

Abstract
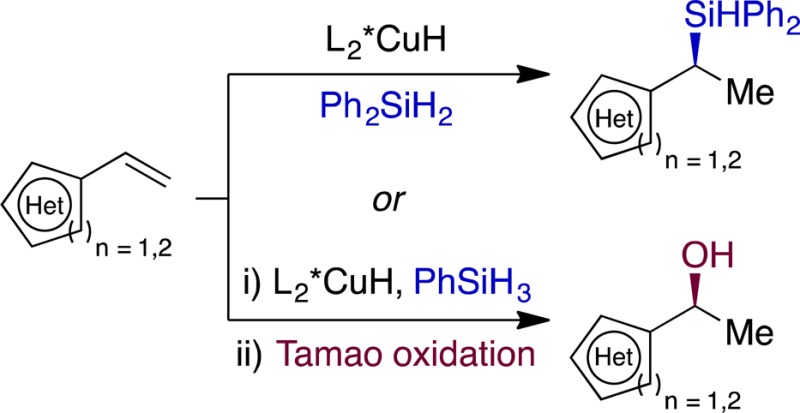
We report a highly enantioselective CuH-catalyzed Markovnikov hydrosilylation of vinylarenes and vinyl heterocycles. This method has a broad scope and enables both the synthesis of isolable silanes and the conversion of crude products to chiral alcohols. Density functional theory calculations support a mechanism proceeding by hydrocupration followed by σ-bond metathesis with a hydrosilane.
Chiral silanes undergo a number of useful transformations1a,1b and possess more desirable toxicological and environmental characteristics than many related main-group reagents.2a,2b However, they can be difficult to prepare due to the scarcity of broadly applicable methods for the construction of C(sp3)–Si bonds in functionalized molecules.3a,3b This limitation also discourages the pursuit of silane drug-candidates, despite the promise organosilicon compounds hold for a variety of therapeutic applications.3a−3c Because functionalized alkylsilanes are additionally important to many modern materials and industrial processes,4a,4b selective new C–Si bond-forming reactions have the potential to be enabling technologies for researchers across a wide range of disciplines.
Late transition metal-catalyzed olefin hydrosilylation (Figure 1a) is one of the core synthetic transformations of organosilicon chemistry.4b,4c The archetypal Pt-catalyzed variant4b is among the highest-volume industrial applications of homogeneous catalysis,4b,5c,5i and the need to replace Pt in this role with more abundant metals has spurred the discovery of many anti-Markovnikov hydrosilylation catalysts based on iron,5a−5f cobalt,5g−5j and nickel.5k−5q In contrast, Markovnikov olefin hydrosilylation catalysts are uncommon, and the synthetic capabilities of the reactions are presently limited.5e,6 Hayashi’s discovery that Pd-MOP complexes catalyze highly enantioselective Markovnikov hydrosilylation with trichlorosilane7a−7e was a milestone achievement in asymmetric catalysis.8 Although this reaction is of exceptional fundamental importance, its reported scope is limited; in particular, we are aware of no applications of this methodology to the hydrosilylation of heterocyclic or appreciably Lewis-basic olefins, presumably because they are incompatible with the sensitive trichlorosilyl group.
Figure 1.
