

Abstract
miRNA plays an important role in tumourgenesis by regulating expression of oncogenes and tumour suppressors. Thus affects cell proliferation and differentiation, apoptosis, invasion and angiogenesis. miRNAs are potential biomarkers for diagnosis, prognosis and therapies of different forms of cancer. However, relationship between response of cancer patients towards targeted therapy and the resulting modifications of the miRNA transcriptome in the context of pathway regulation is poorly understood. With ever-increasing pathways and miRNA-mRNA interaction databases, freely available mRNA and miRNA expression data in multiple cancer therapy have produced an unprecedented opportunity to decipher the role of miRNAs in early prediction of therapeutic efficacy in diseases. Efficient translation of -omics data and accumulated knowledge to clinical decision-making are of paramount scientific and public health interest. Well-structured translational algorithms are needed to bridge the gap from databases to decisions. Herein, we present a novel SMARTmiR algorithm to prospectively predict the role of miRNA as therapeutic biomarker for an anti-EGFR monoclonal antibody i.e. cetuximab treatment in colorectal cancer.
Colorectal cancer (CRC) is one of the most prevalent cancers, with 1.2 million new cases every year. It is the second most commonly diagnosed cancer in females and the third most common in males; the highest incidence occurs in developed countries. By the age of seventy years, one out of every two citizens in the Western world develops benign adenomas that evolve into malignant carcinomas at an estimated yearly rate of 0.1 to 0.25%1. In United States alone, the annual cost of CRC treatment is forecasted to reach $17.7 billion by 2020. However, using simultaneous strategies that reduce risk factors, increasing screening and treatment could avoid 101,353 deaths resulting in $33.9 billion in savings in reduced productivity loss2.
EGFR, a transmembrane receptor tyrosine kinase has been identified as one of the most promising targets for treating metastatic colorectal cancer (mCRC). Among the 20 molecules listed by the National Cancer Institute for the treatment of mCRC (http://www.cancer.gov/cancertopics/treatment/colon-and-rectal), cetuximab is one of the most successful monoclonal antibodies (http://www.croh-online.com/article/S1040-8428(13)00102-9/fulltext)3,4. However, multiple lines of evidence suggest that only 10–20% patients with mCRC benefit from cetuximab treatment4,5. The selective efficacy, side effects and high treatment costs of cetuximab result in the need for focused research to decipher the resistance mechanisms to cetuximab. The response of the scientific community to this need is evident through the increased no. of publications suggesting that the mutational status of KRAS, BRAF and PIK3CA, differential expression of PTEN, EGFR ligand (AREG, EREG) and EGFR gene copy number variation could serve as therapeutic biomarkers for anti-EGFR monoclonal antibody treatment in CRC6,7,8,9,10,11. However, none of these singular molecular changes could accurately predict the response of CRC patients to cetuximab therapy. Because CRC is a systems-level disorder that involves multiple molecular mechanisms to support proliferative signalling, resist cell death, induce angiogenesis and metastasis; molecules such as miRNAs that regulate signalling pathways by affecting the expression of multiple proteins might serve as more potent therapeutic biomarkers12,13. However, little is known regarding the role of miRNAs as a therapeutic biomarker for cetuximab treatment in CRC.
miRNAs are small (18–25 nucletide long) non-coding, highly conserved fragments of RNA. They post-transcriptionally regulate gene expression by complementary binding to the untranslated region of mRNA14,15. As of November 2014; 1881 primary miRNA transcripts in the human genome has been annotated in the miRBase database (http://www.mirbase.org). miRNAs are transcribed from distant regions of genome previously annotated as protein coding part and have an independent transcription machinery14. The primary miRNA transcripts i.e. pri-miRNA are processed inside nucleous by Drosha complex. Resulting precursor miRNA (pre-miRNA), a ~60–70 nucletide long hairpin structure is transported to cytoplasam where it further processed by Dicer. Successively mature miRNA formation and targeting of mRNA is achieved through RICS assembly14.
