Statistical Methods. (Ames, IA: Iowa State University Press; ). [Google Scholar]
- Sparkes I.A., Runions J., Kearns A., Hawes C. (2006). Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1: 2019–2025. [DOI] [PubMed] [Google Scholar]
- Squires J.E., Patel H.R., Nousch M., Sibbritt T., Humphreys D.T., Parker B.J., Suter C.M., Preiss T. (2012). Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40: 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl Y., Simon R. (2010). Plant primary meristems: shared functions and regulatory mechanisms. Curr. Opin. Plant Biol. 13: 53–58. [DOI] [PubMed] [Google Scholar]
- Storey J.D. (2002). A direct approach to false discovery rates. J. R. Stat. Soc. Series B Stat. Methodol. 64: 479–498. [Google Scholar]
- Supek F., Bošnjak M., Škunca N., Šmuc T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M.M., Bird A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9: 465–476. [DOI] [PubMed] [Google Scholar]
- Thompson D.M., Lu C., Green P.J., Parker R. (2008). tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 14: 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuorto F., et al. (2015). The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 34: 2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuorto F., Liebers R., Musch T., Schaefer M., Hofmann S., Kellner S., Frye M., Helm M., Stoecklin G., Lyko F. (2012). RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 19: 900–905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.