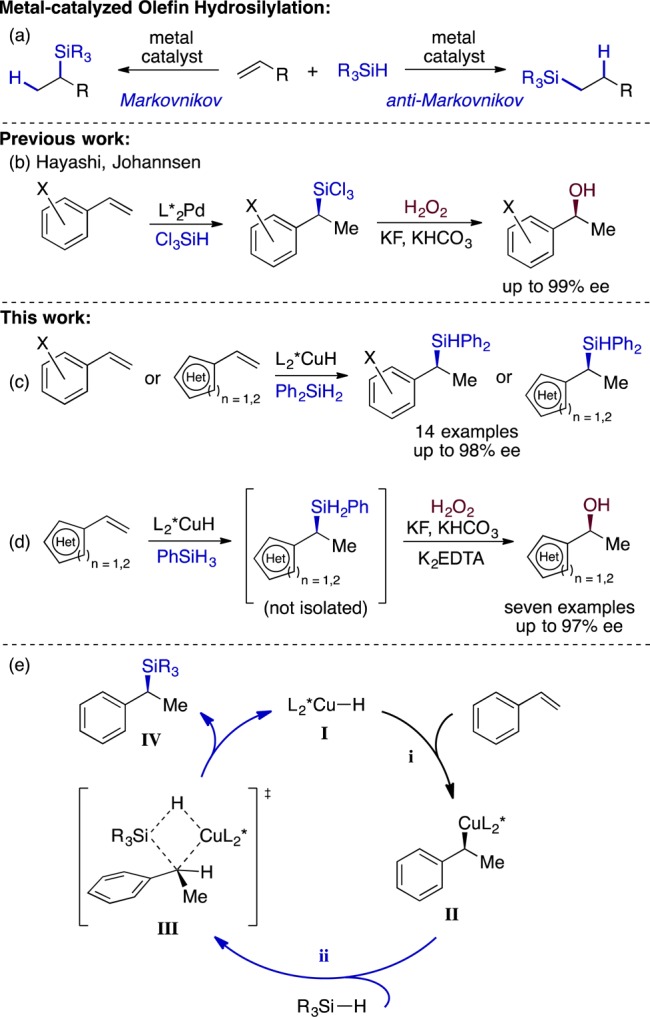
aMetal-catalyzed hydrosilylation of olefins; bPd-catalyzed asymmetric Markovnikov hydrosilylation of vinylarenes; c,dCuH-catalyzed asymmetric hydrosilylation of styrenes; eProposed mechanism for the hydrosilylation.
Our group has recently developed several functional group-tolerant Markovnikov hydrofunctionalization reactions that proceed through electrophilic interception of alkylcopper intermediates generated by asymmetric hydrocupration of vinylarenes9a−9e with an active chiral copper hydride catalyst [I, Figure 1e, step (i)]. We reasoned that an asymmetric Markovnikov hydrosilylation could be achieved if the alkylcopper species II (Figure 1e) could undergo stereoretentive transmetalation with a hydrosilane [Figure 1e, step (ii)].10,11a,11b The hypothesized transformation would constitute an attractive alternative to Pd catalysis if it could provide isolable silanes and silane-derivatives with high generality using readily available catalyst precursors. However, to our knowledge, copper hydride-catalyzed olefin hydrosilylation has not been described in the primary literature,12 despite pervasive use of hydrosilane reductants in copper catalysis.9a,13a−13c This observation suggested that the proposed metathesis step could be challenging and that competitive side reactions might present challenges.
With styrene as our model substrate, we observed clean formation of the desired Markovnikov hydrosilylation product using either PhSiH3 or Ph2SiH2, but the enantioselectivity obtained with Ph2SiH2 was modest (Table 1, entry 1). The selectivity with Ph2SiH2 varied with change in the solvent, although without an obvious correlation with solvent properties. Reducing the concentration of silane was deleterious for enantioselectivity, whereas conducting the reaction neat resulted in excellent levels of asymmetric induction (Table 1, entries 4 and 7). These observations suggest that rapid trapping of the alkylcopper intermediate by the silane may be a key factor for obtaining high levels of enantioselectivity, particularly because the hydrocupration step is known to be highly enantioselective9a−9e in a variety of solvents. This notion was further supported by the observation that the more reactive PhSiH3 underwent hydrosilylation in 96% ee even in THF solution (Table 1, entry 8).
Table 1. Optimization of the CuH-Catalyzed Hydrosilylation of Styrene.