miRNA expression is typically dysregulated in cancer cells and this dysregulation has a high degree of tissue specificity, miRNAs could be used as diagnostic and therapy-related biomarkers. Additionally, miRNAs have an unusually high stability in formalin-fixed tissues, from which they could be extracted with minimal degradation16. Moreover, the techniques of miRNA analysis from a single cell are established, allowing for the analysis of small amounts of miRNAs with increasing sensitivity for potential biomarker assays17. The roles of miRNAs in CRC, EGFR signalling regulation and cetuximab treatment outcome are also evident. Multiple reports regarding the miRNA dysregulation in metastatic colorectal cancer have been published. Some groups reported that major colorectal cancer biomarkers such as EGFR and RAS are regulated by mir-7 and let-7 respectively, thus profoundly affecting downstream signalling18,19,20,21,22. Recently, Bissonnette et al. demonstrated the correlation between EGFR signalling and miR-143, mir-145 in murine colon cancer models23. Another group discovered that cetuximab-mediated EGFR inhibition abrogates the age-related increase of miR21, which is related to age-dependent colorectal cancer24.
These findings cumulatively suggest a potentially significant role of miRNAs in EGFR signalling in CRC. Therefore, these evidences compelled us to create a workflow to identify the most critical miRNAs important for cetuximab resistance in CRC that could be further used for potential biomarker assay development. In this study, we propose ranked miRNA candidates that might contribute to cetuximab resistance in CRC patients. The inference is based on the integration of multiple published and predicted miRNA findings into cellular pathways that lead to oncogenesis and metastasis. We prioritised the biomarker candidates based on a novel algorithm, i.e., SMARTmiR (Scoring-based MARking of Therapeutic MIcroRna), that combines the network parameters and literature-derived evidence. Finally, the significance of our prediction was strongly supported by recently published experimental data that are derived from cetuximab resistant CRC patients. This study provides an actionable insight into the novel mechanism of cetuximab resistance mediated by miRNAs that might lead to identification of miRNAs as biomarkers, thereby predicting optimum responses to the drug.
Results
Construction of molecular pathway maps crucial for cetuximab mode of action in CRC
The efficacy of cetuximab trarement in responsive colorectal cancer is mostly depend on the state of four cellular processes namely apoptosis, proliferation and diffrentiation, angiogenesis, epithelial-to-mesenchymal transition/metastasis25. First a comprehensive framework for the analysis and a structured overview of the bio molecular space for the mechanisms of action of cetuximab therapy in colorectal cancer was created by assembling four pathway maps which are representative of those cellular processes in CRC.
The landscapes of the calculated network parameters for each of the four constructed pathways are presented in Figure 1. The top three nodes of each pathway (Jak2, Akt, p53 in apoptosis; SMAD, TGF-β, Shc in metastasis; VEGFR, VEGF, Src in Angiogenesis; EGFR, c-Fos, c-Raf in proliferation and diffrentiation) are well-known players in the corresponding processes12,25,26,27. The high no. of the nodes representing receptors and kinases in all four pathway maps emphasise the crucial role of these molecules in oncogenesis and metastasis. Our maps also demonstrate that the roles of some molecules, such as transcription factors, in metastasis are better understood and more well known compared with the other three processes.
Figure 1. Landscape of the four cellular processes in terms of the node degree, betweenness centrality, functional category of nodes and edges.

Node degree: no of edges connected a node; Betweenness centrality: the no. of shortest paths from all of the vertices to all of the others that pass through that node.
To understand the degree of the cross-talk and the overlap between the four pathway maps, we integrated all four pathway maps into a resulting integrated map. Table 1 presents a statistical overview of the pathway maps and the percentage of literature validation of the nodes related to the corresponding cellular processes. A list of common nodes in all four pathways are provided in Supplementary Table S1.
