


Abstract
Molecular changes underlying stem cell differentiation are of fundamental interest. scRNA-seq on murine hematopoietic stem cells (HSC) and their progeny MPP1 separated the cells into 3 main clusters with distinct features: active, quiescent, and an un-characterized cluster. Induction of anemia resulted in mobilization of the quiescent to the active cluster and of the early to later stage of cell cycle, with marked increase in expression of certain transcription factors (TFs) while maintaining expression of interferon response genes. Cells with surface markers of long term HSC increased the expression of a group of TFs expressed highly in normal cycling MPP1 cells. However, at least Id1 and Hes1 were significantly activated in both HSC and MPP1 cells in anemic mice. Lineage-specific genes were differently expressed between cells, and correlated with the cell cycle stages with a specific augmentation of erythroid related genes in the G2/M phase. Most lineage specific TFs were stochastically expressed in the early precursor cells, but a few, such as Klf1, were detected only at very low levels in few precursor cells. The activation of these factors may correlate with stages of differentiation. This study reveals effects of cell cycle progression on the expression of lineage specific genes in precursor cells, and suggests that hematopoietic stress changes the balance of renewal and differentiation in these homeostatic cells.
INTRODUCTION
Hematopoietic stem cells (HSCs) capable of long term marrow reconstitution have been separated from other marrow cells prospectively based on differential expression of cell surface markers or differences in the ability to extrude certain dyes. The HSCs give rise to progenitors (MPPs) that are capable of multilineage differentiation without long term marrow reconstitution, although the combinations of markers and the definition of the subsets of MPPs vary between research groups (1–3) (Supplementary Table S1). The pattern of lineage differentiation from MPP cells has been widely studied and correlated with the expression of transcriptional or post-transcriptional regulators, such as transcription factors (4–7), microRNAs (8) and epigenetic regulators (9,10). Alternative lineage roadmaps downstream of MPP cells have been proposed and investigated (11–13).
Heterogeneity of long-term repopulating stem cells is well recognized (12,14,15), including a distinction between replicating and quiescent stem cells (16–19). Changes in stem cells also occur in aging individuals (20) with an accumulation of cycling HSCs in the marrow (21,22) that is strain specific in mice (23). The accumulation of cycling HSCs refers a declining repopulating ability of stem cells from aged animals (24–27) that may be related to increased cycling of the cells (22), epigenetic changes (20,28), a shift towards oligo clonal hematopoiesis (29) and a shift from lymphoid towards myeloid bias (30,31) perhaps due to a decreased ability to lymphoid cells (32) although a recent report has reached a different conclusion about the myeloid/lymphoid ratio of production rates (33). Some HSCs can repopulate specific lineages in the marrow and give rise to long-term repopulating and transplantable progeny that often retain the same lineage bias. These include cells with a bias towards megakaryocytic repopulation (34), sometimes along with erythroid or granulocytic/monocytic lineage production (35). It has also been proposed that platelet-biased stem cells lie at the apex of the hierarchy (34) or that c-kitLo HSCs give rise to c-kitHi HSCs with increasing megakaryocytic lineage bias but decreasing self-renewal capability (36).
To get further understanding of the relation among the various types of early precursors, it is necessary to move beyond the type of limited resolution obtained with cell surface markers (37). Recent technical developments in microfluidics have made it possible to obtain whole transcriptome profile with multiplexed procedures (38), and these methods have been used to study the global transcriptome of single hematopoietic precursor cells (39–43).
In the present study, we performed single cell RNA-seq on HSC and the closely related MPP1 cells from normal mice as well as mice that underwent acute blood loss. Our analyses demonstrated well separated clusters of related cells in standard preparations of HSC and MPP1 cells, and heterogeneity in patterns of gene expression within each cluster. Stimulation of erythropoiesis by acute blood loss caused most HSC cells to shift into the cluster of cycling cells without loss of cell surface markers defining HSC. The HSC and MPP1 cells from anemic mice also increased expression of certain inhibitory transcription factors. We found correlation between the relative expressions of groups of genes specific for certain lineages with stages of the cell cycle. We confirmed stochastic activation of many lineage related transcription factors in HSC and MPP1 but found that a very few key lineage-related factors, perhaps one or two per developmental transition, are essentially silent in these cells, and we speculate that overcoming this inactivity represents a regulatory step in lineage development and maturation.
MATERIALS AND METHODS
Isolation of mouse bone marrow cells
Bone Marrow cells were isolated from 6 to 8 weeks C57BL6 mice (mixed 50% males and 50% females). All mice used in this study were purchased from Charles River and housed at the Yale Animal Resources Center (YARC). Bone marrow cells were flushed from femurs by using Dulbecco's Phosphate Buffered Saline solution (DPBS, 1X, GIBCO) without Calcium and Magnesium, supplemented with 5% fetal bovine serum (GIBCO) (FACS buffer). The harvested cells were gently dispersed by passage through a 21G1 needle attached to a 5 ml syringe. Cells were spun down at 1200 rpm for 10 min, resuspended in 15 ml and filtered through a 40 μm cell strainer (BD Falcon).
Live cell separation
After fresh bone marrow cells were isolated, the apoptotic cells were excluded using Annexin V Apoptosis Detection Kit eFluor® 450, and the Lineage and Flk2 negative cells were obtained with Lineage-biotin cocktail and Flk2-biotin conjured with PEcy7-streptavidin. Then the APC Cy7-ckit and PerCP Cy5.5-Sca-1 double positive cells were gated, and further the FITC-CD48 negative and APC-CD150 positive cells were used to sort HSC and MPP1 combined pure live cell populations for further analysis.
Cell culture
EML cells were obtained and cultured as described previously (44), and cultures in midlog phase were fractionated by preparative FACS using antibodies for lineage markers, CD34 and Sca-1. Human CD34 positive cells were obtained, expanded, and differentiated by the protocols previously described (45). Cells were harvested for RNA-seq at 0 time, 2 and h hours, and 2, 3, 4, 6, 8 and 12 days after initiation of erythropoietin treatment.
Flow cytometry and fluorescence activated cell sorting
Cells were stained with CD62L and CD97 antibodies (Supplementary Table S11) according to the manufacturer's description. Eighteen different populations of mouse BM cells were sorted by FACS AriaII (BD Biosciences) from the Yale cell sorter Core Facility (Supplementary Figure S2). When sorting HSC, progenitors and early stage BM cells, we added Biotin Lineage depletion cocktail (anti-Mac1, anti-CD3ε, anti-B220, anti-Gr-1, anti-CD11b and anti-Ter119) and streptavidin Particles Plus-DM (BD IMag™, mouse hematopoietic progenitor (stem) cell enrichment set). Lineage positive cells were depleted by transferring cells to a magnetic stand. Enriched lineage negative cells were further stained with antibodies listed in Supplementary Table S11. We note that the gate for selecting CD48- cells may have been set slightly higher than that in the literature and in four of seven preparative FACS experiments used for single cell analysis a few outlier cells (average 2.5-3.5% of the CD150+CD48-LSK cells) were excluded. However, our gate was chosen to surround the cluster of similarly stained CD48- cells. The small differences that adding the outlier cells could have made would not have affected our conclusions.
Expression profiling of FACS sorted small numbers of cells by full length mRNA transcriptome
Approximately 1000 cells from each of 18 sorted populations (see Supplementary Figure S1, Supplementary Table S2) were used to amplify cDNA by the PMA method and sequenced as described (46).
Single cell RNA-seq using the fluidigm C1 system
After sorting the HSC and MPP1 population, we measured the size of the cells with a Countess Automated Cell Counter (Invitrogen). After cell counting we loaded cells at a concentration of 2.5 × 105/ml on a C1 Fluidigm 5–10 um RNA-seq chip immediately, and the number of captured cells at each capture site was recorded under a microscope. Harvested amplified cDNAs from a 5 to 10 um C1 Fluidigm chip were transferred to a 96-well PCR plate (Denville, ThermoGrid PCR Plates 96-well standard, Cat# C18096-10). The PCR plate was sealed with Microseal ‘B’ seal Seals (Bio-rad Cat# MSB1001). The concentration of amplified cDNA was measured by using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen Cat# P11496). The libraries were generated by using a Nextera XT DNA sample preparation kit (96 samples) (Illumina, Catalog# FC-131-1096) and Truseq Dual Index sequencing Primer (paired-end) kit (Illumina, Catalog# PE-121-1003), with 96 barcoded samples combined. The libraries were pooled and purified using AMPure XP beads (Beckman Coulter, Inc, item # A63880), with QC evaluated with a bioanalyzer. The combined libraries were sequenced in one lane per plate on the Illumina HiSeq2500 instrument with 101-bp paired end sequencing. On average 1.4 million reads per cell were obtained, sufficient to detect most of the genes in the library (47). A total of eight samples of normal murine precursors, five HSC and three MPP1, were analyzed as were three samples of human CD34+ cells and two samples of EML cells.