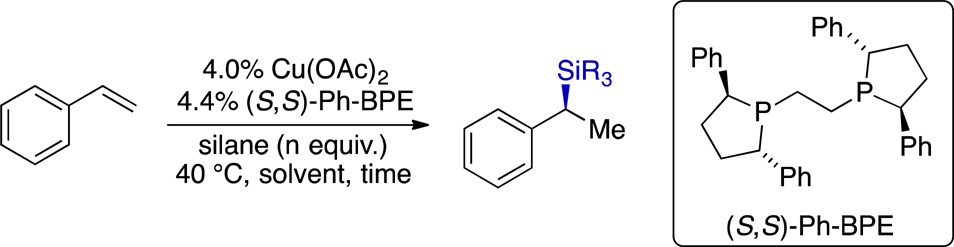
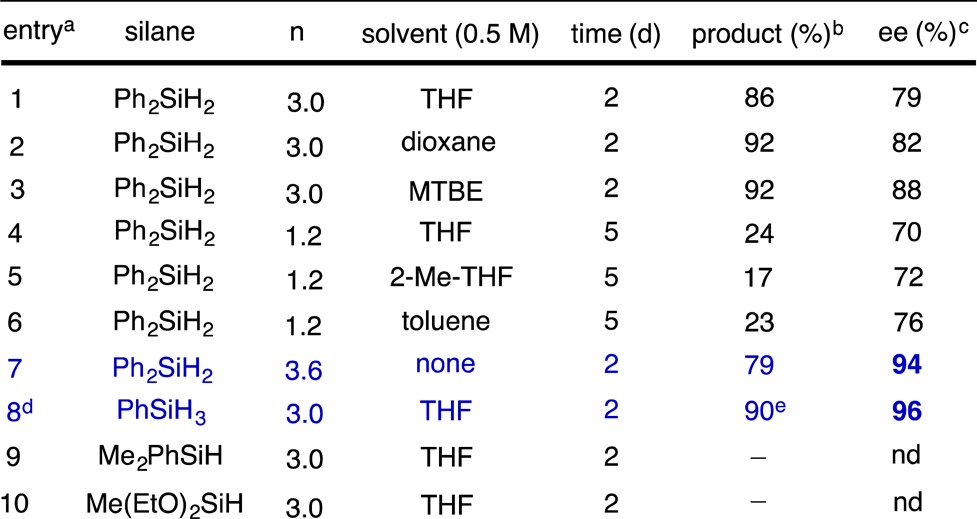
Unless otherwise noted, reactions were conducted on 0.5 mmol scale.
Unless otherwise noted, yields were determined by GC with dodecane internal standard.
Determined by chiral HPLC.
0.2 mmol scale.
Isolated yield.
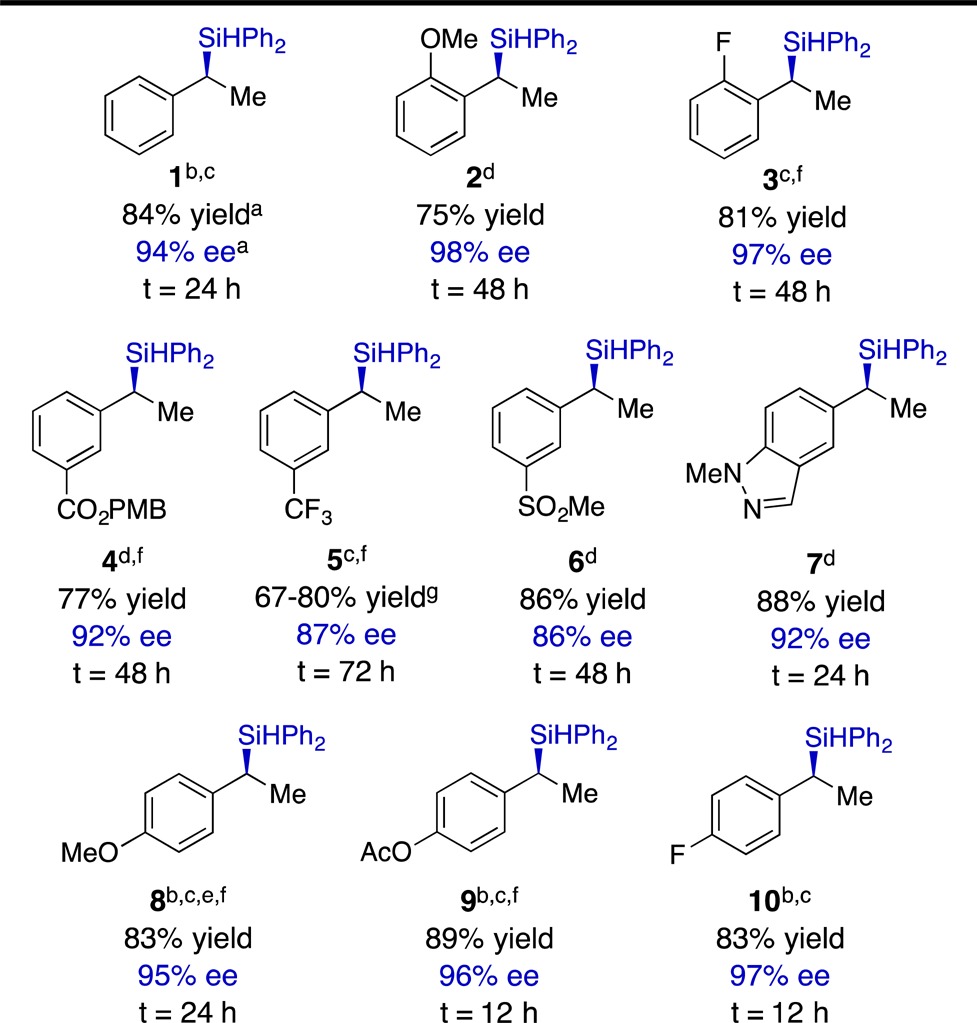
With the goal of generating easily isolable products, we chose to use Ph2SiH2 in our exploration of the vinylarene scope. The results of these studies are presented in Table 2. The hydrosilylation occurred in high yield and with good to excellent enantioselectivity with substrates containing either electron-withdrawing or electron-donating groups and accommodated substituents at any of the three positions on the aryl ring. The highest levels of enantioselectivity were obtained with π-donor substituents at either the para- or ortho-positions (e.g., examples 2 and 9, Table 2). Electron-withdrawing groups were tolerated at the meta-position, and we noted that conducting the reaction at room temperature benefited the selectivity in those cases. Certain functional groups, such as the nitro group and halogens other than fluorine, were incompatible with the reaction, as