Table 1. Statistical overview of the assembled pathway maps representing four cellular processes.
| Cellular Processes | No. of Nodes | No. of Edges | Percentage of published relationships of nodes to the cellular processes |
|---|---|---|---|
| Apoptosis | 179 | 305 | 55.3% (99/179) |
| Proliferation and Differentiation | 151 | 261 | 45% (68/151) |
| Angiogenesis | 201 | 289 | 36.8% (74/201) |
| Metastasis | 245 | 471 | 46% (113/245) |
The high percentage of literature validations (4th column of Table 1) confirms that the constructed pathways represent a good reflection of the current knowledge accumulated in the scientific literature. The metastasis pathway map is the largest in terms of the no. of nodes and edges, followed by angiogenesis, apoptosis, proliferation and diffrentiation. The integrated map consists of 465 nodes and 792 directed edges, demonstrating a large overlap among the molecules involved in the four processes. The literature validations of the node's relationships to the cellular processes are provided in Supplementary Table S2.
To analyse the CRC specificity of the assembled pathway maps, we first explored the published TCGA (The Cancer Genome Atlas) data regarding the differential expression of the nodes' mRNAs in colon and rectum adenocarcinoma28. This analysis demonstrated that 96% of the nodes' mRNAs are differentially expressed (more than two standard deviations from the mean) in at least one of the 244 studied tumour samples. In addition, we examined the association of the pathway maps' nodes to colorectal cancer in the published scientific domain and determined that 45.3% of them (211/465) have been reported in the literature to have a relationship with CRC. All of those relationships are included in Supplementary Table S3. The differential expression of each mRNA in the TCGA tumour samples is provided in Supplementary Table S4.
miRNAome screening for putative candidate biomarker
Upon construction of comprehensive pathway maps and verification of their association with the four cellular processes and in CRC, we screened the published miRNAome for miRNAs targeting at least one node of the four pathway maps, which could serve as potential biomarkers for further analysis. In our screening, we considered experimentally validated miRNA target interactions (MTIs) and computationally predicted MTIs. To increase the chance of the predicted MTIs being biologically relevant, overlapping MTIs between three different miRNA-target prediction algorithms29,30 were used. One method that can be used to compare the quality of MTI prediction is to calculate the percentage of the prediction that has already been experimentally validated. In that direction, the percentage of experimentally validated MTIs is calculated for predictions from three individual algorithms, i.e., Pictar, miRanda, DianaMicroT, and from the intersection of their predictions. Experimentally validated MTIs are obtained from TarBase and Pathway Studio. As demonstrated in Figure 2, the percentage obtained from intersection of the three prediction algorithms is higher than that obtained using any of the three prediction algorithms used individually (Figure 2). Using the intersection of three prediction algorithms, we are able to capture 17.5% of the experimentally validated MTIs (average for four processes). However, the results for each of the prediction algorithms are less impressive: Pictar (3.9%), miRanda (3.4%), DianaMicroT (1.7%). Therefore, the assumption of using the intersection of the three prediction algorithms proved to be a better approach and was used for further analysis. Total number of experimentally validated MTIs for all prediction softwares and their intersect for all four pathway maps are provided in Supplementary Table S5.
Figure 2. Comparative performance of Pictar, miRanda, DianaMicroT and the intersection of the three algorithms in capturing validated miRNA-target interactions.

The miRNAome screening revealed 335 miRNAs that target at least one node of the four assembled pathway maps. We further analysed the 335 miRNAs based on their ability to participate in all four processes, thus having a higher probability to be therapeutic biomarkers for cetuximab treatment in CRC patients. This analysis resulted in the selection of 188 miRNAs that interact with targets in all four of the pathway maps, and these selected miRNAs were used for the ranking and validation (Figure 3).
Figure 3. Quantities of miRNA species targeting each pathway and cross-sections.

To evaluate the relationship of those 188 miRNAs to the four cellular processes and to the neoplasm, we performed a random sampling approach. We randomly selected 20 non-overlapping sets containing 5 miRNAs each (S1, S2,.., S20 in Figure 4). Next the association between each miRNA (from each of those 20 samples) to four cellular processes (apoptosis, proliferation and differentiation, metastasis, angiogenesis) and to neoplasms were searched in the scientific literature. The number of miRNAs from each sample that are published to regulate at least one of those four cellular process and neoplasms are summarised in Figure 4. It is evident from Figure 4 that 74% of the miRNA candidates are known to regulate at least one of the four cellular processes and 51% of those miRNA are linked to different forms of neoplasms.