Processing of bulk RNA-seq data
RNA-seq was performed according to Illumina protocols on the illumine 2500 instrument. For bulk RNA analysis at least ten million reads were obtained per sample. RNA-seq reads were mapped to the mouse genome (mm9 from the UCSC database) by Tophat (v1.3.3) with ‘–r 100’ option (48). Gene expression values were then calculated by Cufflinks (v1.2.1), using RefSeq genes as reference annotation with ‘-G’ option (49).
Differentially expressed genes for each subset were identified (Supplementary Table S3). Principle component analysis (PCA) was performed for genes with an average expression level more than 1 RPKM and gene expression level was transformed to ln(e) (RPKM) value after adding a pseudo count of 1. Principle components (PCs) 1–3 were used to separate cell population in each lineage (Supplementary Figure S3A). PC11 was used to detect DEGs between HSC and MPP1. PCs 8, 9 and 13 were used to identify genes, preferentially expressed in MPP2, 3 and 4, respectively (Supplementary Figure S3B). In this study, 0.4 factor loading (FL) was used as a cutoff of DEGs;for example, genes with FL in PC11 < –0.4 and > 0.4 were selected as HSC and MPP1-specific genes. Enrichment of these DEGs in cell populations sorted by (50) was evaluated by Gene Set Enrichment Analysis (GSEA) software (v2.0.14) with 1,000 permutations of the gene set, weighted enrichment statistic and signal2noise metric without collapse of the dataset (51).
We performed hierarchical clustering with the dist and hclust function in R. Network analysis of 18 cell populations were constructed from a Euclidian distance matrix D of all principle component scores. The distance was then converted into a similarity score matrix S as, 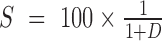 . Two cell populations with S>0.85 were then connected to each other (Supplementary Figure S3H).
. Two cell populations with S>0.85 were then connected to each other (Supplementary Figure S3H).
Processing of single-cell RNA-seq data
Single-cell RNA-seq reads were processed by Tophat and Cufflinks as described above. Single-cell data with mapping ratio >60% was retained for subsequent analyses. The data derived from wells with zero cells or more than one cell was removed (Supplementary Table S4).
Single-cell RNA-seq data was visualized by t-Distributed stochastic neighbor embedding (tSNE) using the RtSNE package (52). This program attempts to minimize the additional information necessary to reconstruct the multidimensional relationship among samples as compared to the information contained in the two dimensional map, and considers each data point as a sampling from a normally distributed variable. Log2(RPKM+1) values were used for tSNE with options ‘initial_dims = 10, perplexity = 10, theta = 0.5, check_duplicates = T’ except as otherwise noted. The tSNE output was used for hierarchical clustering using the hclust function with ‘method = ward.D2’. Smoothed curves were generated by the loess method in R, using default parameters.
DEGs between active and quiescent clusters were identified using two-fold cutoff and P < 0.05 by a one-tailed t test. Gene Ontology (GO) analysis was performed using GO stats in Bioconductor. Multiple hypothesis testing was adjusted using the Benjamini–Hochberg method.
Gene sets of transcriptional modules for hematopoiesis were downloaded from the Stem Site website at the George Daley lab (http://daleystem.hms.harvard.edu/) (53). The cell cycle stage was defined by expression pattern of 30 stage-specific genes, which were shown in (54) and also by expression of manually selected sets of genes known to function in DNA synthesis or in generation of the mitotic spindle, as described in the main text. ‘Lymphocyte’, ‘Granulocyte’, ‘Megakaryocyte’ and ‘Erythrocyte’-specific genes were selected by 4-fold higher average expression in ‘CLP and CLP_Sca-1hi’, ‘CMP, GMP-7 and GMP-11’, ‘Mature_Meg’ and ‘preCFU-E, CFU-E and ProEry’ than the other populations, respectively (Supplementary Table S5). Analysis of myeloid and erythroid gene expression was also performed with gene sets derived from our bulk sequencing of CMP, MEP and GMP as described in the main text. ‘Quiescence’ genes were obtained from (18). Cell cycle, DNA replication, DNA repair and apoptosis-related gene sets were obtained from MSigDB (software.broadinstitute.org). For identification of major changes in gene expression we generally limited the analysis to genes that were expressed, on a log scale, at an average of at least 1 per cell for a relevant subset of cells, and also showed at least two-fold changes in expression between subsets (http://www.broadinstitute.org/gsea/msigdb/index.jsp). GSEA of lineage-specific and quiescence genes was performed in each cluster with comparison to the rest of the clusters. Statistical significance of GSEA was set as FDR <0.1, which is more stringent than default setting of GSEA software. The list of gene sets used for GSEA is presented in Supplementary Table S5.
The tSNE analysis was also performed on HSC and MPP1 from anemic mice combined with those from normal mice. For the classification of HSC and MPP1 from anemic mice, we calculated Euclidean distances from cells into each subgroup (‘G2/M’, ‘G1/S’, ‘early G1’, ‘quiescent’ and ‘unidentified’). Then, we categorized HSC and MPP1 cells from bleeding mice into the subgroup showing the shortest average distance.
Procedure for generating anemic mice
A total of 28 C57/Blk6 wild type mice ranged 6–8 weeks old were used, including nine males and nine females for bleedings, and five male and five female controls. All procedures were performed in compliance with relevant laws and institutional guidelines and were approved by the Yale University Institutional Animal Care and Use Committee. C57BL/6J mice were obtained from Jackson laboratory (Bar Harbor, ME, USA). Mice were kept in microisolator cages, given autoclaved food and water, and handled only with gloves in a biological safety cabinet.
To generate bled, anemic mice, the mice were injected with 2 ml of pre-warmed normal saline IP 2 days before the start of bleeding. At day 2, 500 ul of blood was collected retro-orbitally from ‘bled mice’ to induce anemia. Saline injection was performed on both bled and ‘control mice’. The bleeding and saline injections were repeated on days 5, 8 and 11. On day 12, the mice were analyzed.
Complete blood counts were performed on a Hemavet (Drew Scientific, Waterbury, CT, USA) according to manufacturer's instructions. Peripheral blood was stained with antibody against Ter-119 PE (eBioscience, San Diego, CA, USA) followed by staining with thiazole orange (Sigma-Aldrich, St. Louis, MO, USA) in PBS and assessed by flow cytometry.
RESULTS
Early hematopoietic precursors show multiple levels of molecular heterogeneity
We examined subpopulations (Supplementary Table S2, Supplementary Figure S1) of the hematopoietic cells from mouse bone marrow by separating the cells with preparative FACS (Supplementary Figure S2), and analyzed transcriptional profiles from FACS samples with a thousand cells of each of 18 separate cell fractions (Supplementary Table S3) (1,55). The subpopulations of cells were distinguished by PCA (Supplementary Figure S3A and B). Around 250–300 genes were identified as differentially expressed genes (DEGs) in each population (Supplementary Table S3, Supplementary Figure S3C and D). The additional cell surface markers CD62L (56) and CD97 (57) were negative in HSCs, but a part of the MPP1 (25% and 31% respectively) expressed both proteins on their cell surface (Supplementary Figure S3E and F). FACS analysis of forward scatter and Sca-1 expression also showed somewhat more heterogeneity in MPP1 than in HSC (Supplementary Figure S3G). The sub-populations of cells were clustered in two (Supplementary Figure S3H) or three (Figure 1) dimensions based on Euclidean distance. MPP1 was located closer to the HSCs and separated from the other MPP fractions (Figure 1), consistent with an early observation (50).
Figure 1.

Relationship among 18 murine bone marrow populations. Two and three dimensional plots of grouping 18 populations based on PC1, PC2 and PC3.
To better understand the heterogeneity of the earliest precursors we sequenced single cells of biologic replicate samples of HSC and MPP1 using the Fluidigm C1 system and Smart-seq method (Clontech). Single-cell RNA-seq libraries with >60% of the products mapping to murine cDNA were highly reproducible (Pearson correlation coefficient > 0.9) (Supplementary Figure S4A). An average of 1.4 million and 1.7 million reads per cell, corresponding respectively to 112 HSC and 109 MPP1 single cells of normal mice met our criteria and were used for subsequent analysis (Supplementary Table S4).