were sterically demanding ortho-substituents. Underscoring the utility of the protocol, we conducted a reaction on 10 mmol scale without significant loss of yield or enantioselectivity (eq 1).
Table 2. Hydrosilylation of Vinylarenes.
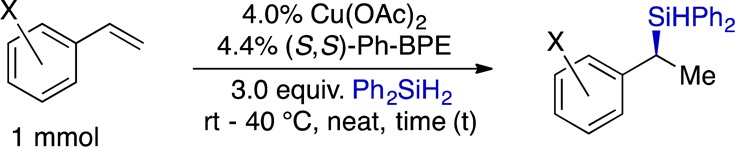
Unless otherwise noted, yields and enantiomeric excesses are the averages for two runs.
Reaction was conducted with 2.0 mol % Cu(OAc)2 and 2.2 mol % (S,S)-Ph-BPE;
Reaction mixture was stirred in a 40 °C oil bath;
Reaction was conducted at ambient temperature;
1.5 equiv. silane were used;
Enantiomeric excesses were determined for the respective silanol derivatives;14
Extrema are the results from two experiments.14
 |
1 |
Table 3 illustrates that a variety of vinyl heterocycles proved to be viable substrates for the reaction. Silanes 11 and 12 (Table 3A) were formed with high enantioselectivity, whereas a less electronically biased heterocycle gave product with modest asymmetric induction (example 13, Table 3A). These results mirrored the enantioselectivity trends evident in Table 2 and those our group has observed in the CuH-catalyzed hydroamination of vinylarenes.9e
Table 3. Hydrosilylation of Vinyl Heterocycles.