Figure 4. The relationships of miRNAs from twenty randomly collected non-overlapping samples (five miRNAs each) to cell processes (angiogenesis, apoptosis, proliferation and differentiation, metastasis) and neoplasms.

The column S1, S2, S3, ……, S20; denote the 20 non overlapping samples having 5 miRNA in each of them. Each sample collected randomly from total 188 miRNA. The corresponding value in the shell for each sample with row starts with “Cell process” denote total no. of miRNA from that sample is linked to any of the four cellular processes and the relation is published. Similarly the corresponding value in the shell for each sample with row starts with “Neoplasm” denote total no. of miRNA from that sample is linked to Neoplasms and the relation is published.
All 20 of the samples and the relationship between the miRNAs from each sample to cellular process and the neoplasm are provided in Supplementary Table S6, Supplementary Table S7 and Supplementary Table S8. Based on the node degree, the top 5 miRNAs from each of the four pathway maps are provided in Supplementary Fig S1.
Prioritisation of the selected miRNAs
Despite the potentially strong impact of the selected miRNAs on the fundamental molecular mechanisms underlying CRC, their relation to cetuximab treatment was not used in previous filtering of the 188 miRNAs, and the resulting candidate list remains too long for experimental validation. To further prioritise miRNA candidates that have the highest likelihood of acting as therapeutic biomarkers for cetuximab treatment, we ranked the miRNA candidates by applying a newly designed SMARTmiR algorithm, as described in the Materials and Methods section. The details of the 10 top-ranked miRNAs, including their scores and evidence of their interaction with cetuximab, and the role of the targets are summarised in Table 2. According to our predictions, those ten miRNAs might serve as the best candidates for therapeutic biomarkers for cetuximab treatment in CRC patients. All 188 miRNA with its SMARTmiR sore and Higo gene id of the targets are provided in Supplementary Fig S2.
Table 2. Top 10 miRNAs along with their scores, expression values, MTI, expression of MTI and miRNA in cetuximab sensitive to resistant CRC patients.
| miRNA | Score | No of Targets | Hugo Gene Id of differentially expressed miRNA Targets in Cetuximab sensitive to resistant CRC patients | No of TF, KNS | miRNA Fold Change |
|---|---|---|---|---|---|
| hsa-miR-21 | 3.57 | 33 | NOTCH1 (0.004868), CD47 (0.031247), BRCA1 (0.013018), APAF1 (0.012285) | 4, 3 | 8.1 |
| hsa-miR-34a | 2.21 | 28 | YY1 (0.029778), NOTCH1 (0.004868), CD47 (0.031247), BRCA1 (0.013018), MYC (0.002497) | 4, 2 | 4.6 |
| hsa-miR-145 | 2.15 | 33 | MKL2 (0.004586), CD47 (0.031247), MYC (0.002497) | 7, 2 | N/A |
| hsa-miR-27a | 2.01 | 48 | MAPK14 (0.009214), APAF1 (0.012285) | 4, 9 | 5.8 |
| hsa-miR-17 | 1.85 | 35 | MAPK14 (0.009214), BRCA1 (0.013018), E2F1 (0.002186), EREG (3.41e-06) | 3, 8 | 9.7 |
| hsa-miR-155 | 1.73 | 29 | CD47 (0.031247), FOXO3 (0.012217) | 6, 4 | N/A |
| hsa-miR-182 | 1.65 | 39 | MKL2 (0.004586), CD47 (0.031247), FOXO3 (0.012217) | 5, 2 | N/A |
| hsa-miR-15a | 1.59 | 25 | BRCA1 (0.013018), PRKAR2A (0.021049) | 1, 4 | 6.2 |
| hsa-miR-96 | 1.53 | 38 | PLCB4 (0.015726), FOXO3 (0.012217) | 4, 5 | N/A |
| hsa-miR-106a | 1.48 | 26 | NOTCH1 (0.004868), BRCA1 (0.013018), E2F1 (0.002186) | 2, 6 | N/A |
1st column: top 10 miRNA candidates; 2nd column: calculated score based on developed scoring function; 3rd column: total no of miRNA targets in the “Integrated map”; 4th column: Hugo gene ID of miRNA targets those were differentially expressed in cetuximab sensitive compared with resistant CRC patients with significant p values; 5th column: no of transcription factors (TFs) and kinases (KNSs) among the targets; 6th column: expression value of the miRNA in cetuximab sensitive compared with resistant CRC patients (KRAS, BRAF wild type) published by Mosakhani et al31.