We used the t-Distributed Stochastic Neighbor Embedding (tSNE) program (52) for display of the results of single-cell RNA sequence data (Figure 2A). The tSNE method gives sensitive separation of closely related groups of objects, although there is variation from run to run in the relative position of remote groups. Unlike principal component analysis (PCA), this method gave a clear separation of pooled HSC and MPP1 single cells into three subsets. This pattern was reproduced in biologic replicates or when the parameters of tSNE were varied (perplexity from 10 to 30 and use of 7 to 15 principal components) (Supplementary Figures S5A and S6A). The majority of HSC cells (55%) and a small fraction of MPP1 cells (7%) were classified into the first cluster (blue ellipse in Figure 2A), which we refer to as the quiescent cluster. Gene signatures significantly enriched in this cluster (Figure 2B, C and Supplementary Table S6) included: (i) quiescent HSC genes (18) (FDR = 0.001); (ii) previously-defined transcriptional modules for definitive HSCs (FDR < 0.058) (53); and (iii) Immunity related genes, external stimuli, interferon response, and cellular defense responses genes (FDR = 0.002) (Figure 2D, Supplementary Figure S5B and C, Supplementary Table S5). The expression level of the immunity related genes was significantly correlated with that of quiescence-related genes (Pearson correlation = 0.873, P < 2.2e-16), and varied markedly between cells within the quiescent cluster (Figure 2D and F, Supplementary Figures S5C and S6C), with a small fraction of the cells expressing high levels of these products (Supplementary Figure S5B). The quiescence related genes were also expressed at considerably different levels in different cells of the quiescent cluster.
Figure 2.

Classification of HSC and MPP1 single cells into three main subpopulations. (A) Dimensional reduction by tSNE separating HSC and MPP1 into three major clusters: U-, quiescent and active clusters. (B) Fraction of HSC and MPP1 cells falling into three major clusters in two biological replicates based on single cell RNA-seq. (C, D, E) GSEA analysis. (C) Quiescence-related genes (18). (D) Genes related to representative GO biological processes in the three major clusters. (E) Different lineages related genes among the three major clusters. (F) Comparison to (A) for the distribution of HSC and MPP1 cells displaying gene expression related to interferon response which mostly are in quiescent cluster. Average expression of ‘interferon gamma response-related’ genes (Supplementary Table S5) are shown. Red represents a higher level of expression and black a lower level. The dots are HSC, the triangles are MPP1, and two replicates are combined. (G) Comparison of the number of detected genes among three clusters.
Most of the MPP1 cells (74.5%) and a small fraction of HSC cells (21.5%) (Figure 2A and B) fell into the second cluster (red ellipse in Figure 2A), which we refer to as the active cluster. Comparison between the cells in active and quiescent clusters showed extensive up and down regulation of genes (Supplementary Table S6). The active group displayed depletion of quiescence-related genes (FDR = 0.001) and enrichment of cell cycle and DNA replication-related genes (FDR = 0.004 and 0.002, respectively), indicating active cell cycling (Figures 2D, 5B, and Supplementary Figure S4D and E), as well as increased expression of erythroid, megakaryocytic and granulocyte/macrophage related genes relative to the quiescent cluster (Figure 2E). Many of the top genes preferentially expressed in the active cluster were related to later phases of the cell cycle including spindle formation, centromere duplication, and DNA replication and repair (Figure 2D, Supplementary Figure S4F).
Figure 5.

Induction of anemia drives more cells into the active stage and especially the G2/M phase of the cell cycle. A, B and C are different tSNE runwith the same parameters and the appearance of the detailed pattern varies (111) (A) The tSNE patterns of the whole population of HSC (purple) and MPP1 (green). Unlike MPP1, the majority of HSC cells are quiescent cells (middle cluster). (B) Comparative analysis of cells expressing high levels of S (red) and G2/M (purple) phase-specific genes in the combined population of HSC and MPP1. The colored cells (red and purple) are 10 cells each expressing highest levels of S or G2/M genes. (C) Both anemic HSC (blue dots) ls shift to the active stage, and the anemic MPP1 (red dots) shift towards the G2/M stage. (D) Three lineage expression levels in HSC/MPP1 cells from the control mice. Cells plotted according to increasing G2/M gene expression. The red, black and purple curves respectively refer to the expression per cell of erythroid precursor genes, of megakaryocyte genes, and of granulocyte/macrophage precursor genes. The curves were generated by loess smoothing of scatter plots. The gene expression levels are plotted on a log scale. (E) Three lineage levels of expression of genes in HSC/MPP1 cells from the anemic mice. Cells plotted as in Figure 5D. Note that a larger fraction of the cells from anemic mice were in the S/G2/M fraction.
The third cluster of cells, the U-cluster (uncharacterized), in the black ellipse (Fig, 2A), was composed of 32 (24%) HSC and 18 (16%) MPP1 cells. This cluster was characterized by lower RNA reads and fewer genes detected per cell (1.3 million reads and 1,045 genes on average) than the other two clusters (2.8 million reads and 3216 genes on average) (Figure 2G), although spike-in RNAs for the most part gave higher reads in cells from the U cluster (Supplementary Figure S7D). The pattern of forward scattering on FACS analysis showed that almost all HSC cells fell into a rather tight cluster, arguing against the possibility that the reduced RNA in the third cluster was due to the presence of a group of cells that were substantially smaller or more shrunken than the remainder. When more cells were analyzed, the U-cluster appeared more compacted (see Figure 5, Supplementary Figures S6A–D, S7F and S10).
The distribution of Sca1 mRNA and surface protein also indicated heterogeneity in the HSC and MPP1 cells. Wilson et al. (40) compared four methods of preparing HSC and looked for expression patterns common to cells obtained by each method. These authors concluded that high Sca-1 (Ly-6A/E) was an effective marker for identifying true long term repopulating cells. We confirmed that there was heterogeneity in the Sca-1 surface marker and mRNA levels in these cells, and found that most of the cells with high levels of Sca-1 mRNA are in the quiescent cluster (Supplementary Figure S10E). However in anemic mice (Supplementary Figure S3G), the levels of Sca-1 are increased even though the precursor cells from such mice have been found to be defective in long term repopulation, so that Sca-1 is a correlated but not a determining factor for long term repopulating cells. Of the 13 genes (B2m, Ndufa3, H2-T9, Ifitm2, Sepw1, Tbxas1, Rps27, Gm12657, Txnip, Ifitm1, Tmem176b, Ifitm3, Shisa5) whose expression correlated most closely with that of Sca-1, three are transmembrane proteins known to be induced by gamma interferon (Ifitm1, 2, and 3), and another of the 13 genes, Shisa5, has also been associated with apoptosis induction (58,59).
To further compare the relative expression of genes, we multiplied cDNA RPKM levels in each cell by a factor such that the total level of counts per cell mapped to known cDNAs was similar in each cluster. After this adjustment the separation of cells into three clusters was still retained and the U cluster cells were enriched for mRNA of genes identified as G0 genes in fibroblasts (60).
We performed a third batch of scRNA-seq in HSCs (replicate 3) with an RNA spike-in (Supplementary Figure S7A, D, G). We filtered out two single-cell libraries flawed by lower efficiency of PCR amplification (Supplementary Figure S7B), and 12 libraries with lower mapping rates (Supplementary Figure S7C). The tSNE analysis showed a similar distribution of three groups to that seen with other replicates of HSC scRNA-seq. The read count for mitochondrial sequences, proposed as an indicator of damaged cells (61), was generally below 12%, although the U-cluster included about 45% cells with a higher number of reads derived from the mitochondria genome (Supplementary Figure S7E). Removal of the cells with over 12% mitochondrial sequences did not alter the pattern of the tSNE display including the detection of three major clusters of cells (Supplementary Figure S7F). The ratio of reads of the RNA spikes to reads of the RNA of the expressed genes was higher in the cells with lower numbers of gene reads (Supplementary Figure S7C), indicating that the cells with low gene reads were a result of lower levels of mRNA per cell rather than a failure of the reverse transcription or downstream reactions.
We also performed FACS in which we omitted CD34 staining, and combined MPP1 and HSC cells, to allow careful exclusion of all cells with surface annexin-V staining. The scRNA-seq data from the annexin-V negative population (live cells) was analyzed by tSNE and showed a pattern resembling that from the preceding experiments except that there was reduction in the number of cells in the U-cluster from about 20–24% to about 12% (Supplementary Figure S8A–D).
Expression of genes characteristic of different lineages changes with the stage of the cell cycle
We mapped the expression pattern of each cell to sets of genes differentially expressed in the G1/S, or G2/M phase of the cell cycle as well as small sets of genes known to have S and G2 phase specific expression (54,62) (Supplementary Table S5).Based on the expression of stage-specific genes, the active cluster was classified into three states: G2/M, G1/S and early G1 (Figure 3A). In the tSNE patterns, cells in the G2/M state lie at the tip of the branch of the graph containing active cells (Figure 3A). Expression of quiescence-related genes was significantly lower in the G2/M state (Figures 3B and 4A). Because not all the genes in the lists in the literature could clearly be related to processes specific to S, G2 or M phases of the cell cycle, we refined the set of G2/M specific genes by selecting 41 genes that corresponded to centromere mitotic, tubulin functions or spindle check point genes (Supplementary Table S5 Refined G2/M). In a repeat tSNE analysis (Supplementary Figure S6A), expression of these genes was highly augmented in 10–15 cells out of the 80 cells in the active cluster (Supplementary Figure S6B). A second subset of cells displayed higher expression level of G1/S and S phase-specific genes but not G2/M and M phase genes, and was located proximal to these G2/M cells in the tSNE map. We separately analyzed the relative expression of each of a number of genes related to DNA synthesis or to the immune response, including Pcna (Supplementary Figure S6D), Ifitm (Supplementary Figure S6C), and MCM genes, subunits of DNA polymerase E, ribonucleotide reductase, and other genes signaling the onset of the S phase. The highest expressing cells for each DNA synthesis gene mapped in the region enriched for the pool of G1/S and S phase genes. However the cells with the highest levels of the individual genes were partially different from one another, and deeper sequencing might detect the sequence of events during the onset of DNA synthesis (Supplementary Figure S5A in green cluster of the red circle). M/G1-specific genes were found in two groups, one near the G1/S genes and the other close to the quiescent cells (Supplementary Figure S5A, the black cluster).We classified the second subset of cells as being in an early G1 state. Although occasionally quiescent cells expressed G1/S and S phase genes, none expressed elevated levels of G2/M genes (Figure 5B). In the anemic mice the HSC (CD34-) were below the S/G2/M region, while the MPP1 (CD34+) fell within that region (Figure 5C). This suggests that on passage through the S/G2/M phases of the cell cycle, cells acquire expression of surface CD34.