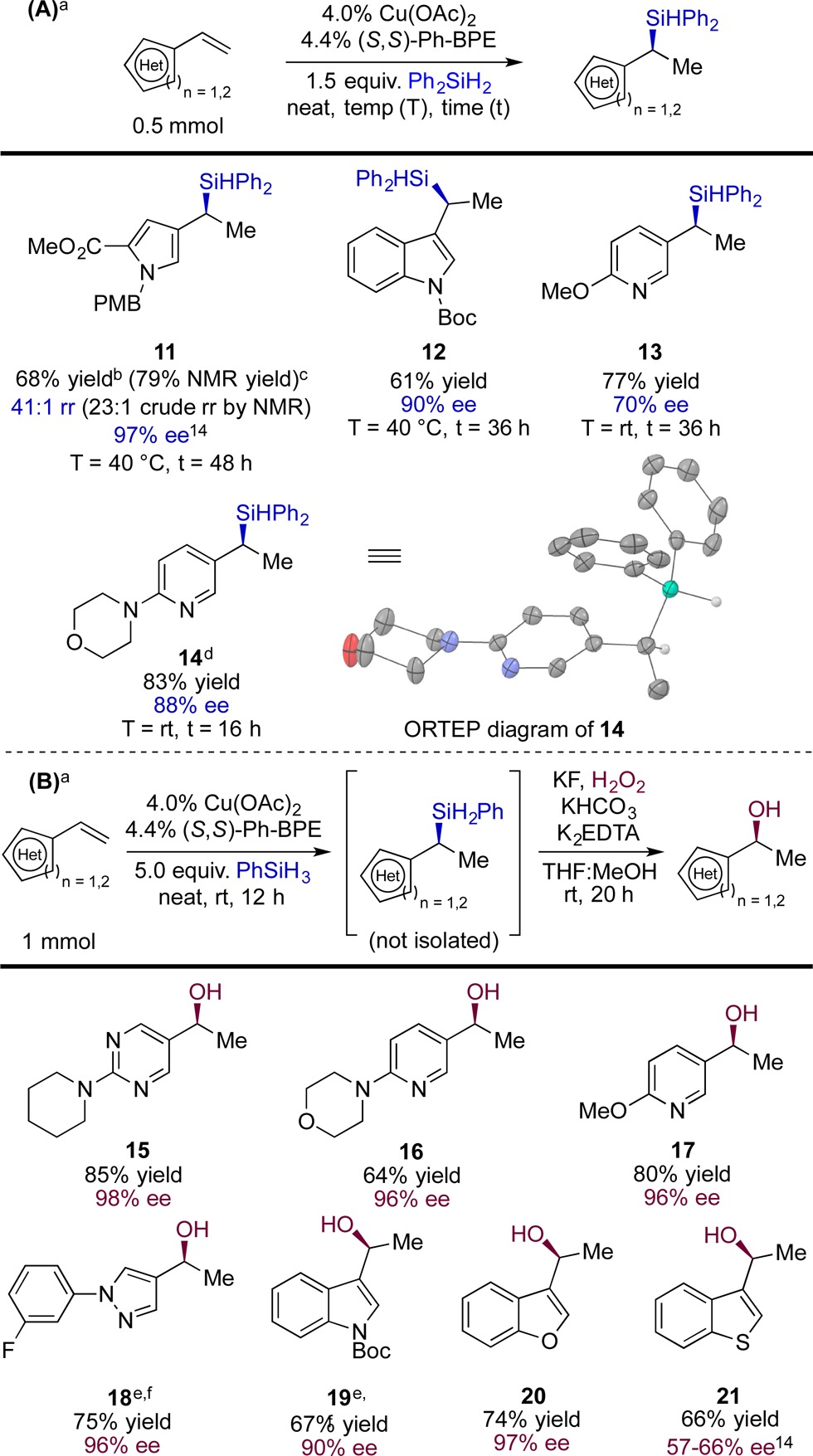
Yields and enantiomeric excess values are averages from two runs.
Isolated yield pertains to the regioisomer mixture.
NMR yield pertains to the major regioisomer.
2.0 equiv. silane used.
MTBE used as the solvent.
Reaction run for 36 h.
A crystal structure of 14 (Table 3A) showed the absolute configuration of the major enantiomer to be (S), which is consistent with the mechanism shown in Figure 1e.9c,9d However, we noted that several entries in Tables 1 and 2 exhibited lower ee’s than were obtained in previously reported reactions thought to proceed by the same hydrocupration step.9c,9d We considered two mechanistic hypotheses that might account for these discrepancies: one posits a racemization step occurring after hydrocupration (i.e., affecting intermediate II of Figure 1e); the second invokes gradual formation of an undesired catalytically active species that undergoes hydrocupration with poor selectivity in the presence of certain substrates. In either case, one would expect that accelerating the transmetalation [Figure 1e, step (ii)], e.g., by employing the more reactive PhSiH3, would result in higher enantioselectivity. We also considered that PhSiH3 might be ideal for derivatization attempts using crude products because it could be easily evaporated beforehand. Hydrosilylation with PhSiH3 occurred in <12 h at room temperature in most examples, and we were able to perform Tamao oxidations15 on the crude products by incorporating EDTA into the reaction mixtures (Table 3B), which suppresses copper-catalyzed disproportionation of hydrogen peroxide. The enantioinduction obtained with PhSiH3 was indeed broadly superior to that observed with Ph2SiH2 and further appeared to be less sensitive to substrate electronic bias (compare 16, 17 [Table 3B] and 13 [Table 3A]).