Validation of the prediction based on published experimental results
The optimal method of validating any systems biology prediction is through experimental results. In our case, the analysis of differentially expressed miRNAs in cetuximab-resistant CRC tumour samples would provide such validation. Recently, a group at the University of Helsinki studied differential miRNA expression patterns of 33 cetuximab-treated patients with metastatic colorectal cancer31. That group tested the association of each miRNA with the overall survival (OS) by applying a Cox proportional hazards regression model, and published a list of the 60 most differentially expressed miRNAs in patients with an extremely poor prognosis (resistant patients). According to our analysis, 85% (51 of 60 miRNAs) of the resistant patient-derived differentially expressed miRNAs are present in our list of the selected 188 miRNAs. Moreover, five of the ten top-ranked predicted miRNAs were found to be highly differentially expressed in resistant patients, exhibiting 4.6- to 9.7-fold changes (Table 2). More studies with greater no. of patients are needed to further validate our prediction; however, we consider this initial evidence to be very encouraging and to prove the applicability of our methodology.
Discussion
In colorectal cancer, the effects of cetuximab are mediated through various molecular pathways, including the Ras-Raf-MAPK, PI3K-AKT, protein kinase C, STAT and SRC pathways25,32. Though the efficacy of cetuximab in responsive CRC patients is mainly manifested through reduction of cell proliferation and diffrentiation, inhibition of angiogenesis, prevention of epithelial to mesenchymal transition (metastasis) and induction of apoptosis33. However deciphering the cetuximab resistance mechanisms a number of alternative molecular pathways were recently reviewed33. The reactivation of pro-angiogenic factors (pMAPK, VEGF) leading to increased angiogenesis in CRC is one such resistant mechanism34. In agreement with those results, our top-ranked miRNA, i.e., miR-21, is a well-known angiogenesis regulator in both in vitro and in vivo models35. Moreover, cetuximab treatment affects the expression of miR-21 in vitro36. Another group suggested that the cetuximab resistance mechanism is a phenomenon caused by an increased rate of EGFR degradation and internalisation; switching towards alternate pathways for growth and survival of CRC resistant tumour cells37. In resistant cells, EGFR is localised in the sub-cellular compartments i.e., endosome, mitochondria and nucleus. The overexpression of nuclear EGFR is linked to SFKs (SRC-family kinase) expression, modulating the up regulation of the PI3K/AKT pathway in cetuximab resistance38. Mutations of KRAS are also connected to the increased activation of SFKs, affecting the MAPK, beta-catenin, STAT and PI3K/AKT pathways in CRC resistant tumours39. However, the mechanism of cetuximab resistance in CRC remain poorly understood.
To address the issue, we attempted to create a comprehensive CRC specific molecular network snapshot leading to the four cellular processes of apoptosis, proliferation and diffrentiation, metastasis and angiogenesis, which are crucial for mode of action of Cetuximab therapy in CRC. Pathways from Metacore were integrated. The Metacore pathway knowledge base was selected because of the high experimental validation of molecular interactions in its pathways40. Of the assembled Integrated map, 96% are differentially expressed in published CRC specific RNASeq data28. Based on the well-accepted node removal algorithm41,42,43, that high percentage makes the Integrated map CRC specific. miRNA-target interactions have been screened from prediction algorithms (miRanda, Pictar and DianaMicroT) and literature based knowledgebase (Tarbase, Pathway Studio). TargetScanS, Pictar and miRanda used individually, or in combination, provide a good balance between precision and recall44. However, Maragkakis et al. demonstrated that Pictar predictions overlap more than 75% of the predictions obtained from TargetsScanS32. Therefore, Pictar, miRanda and DianaMicroT were used. Next, a candidate miRNA was ranked based on its relationship to the four cellular processes and to CRC. The other ranking parameters are a miRNA target's relation to cetuximab; the node degree of the target; and the no. of targets for each candidate miRNA. A high no. of targets of miRNA in a pathway suggests the multi-level regulation of that pathway45,46. miRNAs also preferentially regulate network hubs that participate in complex dynamic processes, and their expression profile is highly dynamic, thereby requiring tighter regulatory control47,48.