Figure 3.

Correlation between cell cycle phase and expression of lineage-related genes. (A) The tSNE patterns of G2/M (red), G1/S (orange) and early G1 states (purple) within the active cluster, along with the quiescent (blue) and U-cluster (black) cluster. (B) Comparative analysis of G1/S, S, G2/M and M phase-specific genes in U-, quiescent- and three subdivisions of the active cluster. (C) GSEA of lineage-specific genes in cells at various stages of the cell cycle in the active cluster. (D) Cells plotted according to increasing G2/M gene expression. The log of expression of red (erythroid) and cyan (G2/M) genes per cell is shown. (E) The relation between expression of S phase and G2/M phase genes in each cell. Cells are plotted according to their rank order of levels of G2/M genes. The lines are loess smoothed representations of the level of expression in each cell. The purple line represents the level of S phase genes and the red line the level of G2/M genes. The green line represents the ratio of S phase to G2/M genes.
Figure 4.

Activation of HSC and MPP1 in anemic mice. (A) The tSNE patterns of HSC and MPP1 from control and anemic mice. Different subsets of cells from the same tSNE display are shown in each panel, with subsets labeled by the color scheme on the right side of the top lane. (B) Ratio of cells falling into each subpopulation in HSC and MPP1 from control and anemic mice. In each image several subsets of the cells are displayed, identified by the color code in the right side upper rectangle. bHSC and bMPP1 are HSC and MPP1 obtained from mice made anemic by bleeding. (C) Schematic graph of correlation between lineage specificity and the phase of the cell cycle. After induction of anemia by bleeding, HSC shift into the active cluster in map regions occupied by MPP1 cells in normal marrow.
We selected signatures of groups of genes that were specifically expressed in either the erythroid, the megakaryocytic, the lymphocytic or the granulocytic lineages from our RNA-seq data (Supplementary Tables S3 and S5). For granulocyte/ monocyte and for lymphoid cells we selected genes expressed at 4-fold higher levels in the appropriate precursors than in the average of precursors for other lineages. For the erythroid and megakaryocytic clusters we selected genes that were expressed 4-fold higher in erythroid precursors than in megakaryocytes or other lineages, and vice versa. These gene sets were projected across the tSNE groups (Supplementary Figure S10). The cells expressing higher levels of lymphoid related genes were separated from those expressing other lineages, and fell into the quiescent cluster (FDR = 0.012) (Figure 2E). In contrast, cells expressing higher levels of erythroid, granulocytic, and megakaryocytic genes tended to fall into the active cluster (Figure 2E, FDR = 0.001, 0.09 and 0.054, respectively). Genes of multiple lineages were often expressed in the same cell, but the average relative levels of expression of the lineage-specific genes varied with the stage of the cell cycle (Figure 3C).
We also plotted expression data in cells ordered according to the rank order of levels of expression of G2/M genes (Figure 3D), and compared the pattern with that of S phase and G2M phase genes (Figures 3E and 5D, E). This showed that the cells with high expression of erythroid genes were in the G2/M phase. Megakaryocyte and granulocyte precursor genes showed similar levels of expression relative to erythroid genes in the early G1 state of the active cluster (FDR = 0.031 and 0.086, respectively) and lower relative expression in G1/S and G2/M state (FDR>0.198) (Figure 3C) although the megakaryocyte genes rose more than the granulocyte precursors genes in G2/M cells (Figure 5D and E). The erythroid genes showed prominent up regulation in the G2/M (FDR = 0.003) and some up-regulation in G1/S state cells (FDR = 0.083) (Figure 3C, D and E). A composite PCA analysis (Supplementary Figure S5E) showed that, relative to megakaryocyte genes, granulocyte/macrophage genes were more highly expressed in many MPP1 cells than in HSC, demonstrating further levels of heterogeneity in these precursor cells. Single cell RNA-seq of total human CD34+ cells expanded for 6 days without erythropoietin (50) also showed a striking rise of erythroid genes in cells in the S/G2/M phase (Supplementary Figure S5D), so that the elevation of erythroid mRNA in G2/M phase early precursors is not species-specific.
To confirm the results we performed a separate analysis after preparing sufficient numbers of CMP, MEP, and GMP cells to be able to doRNA-seq without amplification. From this data, we then selected genes that were more than two-fold over expressed in either MEP or GMP relative to the average level of expression for the three cell types and were also more than four-fold over expressed in MEP versus GMP or vice versa. Again the pattern for the level of expression of granulocyte/macrophage genes was different from that for the erythroid genes, with a marked increase in erythroid gene expression in cells with the highest levels of G2/M genes (Supplementary Figure S10C).
Stimulation of erythropoiesis shifts HSC into active states
To confirm that particular clusters represent quiescent cells that can become activated, we performed single-cell RNA-seq on HSC (bHSC) and MPP1 (bMPP1) from mice whose erythropoiesis had been stimulated by bleeding (Figure 4, Supplementary Figure S9). The tSNE analysis with 99 single cells with acceptable sequencing quality revealed that a higher fraction of HSC and MPP1 cells were in active states in acute blood loss mice than in control mice (Figure 4A). Approximately 80% of HSC were in either the U or quiescent clusters in normal mice, but more than 70% of the HSC fell into the early G1 and G1/S stages of active cells in the anemic mice (Figure 4B, Supplementary Figure S10B), i.e. only 20% of the cells were in the Uor quiescent state. In contrast, the proportion of MPP1 in the active state was not different between control and anemic mice (74.5% and 75.5%, respectively) although the MPP1 cells in the active cluster from anemic mice were shifted towards the S and G2/M enriched regions of the active cluster. This data indicates that the stress of anemia caused most of the quiescent cells to shift into the active state, and promoted progression through the cell cycle within the MPP1 population (Supplementary Figure S10A and B). There was a reduction of cells in the U cluster from 24% to 8%, P = 0.0009 (one-sided t-test), with Malat1 remaining higher per cell in the U cluster (Supplementary Figure S10D), and Sca1 was high in the active cluster HSC (Supplementary Figure S10E). HSC increased gene expression for all lineages in the anemic mice (P < 1.42e–5 by the t test) but this was due to the decreased fraction of inactive cells (Figure 5C) rather than increase per cell among the more active HSC. HSC showed a higher level of Sca-1 expression than in control mice (Supplementary Figure S3G) as well as expression of relatively high levels of a number of immune response genes (Supplementary Tables S7–S9). MPP1 did not show any significant difference in lymphoid genes, but significantly induced granulocytic, megakaryocytic and erythroid genes (P < 0.03 by the t-test). The effects of stage of the cell cycle on erythroid gene expression was similar in the anemic and control mice (Figure 5D, E and Supplementary Figure S10C). Overall the effect of bleeding was depletion of cells from the U and quiescent clusters, and augmentation of the fraction of cells in the cycling cluster.
Among the most significantly induced genes in bHSC as compared to resting HSC and MPP1 (Supplementary Tables S7 and S8) were a series of transcription factors (7 out of the 10 most strongly induced genes) that were generally not specific to any one lineage. Of these all except Id1 and Hes1 were significantly increased in normal MPP1 compared with normal HSC, so that activation of the factors other than Hes1 and Id1 may reflect shifting of HSC into the active cluster. A small group of genes including Hes1, Id1 and Lst1 were more highly expressed in both MPP1 and HSC from anemic mice than in either cell type in control mice and these are briefly discussed below.