To sharpen our mechanistic hypothesis,16 we performed density functional theory (DFT) calculations on the CuH-catalyzed hydrosilylation of styrene with PhSiH3 using bis(dicyclohexylphosphino)ethane (DCyPE) as the model ligand (Figure 2). After hydrocupration, we located a σ-complex C upon interaction of copper with phenylsilane. From here, σ-bond metathesis may proceed irreversibly through a thermally accessible four-membered transition state TS-C (+30.9 or +35.8 kcal/mol relative to B). The net reaction is energetically favorable (A vs D).
Figure 2.
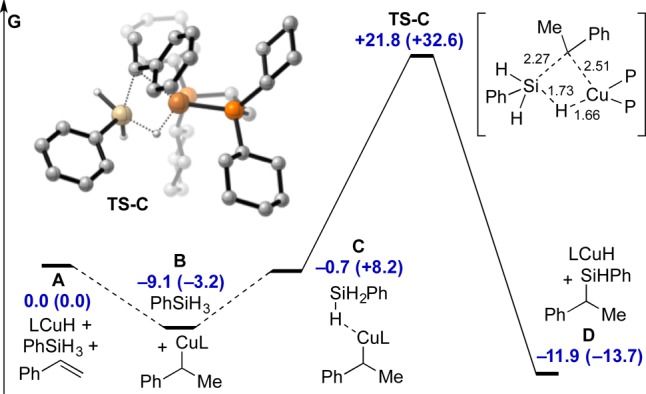
DFT model for copper-catalyzed hydrosilylation of styrene (L = DCyPE). M06/6-311+G(d,p) SDD/SMD(THF)//B3LYP/6-31G(d)-SDD Gibbs free energy values displayed in kcal/mol. Corresponding PBE0/6-311+G(d,p)-SDD/SMD(THF)//B3LYP/6-31G(d)-SDD values shown in brackets. Key bond distances shown in units of Å. Carbon-bonded hydrogen atoms are omitted for clarity.
In summary, we have developed a broadly applicable base-metal-catalyzed asymmetric hydrosilylation that provides access to bench-stable silanes and chiral alcohol derivatives. The method uses mild conditions, employs commerically available catalyst precursors, and enables the functionalization of a variety of medicinally relevant heterocyclic olefins. Although our mechanistic hypotheses remain speculative at this time, we propose that they provide a useful framework for rationalizing the observed selectivity trends. We also postulate that a thorough mechanistic investigation will be of value to our group’s ongoing efforts to develop new CuH-catalyzed transformations.
Acknowledgments
The National Institutes of Health under award number GM46059 supported research reported in this publication. M.T.P. and J.S.B. thank the National Institutes of Health for postdoctoral fellowships (1F32GM113311 [M.T.P], GM112197 [J.S.B.]). We thank Dr. Yi-Ming Wang (MIT) for advice on the preparation of this paper, Jonathan Becker (MIT) for X-ray crystallographic analysis, and the National Institutes of Health for a supplemental grant for the purchase of supercritical fluid chromatography equipment (GM058160-17S1).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b13029.