miR-21 is the top-ranked candidate miRNA (Table 2) according to the scoring function. Although no relation between miRNA to cetuximab was incorporated into the scoring function, a literature search confirmed that miR-21 is a well-known biomarker for cetuximab therapy36. Our methodology has successfully predicted possible relationships between a miRNA and cetuximab; in the case of a top-ranked miRNA, i.e., miR-21, the relationship has previously been experimentally validated. This result again demonstrates the novelty and applicability of our methodology.
Genome-wide miRNA and mRNA expression profiles of cetuximab-sensitive and cetuximab-resistant mCRC patients and PLS regression/Pearson's correlation of significantly differentially expressed miRNAs and target mRNAs followed by pathway-centric interpretation could be another approach to discover the role of miRNAs in cetuximab resistance mechanisms in mCRC. However, due to a lack of such experimental data, the SMARTmiR algorithm utilised existing resources to predict crucial miRNAs as therapeutic biomarkers for cetuximab treatment in CRC patients.
The accurate prediction of miRNAs that might serve as potential therapeutic biomarkers would be of great importantance for patients and for the pharmaceutical industry. Herein, we developed a novel algorithm that facilitates the prediction of potential miRNA biomarkers based on the knowledge accumulated in the public domain. To our knowledge, there is only one published alternative methodology that uses literature-based evidence for drug-associated miRNA predictions. Recently, Rukov et al. launched PharmacomiR, a miRNA Pharmacogenomics database that uses the triplet sets consisting of a miRNA, a target gene and a drug associated with the gene to predict miRNAs that could serve as potential therapeutic biomarkers13. We compared the performance of PharmacomiR to that of our methodology in predicting the pharmacogenomics role of miRNAs in cetuximab treatment. PharmacoMIR predicted 1102 unique miRNAs (6975 redundant miRNAs as the initial output). Next, we calculated the overlap between the no. of predicted miRNAs with the published differentially upregulated miRNAs by Mosakhani et al.31 in cetuximab-sensitive and cetuximab-resistant CRC patients. Clearly, our methodology has a higher prediction accuracy, with 27.1% (51 of 188) compared with 4.44% (49 of 1102) for PharmacomiR, in identifying experimentally validated potential miRNAs as therapeutic biomarkers for cetuximab therapy in CRC patients. Unlike PharmacomiR, SMARTmiR could also rank each candidate miRNA based on a novel disease specific score, making our methodology more advanced in prioritising candidate miRNAs for further experimental validation. A methodological comparision SMARTmiR and PharmacomiR in Supplementary Table S9.
Conclusion
This novel knowledge-based modelling and mining algorithm, i.e., SMARTmiR, has provided a pathway-centric methodology that facilitates the prediction of pharmacogenomic role of miRNA. However, this algorithm has limitations. The inherent issue of pathway map building is that its completeness is limited to valid and available data. Therefore, the sensitivity and specificity of the translational methodology is a function of completeness of the presented interactome. Additionally, the developed pathway maps could not integrate the dynamic nature of miRNA regulations. Annotations and encoding of the transcripts of the genes in the pathway maps are absent. However, these are outstanding challenges in representing dynamic biological systems, and the scientific community must act to solve these issues. The method is generic and can be applied to model the role of miRNA as a therapeutic biomarker for targeted therapy in other disease indications. The authors are optimistic that the application of an optimised and fully automated version of the algorithm has the potential to be used as clinical decision support tool. Moreover, this research will also provide a valuable background for colorectal cancer investigators to identify critical miRNA regulatory elements for better understanding regulatory mechanisms or designing future experimental studies in colorectal cancer.