For most lineages one or two key lineage related transcription factors are not stochastically activated in HSC and MPP1
Consistent with earlier reports (63), also in the discussion in (64), single HSC or MPP1 cells expressed, in an apparently stochastic manner, a range of genes characteristic of various differentiated hematopoietic lineages. However this promiscuity was selective. We compiled lists of transcription factors characteristic of the precursors of lymphocytes, megakaryocytes, erythroid cells, or granulocytes/macrophages and added to these lists a number of transcription factors that function in specific lineages of hematopoietic cells (Supplementary Table S10). Most genes in each list were expressed in over 5% of the cells, often over 10%. However a small set of transcription factors were expressed either not at all, or only in a very few cells and at low levels (Figure 6). Among the silent factors were several that are known to be critical and highly expressed during specific lineage development: Cebpa (Figure 6B) for granulcoyte/macrophage and, at higher levels, specifically granulocyte (65); Klf1 (Figure 6A) for erythropoiesis, at late stages of maturation of a lineage(Cebpe for granulocytes (66) (Figure 6B), or to specify sub-lineages from a differentiated precursor (for example, Foxp3 for Treg cells (67). Several of the 55 lymphocyte specific transcription factors were not detected (Figure 6C). This may reflect the multiple branches of lymphocyte differentiation. All megakaryocyte transcription factors were expressed in HSC and MPP1 (Supplementary Figure S6D), so that, in this sense, as has recently been discussed (68), megakaryocyte development is a default process.
Figure 6.

The expression of the transcription factors (TFs) specifically elevated in different lineages in mouse HSC and MPP1 single cells. The X axis presents the different TFs (Supplementary Table S9); the Y axis represents the log10 of the number of cells in which the TF mRNA was detected (plus 1 to avoid logs of numbers less than 0). In total 228 single cells were analyzed, combining replicate 1 and 2. (A) Klf1 is the least expressed among 55 TFs for the erythroid lineage although it is one of the most highly expressed factors in maturing erythroid cells. (B) Cebpa and Cebpe were depleted in contrast to the other 41 TFs for granulocyte lineage. (C) Seven TFs were not detected and several other TFs are very low among 55 TFs specific for lymphocyte lineage. (D) All of the list of 22 TFs expressed selectively in maturing megakaryocytes are expressed in a substantial fraction of the HSC/MPP1 cells.
Klf1 is a well-studied erythroid transcription factor that is highly expressed during the course of erythroid cell maturation (69) and coordinates many aspects of erythropoiesis (70,71). To confirm the particular features of expression of this gene, we also examined Klf1 expression during erythropoietic development in the murine multipotent hematopoietic cell line EML (72), and in human peripheral blood mobilized CD34+ precursor cells.
Treatment of human peripheral blood mobilized CD34+ precursor cells with erythropoietin causes erythroid differentiation and maturation (Supplementary Figures S11C and S12A, B, C). Among the transcription factors strongly expressed in maturing erythroid human CD34+ cells, KLF1 showed by far the greatest amplitude of induction (Supplementary Figure S11C). During erythroid development from human CD34+ cells, KLF1 induction preceded or paralleled the induction of other most strongly induced genes (Supplementary Figure S12A). In the first few days after stimulation of the human cells with erythropoietin, a set of megakaryocyte specific genes including Pf4, Ppbp, Itga2b, Gpiba and Vwf were activated, but these were progressively silenced when Klf1 mRNA approached its peak levels (Supplementary Figure S12B).
EML suspension cultures contain CD34+, Sca-1+, Lin− precursors, which spontaneously differentiate into intermediate erythroid precursors (44). Klf1 was essentially silent in the FACS sorted EML precursor cells, unlike most other erythroid transcription factors (Supplementary Figure S11B), but was the most strongly induced transcription factor in EML erythroid cells.
Knockout of Klf1 in mice produces early embryonic lethality due to a failure to form properly hemoglobinized red cells, and Klf1 deficient cells also fail to normally express a variety of other erythroid genes (73). Nevertheless erythroid progenitors are formed in Klf1 knockout embryos (74) so that initial steps of erythropoiesis are not exclusively dependent on Klf1.Mutations in KLF1 are associated with a range of red blood cell disorders (75). The results summarized above suggest that full Klf1 activation contributes to or controls a stage transition during erythroid differentiation. The sequences in the first 900 bp upstream of the transcription start site direct cell type and state specific Klf1 transcription (76,77). Histone 3 lysine 27 acetylation is a mark for active promoters and enhancers, and is very much reduced in the 900 bp upstream of the Klf1 gene in EML precursors compared to the spontaneously emerging erythroid cells (Supplementary Figure S12D), so that the regulation of Klf1 mRNA occurs at the transcriptional level.
The mechanisms differentially regulating Klf1 expression are of special interest. The ratio of Gata2 to Gata1 declines during erythroid differentiation (78–80), and may regulate the levels of Klf1 expression. This is consistent with the results from human CD34+ cells (Supplementary Figure S12C). However, this ratio changed only 3.4 fold during differentiation of the EML cells, based on bulk RNA-seq (in the same experiment as Supplementary Figure S11). In addition to Gata1 and Gata2, other factors including Smad and Dek oncoprotein (77) also regulate the transcription of Klf1, but the levels of DEK and SMAD proteins during erythroid development from human CD34+ cells did not increase with the levels of Klf1 (Supplementary Figure S12C).
Another factor, Bcl11a, in EML cells, unlike in mouse HSC and MPP1, was also silenced similarly to Klf1 (Supplementary Figure S11B). Bcl11a is a major player in regulating the switch from fetal to adult hemoglobin in humans (81) and its transcription is mediated in part by Klf1 (82). Therefore, Bcl11a is another example of a selectively silenced regulatory gene at a developmental transition state. A recent report confirmed that both Klf1 and Bcl11A are needed for substantial expression of fetal and adult hemoglobin in human fetal erythroblasts (71).
Overall, there are transcription factors acting at transition steps during hematopoietic development that are under more stringent or specific control in HSC/MPP1 than most lineage-specific transcription factors and are essentially silent in early precursors. This may offer an empirical test for identifying a subset of transcription factors or other genes that are uniquely required for particular developmental transitions in other systems.
DISCUSSION
Early hematopoietic precursors show multiple levels of heterogeneity
Defining the differences in composition between closely related HSC and MPP1 populations that cause their differences in long term repopulation of marrow is an approach towards understanding the basis for this fundamental property. Surface CD34 in mice has been associated with lack of long term repopulating activity (32,83). In the present studies, cells that were at the S/G2/M phases of the cycle were CD34 positive. Because CD34+ cells are less efficient at long term repopulation this is consistent with the suggestion that cells that are actively replicating lack long term repopulating activity in standard assays.
Our bulk RNA-seq data are in agreement with earlier results (18,19,50,84–87), and showed that the lineage-related precursors fell into different volumes of the first three dimensions of PCA space, and that the MPP population was heterogeneous (88). The number of genes detected in a single cell by current methods may vary over as much as an order of magnitude between vigorously growing or large cells and relatively inactive small cells. Cutoff levels of 2300–4000 genes per cell have been used by others, with partly different goals. However, resting T cells show an average of <1300 genes per cell in Fluidigm C1 analyses. There could be a risk of biasing the result against some interesting cells if we had used a high cutoff for the genes identified per cell. We therefore chose to accept all capture sites that contained a single cell with over 60% mappable reads and >800 genes expressed/cell. 80% of the empty wells showed <700 genes, with an average of 180 genes per empty well. The remaining empty wells gave results resembling wells with a single cell and may have either trapped single cells that were missed by visual inspection or trapped cytoplasmic fragments of cells. We emphasize that it is unknown whether the cells in the U-cluster are a technical artifact or whether they have some biologic significance although we note that other groups have identified a biologic function for cells with low read counts in other systems (90,91).
The tSNE analysis displayed heterogeneity within HSC and MPP1. The major component of HSC is clearly separated from the MPP1 cells and displays gene signatures of quiescence and elevation of mRNAs for Sca-1 and a subset of interferon responsive genes. The main cluster of MPP1 is characterized by higher expression of cell cycle and DNA replication-related genes.
The subsets of HSC and MPP1 were defined by the destructive procedure of transcriptome analysis, limiting our ability to apply concurrent functional analyses to these cells. Further understanding of these clusters would be greatly expedited by development of a method for selectively isolating viable subsets of cells in a non-destructive protocol. Assignment of other molecular characteristics such as epigenomic changes to subgroups of HSC would require the application of demanding and low throughput techniques such as simultaneous analysis of the RNA and DNA of the same cell (89–93).
The relative expression of genes of megakaryocytic, erythroid or other lineage precursors changes during progression through the cell cycle
Lineage choice in precursor cells may require cells to go through a replicative cycle (94–98). Recent studies have suggested that the lineage biases in differentiation of hematopoietic (99) or embryonic stem cells (100,101) may vary with progression of the cell cycle. In particular, studies of colony formation from HSC expanded in a cytokine mixture in vitro indicated that megakaryocytes may preferentially be produced from cells early in the S phase at about the same time as proliferative granulocytes, while non-proliferative granulocytes are produced by cells in mid-S phase (102). Socolovsky et al. (94) have found that a key commitment step for maturation of erythropoiesis is linked to progression through the cell cycle. The present results show that there was an erythroid specific augmentation of gene expression that occurs in cells as they passed through the G2/M phase of the cycle, and this could explain the effect of cell cycling on differentiation. There are a number of possible mechanisms for the G2/M phase relative elevation of erythroid related transcripts in G2/M including the speculation that this may be related to the observation that Gata1 remains associated with chromatin throughout mitosis (103) and therefore may not be removed from chromatin during S phase.