Author Contributions
† M.T.P. and J.S.B. made equal contributions to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Chan T. H.; Wang D. Chem. Rev. 1992, 92, 995–1006. 10.1021/cr00013a012. [DOI] [Google Scholar]; b Fleming I.; Barbero A.; Walter D. Chem. Rev. 1997, 97, 2063–2192. 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]
- a Denmark S. E.; Ambrosi A. Org. Process Res. Dev. 2015, 19, 982–994. 10.1021/acs.oprd.5b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nakao Y.; Hiyama T. Chem. Soc. Rev. 2011, 40, 4893–4901. 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]
- a Min G. K.; Hernández D.; Skrydstrup T. Acc. Chem. Res. 2013, 46, 457–470. 10.1021/ar300200h. [DOI] [PubMed] [Google Scholar]; b Bo Y.; Singh S.; Duong H. Q.; Cao C.; Sieburth S. M. Org. Lett. 2011, 13, 1787–1789. 10.1021/ol2002978. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Franz A. K.; Wilson S. O. J. Med. Chem. 2013, 56, 388–405. 10.1021/jm3010114. [DOI] [PubMed] [Google Scholar]
- a See volumes III–VI of Organosilicon Chemistry: From Molecules to Materials; Auner N., Weis J., Eds.; Wiley-VCH: Weinheim, Germany, 1996 (III), 2000 (IV), 2004 (V), 2005 (VI). [Google Scholar]; b Troegel D.; Stohrer J. Coord. Chem. Rev. 2011, 255, 1440–1459. 10.1016/j.ccr.2010.12.025. [DOI] [Google Scholar]; c Nakajima Y.; Shimada S. RSC Adv. 2015, 5, 20603–20616. 10.1039/C4RA17281G. [DOI] [Google Scholar]
- a Nesmeyanov A. N.; Freidlina R. K.; Chukovskaya E. C.; Petrova R. G.; Belyavsky A. B. Tetrahedron 1962, 17, 61–68. 10.1016/S0040-4020(01)99004-0. [DOI] [Google Scholar]; b Bart S. C.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2004, 126, 13794–13807. 10.1021/ja046753t. [DOI] [PubMed] [Google Scholar]; c Tondreau A. M.; Atienza C. C. H.; Weller K. J.; Nye S. A.; Lewis K. M.; Delis J. G. P.; Chirik P. J. Science 2012, 335, 567–570. 10.1126/science.1214451. [DOI] [PubMed] [Google Scholar]; d Chen J.; Cheng B.; Cao M.; Lu Z. Angew. Chem., Int. Ed. 2015, 54, 4661–4664. 10.1002/anie.201411884. [DOI] [PubMed] [Google Scholar]; e Du X.; Zhang Y.; Peng D.; Huang Z. Angew. Chem., Int. Ed. 2016, 55, 6671–6675. 10.1002/anie.201601197. [DOI] [PubMed] [Google Scholar]; f Noda D.; Tahara A.; Sunada Y.; Nagashima H. J. Am. Chem. Soc. 2016, 138, 2480–2483. 10.1021/jacs.5b11311. [DOI] [PubMed] [Google Scholar]; g Harrod J. F.; Chalk A. J. J. Am. Chem. Soc. 1965, 87, 1133. 10.1021/ja01083a034. [DOI] [Google Scholar]; h Mo Z.; Liu Y.; Deng L. Angew. Chem., Int. Ed. 2013, 52, 10845–10849. 10.1002/anie.201304596. [DOI] [PubMed] [Google Scholar]; i Chen C.; Hecht M. B.; Kavara A.; Brennessel W. W.; Mercado B. Q.; Weix D. J.; Holland P. L. J. Am. Chem. Soc. 2015, 137, 13244–13247. 10.1021/jacs.5b08611. [DOI] [PubMed] [Google Scholar]; j Schuster C. H.; Diao T.; Pappas I.; Chirik P. J. ACS Catal. 2016, 6, 2632–2638. 10.1021/acscatal.6b00304. [DOI] [Google Scholar]; k Chen Y.; Sui-Seng C.; Boucher S.; Zargarian D. Organometallics 2005, 24, 149–155. 10.1021/om0494420. [DOI] [Google Scholar]; l Lipschutz M. I.; Tilley T. D. Chem. Commun. 2012, 48, 7146–7148. 10.1039/c2cc32974c. [DOI] [PubMed] [Google Scholar]; m Kuznetsov A.; Gevorgyan V. Org. Lett. 2012, 14, 914–917. 10.1021/ol203428c. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Kuznetsov A.; Onishi Y.; Inamoto Y.; Gevorgyan V. Org. Lett. 2013, 15, 2498–2501. 10.1021/ol400977r. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Buslov I.; Becouse J.; Mazza S.; Montandon-Clerc M.; Hu X. Angew. Chem., Int. Ed. 2015, 54, 14523–14526. 10.1002/anie.201507829. [DOI] [PubMed] [Google Scholar]; p Pappas I.; Treacy S.; Chirik P. J. ACS Catal. 2016, 6, 4105–4109. 10.1021/acscatal.6b01134. [DOI] [Google Scholar]; q Nakajima Y.; Sato K.; Shimada S. Chem. Rec. 2016, 16, 2379–2387. 10.1002/tcr.201600056. [DOI] [PubMed] [Google Scholar]