Methods
Herein, we propose a predictive algorithm, i.e., SMARTmiR, that combines knowledge and data-driven approaches to identify miRNAs contributing to the therapeutic effects of cetuximab in CRC patients. The algorithm consists of the following four steps (Figure 5):
Figure 5. SMARTmiR workflow for the selection of miRNAs as candidate biomarkers conferring cetuximab resistance in colorectal cancer.

Step 1: Construction of pathway maps leading to oncogenesis and metastasis in CRC.
Step 2: Identification of miRNA candidate biomarkers via miRNAome screening.
Step 3: Ranking of miRNAs based on accumulated evidence and the effects on the cellular process of CRC patients treated with cetuximab.
Step 4: Validation of the prediction based on experimental data.
Construction of molecular pathway maps leading to CRC oncogenesis and metastasis
Following the first step in the workflow as illustrated in Figure 5, we created a comprehensive bio molecular space for the mechanisms of action of cetuximab therapy in colorectal cancer. In doing so we assembled four pathway maps linked to four fundamental cellular processes in oncogenesis and metastasis; namely apoptosis, proliferation and diffrentiation, angiogenesis, and metastasis. These pathway maps integrate known pathways from Metacore (Thomson Reuters, New York, USA) that lead to the four cellular processes and protein-protein interactions from IntAct, BioGRID and HPRD (Human Protein Reference Database) Databases49,50. The pathways that were integrated to build the four pathway maps leading to the cellular processes are listed in Table 3. All of the molecules in the maps (nodes) are annotated with the Entrez gene ID, HGNC gene symbols and corresponding UniProt IDs. The distinct features of the pathways are as follows: (A) Node shapes and colours correspond to their functional category (e.g., receptor, ligand, transcription factor, kinase); (B) All of the edges have directionality; (C) The edges are differentiated by shapes and colour corresponding to the type of interaction (such as binding, catalysis, phosphorylation, transcription regulation, transformation etc.); (D) Reactions (edges) are tagged to PubMed IDs as evidence. Four pathway maps corresponding to proliferation and differentiation, apoptosis, angiogenesis, and metastasis are available in standardised SBML (Systems Biology Markup Language)51 for file exchange between different tools. The four pathways are provided as four Supplementary Pathway maps (i.e., Apoptosis.xml, Proliferation_Diffrentiation.xml, Angiogenesis.xml and Metastasis.xml). The Pubmed IDs supporting an edge can be found by selecting respective edge in the pathway map specific xml files after opening those in Cytoscape.
Table 3. Detailed listing of the pathways used for assembling the proliferation and differentiation, apoptosis, angiogenesis, and metastasis processes.
| Cellular processes | Pathways assembled |
|---|---|
| Proliferation and Differentiation | EGFR signalling pathway, EGFR signalling via small GTPases, EGFR signalling via PIP3, EPO-induced MAPK pathway, ERK5 in cell proliferation, PDGF signalling via MAPK cascades, PDGF signalling via STATs and NF-kB |
| Apoptosis | Apoptosis and survival Anti-apoptotic TNFs/NF-kB/Bcl-2 pathway, Apoptosis and survival_Anti-apoptotic TNFs/NF-kB/IAP pathway, Apoptosis and survival p53-dependent apoptosis, EPO-induced Jak-STAT pathway, EPO-induced PI3K, AKT pathway and Ca(2+) influx, Signal transduction of AKT signalling, Signal transduction of PTEN pathway |
| Angiogenesis | VEGF-family signalling and activation, Angiopoietin - Tie2 signalling, VEGF signalling via VEGFR2 - generic cascades, S1P1 signalling pathway Development Role of IL-8 in angiogenesis, Thrombospondin-1 signalling |
| Metastasis | TGF-beta receptor signalling, TGF-beta-dependent induction of EMT via MAPK, Regulation of epithelial-to-mesenchymal transition (EMT), Immune response of Oncostatin M signalling via JAK-Stat in human cells FGF2-dependent induction of EMT, TGF-beta-induction of EMT via ROS, NOTCH-induced EMT, HGF-dependent inhibition of TGF-beta-induced EMT, TGF-beta- dependent induction of EMT via RhoA, PI3K and ILK, Immune response of Oncostatin M signalling via MAPK in human cells, HGF signalling pathway |
TCGA RNA Seq data in colon and rectal adenocarcinoma (2012) has been analysed to demonstrate the CRC specificity of the constructed maps28.