This data suggests a model in which lineage choice is influenced in part by the stage of the cell cycle at which the cell receives signals for lineage activation, perhaps different from signals needed for self-renewal or survival (104). If lineage selection is based on stochastic changes in the balance of regulatory factors, then differences in the relative amounts of erythroid and myeloid factors during the cell cycle might bias stochastic activation of one or the other lineage. Coupling of lineage bias with stages of the cell cycle could play a role in maintaining the balance between productions of different types of cells. Alternatively, the high ratio of erythroid to granulocytic gene expression in the G2/M cells could reflect the relative rates of production needed for the different lineages, if differentiation occurred in the early precursors during G2/M.
Stimulation of erythropoiesis by acute anemia shifts HSC and MPP1 into active states and produces specific changes in their transcriptome
Induction of anemia caused HSC to shift into the active cluster together with MPP1, and increased the fraction of MPP1 cells in cycle. However, the transcriptome of activated cells did not show any bias towards erythroid or megakaryocytic development beyond that seen in normal cells at the same stage of the cell cycle although the production of downstream progeny would be directed towards the erythroid lineage, so it appears that lineage specific amplification occurs downstream of these earliest precursor cells.
Id1 and Hes1 are two transcription factors whose mRNAs were elevated on average over 10-fold in HSC and MPP1 from anemic animals above the levels seen in either normal HSC or MPP1, although the induction is partly a consequence of more cells expressing these genes. Id1 is a transcription factor that antagonized the action of various basic HLH factors, inhibits differentiation in other systems (105), and functions to maintain stem cells (106–108). Hes1 is also a negative regulator of transcription that functions in the maintenance of cancer stem cells (109) and is an essential regulator of HSC development (110). Elevated Id1 and Hes1 in the HSC from anemic animals may play a role in retaining HSC or in promoting expansion of early hematopoietic precursors by limiting their rate of differentiation.
Resistance to stochastic activation marks a subset of transcription factors key to passage through specific stages during lineage development
The earliest hematopoietic precursors show an apparently stochastic activation of transcription of a variety of genes characteristic of progeny cell lineages (63). However, a few transcription factors, perhaps as few as one per developmental transition, are silent or only very weakly expressed. Examining one of these factors, the erythroid factor Klf1, in more detail, we found that it is the transcription factor with the greatest amplitude of change in expression level in both mouse EML and human CD34+ cells modeling erythroid development. Klf1 is an example of a subset of transcription factors coupled to and regulating distinct transitions during development of various lineages. Such transitions represent discrete developmental steps subsequent to initial choice of lineage predilection. Understanding the basis for the failure to stochastically activate Klf1 in precursor cells might have more general significance for understanding the staging of differentiation events in other cell types.
CONCLUSIONS
In summary single cell RNA sequencing showed distinct clusters of cells in both the standard HSC and MPP1 population, including active cells and quiescent cells, and a new, undefined fraction of cells. HSC express a number of interferon response genes that may contribute to quiescence. However, when erythropoiesis was stimulated by induction of anemia, most HSC cells enter into active phases of the cell cycle without silencing of the interferon response genes, so that induction of cell cycling occurs despite continuing presence of mRNA for genes corresponding to interferon-like cell responses. More precise information would become available as it becomes feasible to directly study the protein content of the cells.
The relative levels of expression of mRNAs characteristic of early stages of differentiation of various lineages correlates with stage of the cell cycle in individual cells, possibly contributing to maintenance of balanced multipotency. Strikingly, erythroid lineage genes are relatively highly expressed in G2/M stages. Meanwhile, a small subset of lineage specific transcription factors, perhaps one factor per developmental step, such as Klf1 in erythroid lineage, are largely silent in the HSC and MPP1, and these may mediate limiting steps in the progression of differentiation along various lineages. Megakaryocyte differentiation is an exception, as the megakaryocyte related transcription factors we detected were all expressed in a number of HSC and MPP1 cells. Therefore, the oscillation or burst of gene expression in different single cells of early hematopoietic precursors, at least for some genes correlated to cell cycling, are functionally associated to lineage specific-expression. The stress of anemia causes HSC to shift to the active cluster and activates expression of several genes including Id1 and Hes1 in both HSC and MPP1, perhaps shifting the balance between precursor renewal and differentiation.
ACCESSION NUMBER
RNA-seq data sets supporting the results of this article are registered in the GEO database. The accession number is GSE68981.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Nenad Sestan for support single cell RNA-seq, Candace Bichsel Tebbenkamp for assistance in single cell experiment arrangement, Lucia Ramirez, Nathaniel Watson and Nathan Hammond for sequencing, Zhao Zhao for FACS sorting. Computation time was provided by the Yale University Biomedical High Performance Computing Center.
Author contributions: S.M.W. and X.P, designed and coordinated the study. J.Y. and M.S. prepared the cells. S.H. and A.T. prepared anemia mice. J.Y. performed cell fractionation and FACS analyses, J.Y. performed bulk RNA-seq. J.Y. and Z.L. performed single cell RNA-seq. Y.T., S.M.W., and X.P analyzed RNA-seq data. S.M.W., X.P., Y.T., J.Y., L.W., I.H.P., S.H. wrote and/or edited the manuscript. G.E performed illumine high-throughput sequencing. M.P.S. supervised the sequencing. L.X.G., Y.K. and X.Z. performed additional clustering analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [1P01GM099130-01 and R01DK100858]. Funding for open access charge: NIH [R01DK100858].
Conflict of interest statement. None declared.
REFERENCES
- 1.Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E. et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008; 135:1118–1129. [DOI] [PubMed] [Google Scholar]
- 2.Park I.K., He Y., Lin F., Laerum O.D., Tian Q., Bumgarner R., Klug C.A., Li K., Kuhr C., Doyle M.J. et al. Differential gene expression profiling of adult murine hematopoietic stem cells. Blood. 2002; 99:488–498. [DOI] [PubMed] [Google Scholar]
- 3.Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J.. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005; 121:1109–1121. [DOI] [PubMed] [Google Scholar]
- 4.Orkin S.H., Zon L.I.. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008; 132:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeVilbiss A.W., Sanalkumar R., Johnson K.D., Keles S., Bresnick E.H.. Hematopoietic transcriptional mechanisms: from locus-specific to genome-wide vantage points. Exp. Hematol. 2014; 42:616–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moignard V., Macaulay I.C., Swiers G., Buettner F., Schutte J., Calero-Nieto F.J., Kinston S., Joshi A., Hannah R., Theis F.J. et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat. Cell Biol. 2013; 15:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurenti E., Doulatov S., Zandi S., Plumb I., Chen J., April C., Fan J.B., Dick J.E.. The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nat. Immunol. 2013; 14:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monticelli S., Ansel K.M., Xiao C., Socci N.D., Krichevsky A.M., Thai T.H., Rajewsky N., Marks D.S., Sander C., Rajewsky K. et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005; 6:R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tadokoro Y., Ema H., Okano M., Li E., Nakauchi H.. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 2007; 204:715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broske A.M., Vockentanz L., Kharazi S., Huska M.R., Mancini E., Scheller M., Kuhl C., Enns A., Prinz M., Jaenisch R. et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009; 41:1207–1215. [DOI] [PubMed] [Google Scholar]
- 11.Adolfsson J., Mansson R., Buza-Vidas N., Hultquist A., Liuba K., Jensen C.T., Bryder D., Yang L., Borge O.J., Thoren L.A. et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005; 121:295–306. [DOI] [PubMed] [Google Scholar]
- 12.Ema H., Morita Y., Suda T.. Heterogeneity and hierarchy of hematopoietic stem cells. Exp. Hematol. 2014; 42:74–82. [DOI] [PubMed] [Google Scholar]
- 13.Heffner G.C., Clutter M.R., Nolan G.P., Weissman I.L.. Novel hematopoietic progenitor populations revealed by direct assessment of GATA1 protein expression and cMPL signaling events. Stem Cells. 2011; 29:1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita Y., Ema H., Nakauchi H.. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J. Exp. Med. 2010; 207:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copley M.R., Beer P.A., Eaves C.J.. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012; 10:690–697. [DOI] [PubMed] [Google Scholar]