- During the preparation of our paper, the Lu group described an alternative approach to the synthesis of chiral benzylic silanes by Co-catalyzed alkyne hydrosilylation followed by in situ enantioselective hydrogenation. See:Guo J.; Shen X.; Lu Z. Angew. Chem., Int. Ed. 2017, 56, 615–618. 10.1002/anie.201610121. [DOI] [PubMed] [Google Scholar]
- a Han J. W.; Hayashi T. Tetrahedron: Asymmetry 2014, 25, 479–484. 10.1016/j.tetasy.2014.03.002. [DOI] [Google Scholar]; b Uozumi Y.; Hayashi T. J. Am. Chem. Soc. 1991, 113, 9887–9888. 10.1021/ja00026a044. [DOI] [Google Scholar]; c Kitayama K.; Uozumi Y.; Hayashi T. J. Chem. Soc., Chem. Commun. 1995, 1533–1534. 10.1039/c39950001533. [DOI] [Google Scholar]; d Hayashi T.; Hirate S.; Kitayama K.; Tsuji H.; Torii A.; Uozumi Y. J. Org. Chem. 2001, 66, 1441–1449. 10.1021/jo001614p. [DOI] [PubMed] [Google Scholar]; Also see:; e Jensen J. F.; Svendsen B. Y.; la Cour T. V.; Pedersen H. L.; Johannsen M. J. Am. Chem. Soc. 2002, 124, 4558–4559. 10.1021/ja025617q. [DOI] [PubMed] [Google Scholar]
- The Nishiyama group has described a rhodium-catalyzed asymmetric synthesis of bench-stable silanes, but the regioselectivity of this reaction is modest in many cases:Naito T.; Yoneda T.; Ito J.-I.; Nishiyama H. Synlett 2012, 23, 2957–2960. 10.1055/s-0032-1317677. [DOI] [Google Scholar]
- See, for example:; a Pirnot M. T.; Wang Y.-M.; Buchwald S. L. Angew. Chem., Int. Ed. 2016, 55, 48–57. 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bandar J. S.; Ascic E.; Buchwald S. L. J. Am. Chem. Soc. 2016, 138, 5821–5824. 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang Y.-M.; Buchwald S. L. J. Am. Chem. Soc. 2016, 138, 5024–5027. 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang Y.; Perry I. B.; Buchwald S. L. J. Am. Chem. Soc. 2016, 138, 9787–9790. 10.1021/jacs.6b06299. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bandar J. S.; Pirnot M. T.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 14812–14818. 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks has proposed this mechanism for a lanthanide-catalyzed hydrosilylation of styrenes. See:Fu P.-F.; Brard L.; Li Y.; Marks T. J. J. Am. Chem. Soc. 1995, 117, 7157–7168. 10.1021/ja00132a015. [DOI] [Google Scholar]; Also see ref (5k).
- For a mechanistically analogous Cu-catalyzed hydroboration, see:; a Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062–6064. 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]; b Noh D.; Yoon S. K.; Won J.; Lee J. Y.; Yun J. Chem. - Asian J. 2011, 6, 1967–1969. 10.1002/asia.201100146. [DOI] [PubMed] [Google Scholar]
- Dow Corning holds a patent claiming certain copper-based catalysts for the hydrosilylation of olefins.Brandstadt K.; Cook S.; Nguyen B. T.; Surgenor A.; Taylor R.; Tzou M.-S.. Copper Containing Hydrosilylation Catalysts and Compositions Containing the Catalysts. WO 2013043792 A, March 28, 2013.
- See, for example:; a Deschamp J.; Chuzel O.; Hannedouche J.; Riant O. Angew. Chem., Int. Ed. 2006, 45, 1292–1297. 10.1002/anie.200503791. [DOI] [PubMed] [Google Scholar]; b Rendler S.; Oestreich M. Angew. Chem., Int. Ed. 2007, 46, 498–504. 10.1002/anie.200602668. [DOI] [PubMed] [Google Scholar]; c Deutsch C.; Krause N.; Lipshutz B. H. Chem. Rev. 2008, 108, 2916–2927. 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- See Supporting Information.
- Tamao K.; Ishida N.; Tanaka T.; Kumada M. Organometallics 1983, 2, 1694–1696. 10.1021/om50005a041. [DOI] [Google Scholar]
- For a discussion of metal-catalyed hydrosilylation mechansims, see refs (4b) and (4c).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.