- 16.Anjos-Afonso F., Bonnet D.. Forgotten gems: human CD34(-) hematopoietic stem cells. Cell Cycle. 2014; 13:503–504. [DOI] [PubMed] [Google Scholar]
- 17.Anjos-Afonso F., Currie E., Palmer H.G., Foster K.E., Taussig D.C., Bonnet D.. CD34(-) cells at the apex of the human hematopoietic stem cell hierarchy have distinctive cellular and molecular signatures. Cell Stem Cell. 2013; 13:161–174. [DOI] [PubMed] [Google Scholar]
- 18.Qiu J., Papatsenko D., Niu X., Schaniel C., Moore K.. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2014; 2:473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg E.C., Passegue E., Prohaska S.S., Wagers A.J., Koeva M., Stuart J.M., Weissman I.L.. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS One. 2010; 5:e8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun D., Luo M., Jeong M., Rodriguez B., Xia Z., Hannah R., Wang H., Le T., Faull K.F., Chen R. et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014; 14:673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison S.J., Wandycz A.M., Akashi K., Globerson A., Weissman I.L.. The aging of hematopoietic stem cells. Nat. Med. 1996; 2:1011–1016. [DOI] [PubMed] [Google Scholar]
- 22.Sudo K., Ema H., Morita Y., Nakauchi H.. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000; 192:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Haan G., Nijhof W., Van Zant G.. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997; 89:1543–1550. [PubMed] [Google Scholar]
- 24.Liang Y., Van Zant G., Szilvassy S.J.. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005; 106:1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim M., Moon H.B., Spangrude G.J.. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann. N. Y. Acad. Sci. 2003; 996:195–208. [DOI] [PubMed] [Google Scholar]
- 26.Dykstra B., Kent D., Bowie M., McCaffrey L., Hamilton M., Lyons K., Lee S.J., Brinkman R., Eaves C.. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007; 1:218–229. [DOI] [PubMed] [Google Scholar]
- 27.Dykstra B., Olthof S., Schreuder J., Ritsema M., de Haan G.. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011; 208:2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beerman I., Rossi D.J.. Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease. Cell Stem Cell. 2015; 16:613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014; 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller-Sieburg C.E., Sieburg H.B., Bernitz J.M., Cattarossi G.. Stem cell heterogeneity: implications for aging and regenerative medicine. Blood. 2012; 119:3900–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambers S.M., Goodell M.A.. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 2007; 3:201–211. [DOI] [PubMed] [Google Scholar]
- 32.Eaves C.J. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015; 125:2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Busch K., Klapproth K., Barile M., Flossdorf M., Holland-Letz T., Schlenner S.M., Reth M., Hofer T., Rodewald H.R.. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015; 518:542–546. [DOI] [PubMed] [Google Scholar]
- 34.Sanjuan-Pla A., Macaulay I.C., Jensen C.T., Woll P.S., Luis T.C., Mead A., Moore S., Carella C., Matsuoka S., Bouriez Jones T. et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013; 502:232–236. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto R., Morita Y., Ooehara J., Hamanaka S., Onodera M., Rudolph K.L., Ema H., Nakauchi H.. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013; 154:1112–1126. [DOI] [PubMed] [Google Scholar]
- 36.Shin J.Y., Hu W., Naramura M., Park C.Y.. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 2014; 211:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oguro H., Ding L., Morrison S.J.. SLAM Family Markers Resolve Functionally Distinct Subpopulations of Hematopoietic Stem Cells and Multipotent Progenitors. Cell Stem Cell. 2013; 13:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Streets A.M., Huang Y.. Chip in a lab: Microfluidics for next generation life science research. Biomicrofluidics. 2013; 7:11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kowalczyk M.S., Tirosh I., Heckl D., Rao T.N., Dixit A., Haas B.J., Schneider R.K., Wagers A.J., Ebert B.L., Regev A.. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015; 25:1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson N.K., Kent D.G., Buettner F., Shehata M., Macaulay I.C., Calero-Nieto F.J., Sanchez Castillo M., Oedekoven C.A., Diamanti E., Schulte R. et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell. 2015; 16:712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsang J.C., Yu Y., Burke S., Buettner F., Wang C., Kolodziejczyk A.A., Teichmann S.A., Lu L., Liu P.. Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol. 2015; 16:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paul F., Arkin Y., Giladi A., Jaitin D.A., Kenigsberg E., Keren-Shaul H., Winter D., Lara-Astiaso D., Gury M., Weiner A. et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015; 163:1663–1677. [DOI] [PubMed] [Google Scholar]
- 43.Klimmeck D., Cabezas-Wallscheid N., Reyes A., von Paleske L., Renders S., Hansson J., Krijgsveld J., Huber W., Trumpp A.. Transcriptome-wide profiling and posttranscriptional analysis of hematopoietic stem/progenitor cell differentiation toward myeloid commitment. Stem Cell Rep. 2014; 3:858–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye Z.J., Kluger Y., Lian Z., Weissman S.M.. Two types of precursor cells in a multipotential hematopoietic cell line. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18461–18466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahajan M.C., Karmakar S., Newburger P.E., Krause D.S., Weissman S.M.. Dynamics of alpha-globin locus chromatin structure and gene expression during erythroid differentiation of human CD34(+) cells in culture. Exp. Hematol. 2009; 37:1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan X., Durrett R.E., Zhu H., Tanaka Y., Li Y., Zi X., Marjani S.L., Euskirchen G., Ma C., Lamotte R.H. et al. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu A.R., Neff N.F., Kalisky T., Dalerba P., Treutlein B., Rothenberg M.E., Mburu F.M., Mantalas G.L., Sim S., Clarke M.F. et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods. 2014; 11:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cabezas-Wallscheid N., Klimmeck D., Hansson J., Lipka D.B., Reyes A., Wang Q., Weichenhan D., Lier A., von Paleske L., Renders S. et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014; 15:507–522. [DOI] [PubMed] [Google Scholar]
- 51.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Maaten L, Hinton G.. Visualizing high-dimensional data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 53.McKinney-Freeman S., Cahan P., Li H., Lacadie S.A., Huang H.T., Curran M., Loewer S., Naveiras O., Kathrein K.L., Konantz M. et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell. 2012; 11:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitfield M.L., Sherlock G., Saldanha A.J., Murray J.I., Ball C.A., Alexander K.E., Matese J.C., Perou C.M., Hurt M.M., Brown P.O. et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002; 13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pronk C.J., Rossi D.J., Mansson R., Attema J.L., Norddahl G.L., Chan C.K., Sigvardsson M., Weissman I.L., Bryder D.. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 2007; 1:428–442. [DOI] [PubMed] [Google Scholar]
- 56.Cho S., Spangrude G.J.. Enrichment of functionally distinct mouse hematopoietic progenitor cell populations using CD62L. J. Immunol. 2011; 187:5203–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Pel M., Hagoort H., Hamann J., Fibbe W.E.. CD97 is differentially expressed on murine hematopoietic stem-and progenitor-cells. Haematologica. 2008; 93:1137–1144. [DOI] [PubMed] [Google Scholar]
- 58.Daniel-Carmi V., Makovitzki-Avraham E., Reuven E.M., Goldstein I., Zilkha N., Rotter V., Tzehoval E., Eisenbach L.. The human 1-8D gene (IFITM2) is a novel p53 independent pro-apoptotic gene. Int. J. Cancer. 2009; 125:2810–2819. [DOI] [PubMed] [Google Scholar]
- 59.Gupta R.K., Tripathi R., Naidu B.J., Srinivas U.K., Shashidhara L.S.. Cell cycle regulation by the pro-apoptotic gene Scotin. Cell Cycle. 2008; 7:2401–2408. [DOI] [PubMed] [Google Scholar]
- 60.Oki T., Nishimura K., Kitaura J., Togami K., Maehara A., Izawa K., Sakaue-Sawano A., Niida A., Miyano S., Aburatani H. et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0-G1 transition. Sci. Rep. 2014; 4:4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilicic T., Kim J.K., Kolodziejczyk A.A., Bagger F.O., McCarthy D.J., Marioni J.C., Teichmann S.A.. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 2016; 17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grant G.D., Brooks L. 3rd, Zhang X., Mahoney J.M., Martyanov V., Wood T.A., Sherlock G., Cheng C., Whitfield M.L.. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell. 2013; 24:3634–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu M., Krause D., Greaves M., Sharkis S., Dexter M., Heyworth C., Enver T.. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997; 11:774–785. [DOI] [PubMed] [Google Scholar]
- 64.Teles J., Pina C., Eden P., Ohlsson M., Enver T., Peterson C.. Transcriptional regulation of lineage commitment–a stochastic model of cell fate decisions. PLoS Comput. Biol. 2013; 9:e1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma O., Hong S., Guo H., Ghiaur G., Friedman A.D.. Granulopoiesis requires increased C/EBPalpha compared to monopoiesis, correlated with elevated Cebpa in immature G-CSF receptor versus M-CSF receptor expressing cells. PLoS One. 2014; 9:e95784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lekstrom-Himes J.A. The role of C/EBP(epsilon) in the terminal stages of granulocyte differentiation. Stem Cells. 2001; 19:125–133. [DOI] [PubMed] [Google Scholar]
- 67.Feuerer M., Hill J.A., Mathis D., Benoist C.. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat. Immunol. 2009; 10:689–695. [DOI] [PubMed] [Google Scholar]
- 68.Woolthuis C.M., Park C.Y.. Hematopoietic stem/progenitor cell commitment to the megakaryocyte lineage. Blood. 2016; 127:1242–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yien Y.Y., Bieker J.J.. EKLF/KLF1, a tissue-restricted integrator of transcriptional control, chromatin remodeling, and lineage determination. Mol. Cell. Biol. 2013; 33:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tallack M.R., Perkins A.C.. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. IUBMB Life. 2010; 62:886–890. [DOI] [PubMed] [Google Scholar]
- 71.Vinjamur D.S., Alhashem Y.N., Mohamad S.F., Amin P., Williams D.C. Jr, Lloyd J.A.. Kruppel-like transcription factor KLF1 is required for optimal gamma- and beta-globin expression in human fetal erythroblasts. PLoS One. 2016; 11:e0146802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai S., Bartelmez S., Sitnicka E., Collins S.. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994; 8:2831–2841. [DOI] [PubMed] [Google Scholar]
- 73.Drissen R., von Lindern M., Kolbus A., Driegen S., Steinlein P., Beug H., Grosveld F., Philipsen S.. The erythroid phenotype of EKLF-null mice: defects in hemoglobin metabolism and membrane stability. Mol. Cell. Biol. 2005; 25:5205–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perkins A.C., Sharpe A.H., Orkin S.H.. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995; 375:318–322. [DOI] [PubMed] [Google Scholar]
- 75.Perkins A., Xu X., Higgs D.R., Patrinos G.P., Arnaud L., Bieker J.J., Philipsen S., Workgroup K.L.F.C.. Kruppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood. 2016; 127:1856–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xue L., Chen X., Chang Y., Bieker J.J.. Regulatory elements of the EKLF gene that direct erythroid cell-specific expression during mammalian development. Blood. 2004; 103:4078–4083. [DOI] [PubMed] [Google Scholar]
- 77.Lohmann F., Dangeti M., Soni S., Chen X., Planutis A., Baron M.H., Choi K., Bieker J.J.. The DEK oncoprotein is a critical component of the EKLF/KLF1 enhancer in erythroid cells. Mol. Cell. Biol. 2015; 35:3726–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bresnick E.H., Lee H.Y., Fujiwara T., Johnson K.D., Keles S.. GATA switches as developmental drivers. J. Biol. Chem. 2010; 285:31087–31093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dore L.C., Chlon T.M., Brown C.D., White K.P., Crispino J.D.. Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood. 2012; 119:3724–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lohmann F., Bieker J.J.. Activation of Eklf expression during hematopoiesis by Gata2 and Smad5 prior to erythroid commitment. Development. 2008; 135:2071–2082. [DOI] [PubMed] [Google Scholar]
- 81.Bauer D.E., Orkin S.H.. Hemoglobin switching's surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015; 33:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou D., Liu K., Sun C.W., Pawlik K.M., Townes T.M.. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat. Genet. 2010; 42:742–744. [DOI] [PubMed] [Google Scholar]
- 83.Challen G.A., Boles N., Lin K.K., Goodell M.A.. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009; 75:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chambers S.M., Boles N.C., Lin K.Y., Tierney M.P., Bowman T.V., Bradfute S.B., Chen A.J., Merchant A.A., Sirin O., Weksberg D.C. et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007; 1:578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gazit R., Garrison B.S., Rao T.N., Shay T., Costello J., Ericson J., Kim F., Collins J.J., Regev A., Wagers A.J. et al. Transcriptome analysis identifies regulators of hematopoietic stem and progenitor cells. Stem Cell Rep. 2013; 1:266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Terskikh A.V., Miyamoto T., Chang C., Diatchenko L., Weissman I.L.. Gene expression analysis of purified hematopoietic stem cells and committed progenitors. Blood. 2003; 102:94–101. [DOI] [PubMed] [Google Scholar]
- 87.Chen L., Kostadima M., Martens J.H., Canu G., Garcia S.P., Turro E., Downes K., Macaulay I.C., Bielczyk-Maczynska E., Coe S. et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014; 345:1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo G., Luc S., Marco E., Lin T.W., Peng C., Kerenyi M.A., Beyaz S., Kim W., Xu J., Das P.P. et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell. 2013; 13:492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han L., Zi X., Garmire L.X., Wu Y., Weissman S.M., Pan X., Fan R.. Co-detection and sequencing of genes and transcripts from the same single cells facilitated by a microfluidics platform. Sci Rep. 2014; 4:6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A.. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015; 33:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Macaulay I.C., Haerty W., Kumar P., Li Y.I., Hu T.X., Teng M.J., Goolam M., Saurat N., Coupland P., Shirley L.M. et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods. 2015; 12:519–522. [DOI] [PubMed] [Google Scholar]
- 92.Angermueller C., Clark S.J., Lee H.J., Macaulay I.C., Teng M.J., Hu T.X., Krueger F., Smallwood S.A., Ponting C.P., Voet T. et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods. 2016; 13:229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hou Y., Guo H., Cao C., Li X., Hu B., Zhu P., Wu X., Wen L., Tang F., Huang Y. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016; 26:304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pop R., Shearstone J.R., Shen Q., Liu Y., Hallstrom K., Koulnis M., Gribnau J., Socolovsky M.. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol. 2010; 8:e1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu L., Skoultchi A.I.. Coordinating cell proliferation and differentiation. Curr. Opin. Genet. Dev. 2001; 11:91–97. [DOI] [PubMed] [Google Scholar]
- 96.Miller J.P., Yeh N., Vidal A., Koff A.. Interweaving the cell cycle machinery with cell differentiation. Cell Cycle. 2007; 6:2932–2938. [DOI] [PubMed] [Google Scholar]
- 97.Wells A.D., Morawski P.A.. New roles for cyclin-dependent kinases in T cell biology: linking cell division and differentiation. Nat. Rev. Immunol. 2014; 14:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Furukawa Y., Kikuchi J., Nakamura M., Iwase S., Yamada H., Matsuda M.. Lineage-specific regulation of cell cycle control gene expression during haematopoietic cell differentiation. Br. J. Haematol. 2000; 110:663–673. [DOI] [PubMed] [Google Scholar]
- 99.Colvin G.A., Berz D., Liu L., Dooner M.S., Dooner G., Pascual S., Chung S., Sui Y., Quesenberry P.J.. Heterogeneity of non-cycling and cycling synchronized murine hematopoietic stem/progenitor cells. J. Cell. Physiol. 2010; 222:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pauklin S., Vallier L.. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013; 155:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li V.C., Kirschner M.W.. Molecular ties between the cell cycle and differentiation in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9503–9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Colvin G.A., Dooner M.S., Dooner G.J., Sanchez-Guijo F.M., Demers D.A., Abedi M., Ramanathan M., Chung S., Pascual S., Quesenberry P.J.. Stem cell continuum: directed differentiation hotspots. Exp. Hematol. 2007; 35:96–107. [DOI] [PubMed] [Google Scholar]
- 103.Kadauke S., Udugama M.I., Pawlicki J.M., Achtman J.C., Jain D.P., Cheng Y., Hardison R.C., Blobel G.A.. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012; 150:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wohrer S., Knapp D.J., Copley M.R., Benz C., Kent D.G., Rowe K., Babovic S., Mader H., Oostendorp R.A., Eaves C.J.. Distinct stromal cell factor combinations can separately control hematopoietic stem cell survival, proliferation, and self-renewal. Cell Rep. 2014; 7:1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aloia L., Gutierrez A., Caballero J.M., Di Croce L.. Direct interaction between Id1 and Zrf1 controls neural differentiation of embryonic stem cells. EMBO Rep. 2015; 16:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang N., Yantiss R.K., Nam H.S., Chin Y., Zhou X.K., Scherl E.J., Bosworth B.P., Subbaramaiah K., Dannenberg A.J., Benezra R.. ID1 is a functional marker for intestinal stem and progenitor cells required for normal response to injury. Stem Cell Rep. 2014; 3:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jankovic V., Ciarrocchi A., Boccuni P., DeBlasio T., Benezra R., Nimer S.D.. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perry S.S., Zhao Y., Nie L., Cochrane S.W., Huang Z., Sun X.H.. Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood. 2007; 110:2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu Z.H., Dai X.M., Du B.. Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 2015; 16:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guiu J., Shimizu R., D'Altri T., Fraser S.T., Hatakeyama J., Bresnick E.H., Kageyama R., Dzierzak E., Yamamoto M., Espinosa L. et al. Hes repressors are essential regulators of hematopoietic stem cell development downstream of Notch signaling. J. Exp. Med. 2013; 210:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reichert D., Scheinpflug J., Karbanova J., Freund D., Bornhauser M., Corbeil D.. Tunneling nanotubes mediate the transfer of stem cell marker CD133 between hematopoietic progenitor cells. Exp. Hematol. 2016; 44, doi:10.1016/j.exphem.2016.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.