Identification of miRNAs candidate biomarkers via miRNAome screening
In the second step, we identified miRNA-target interactions (MTIs) for all of the nodes in the four constructed pathway maps. The exhaustive search for these miRNAs included both experimentally validated miRNAs and predicted miRNAs.
Experimentally validated and literature reported miRNAs
Through screening public and commercial sources for experimentally validated MTIs, we identified two major resources that are widely used because of their extensive coverage and the quality of scientific evidence, i.e., TarBase and Pathway Studio (Elsevier, Amsterdam, Netherlands)52. TarBase hosts manually curated MTIs that are experimentally validated53. It also incorporates entries from other well-known databases, such as miRecords54, miRTarBase55 and miR2Disease56. All of the literature-derived MTIs that were extracted for our study from the two sources described above are included in Supplementary Table S10.
Predicted miRNAs
Several computer-aided algorithms are available for the identification of MTIs. We used Diana-Micro T 3.0, Pictar and TargetScanS to identify predicted MTIs for the four developed pathways, focusing in particular on their overlapping predictions. These identified miRNAs have not been encoded into the pathway maps and are used to further filter and rank potential candidate miRNAs (described below).
Ranking of miRNAs based on accumulated evidence and their effect on the system
A novel raking formula was developed to rank the miRNAs for their potential to serve as therapeutic biomarkers for cetuximab treatment in CRC patients (Equation 1). To enhance the accuracy of the ranking function, the four assembled processes were integrated in one Integrated map, representing a comprehensive knowledge space of functional molecular networks that lead to the cellular process those are mostly dysregulated in colorectal cancer. Each miRNA is ranked based on its topological properties, network properties of its targets and based on literature-derived evidence of miRNA regulating signalling pathways that are important for colorectal carcinogenesis, miRNA target's (i.e., nodes in the Integrated map) relations to cetuximab. All of the literature-derived evidence for the candidate miRNA's relationships to cellular processes, CRC and the target's relations to cetuximab are provided in Supplementary Table S11, Supplementary Table S12 and Supplementary Table S13; respectively. The network parameters (Betweeness centrality, Node degree) were calculated using the CentiScaPe plugin57.
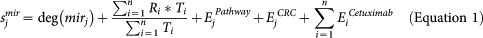 |
Sjmir is the score of jth miRNA. Each feature is given equal weightage due its natural importance and is normalised between 0 and 1 such that the calculated score could have a maximum value of 5. The overall scoring function was implemented in the Perl programming language.
The first feature of miRNA ranking is the node degree of the jth miRNA, i.e., the no. of targets of the jth miRNA in the Integrated map, as defined as the deg(mirj). The node degree corresponds to the no. of nodes adjacent to a given node v. The degree allows for an immediate evaluation of the regulatory relevance of the node. For example, in signalling networks, proteins with a very high degree are interacting with several other signalling proteins and are likely to be regulatory hubs. The second parameter was calculated based on the weighted node degree of all of the targets of jth miRNA, depicted as 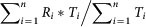 , where Ti is the node degree, and Ri is rank of ith target based on node degree, targeted by jth miRNA. Additionally, betweenness is calculated considering couples of nodes (v1, v2) and by counting the no. of shortest paths linking v1 and v2 and passing through a node n. The betweenness of a node in a biological network, such as a protein-signalling network, could indicate the relevance of a protein as functionally capable of holding together communicating proteins. Betweeness centrality was applied to identify the importance of a node in each of the four pathways (see the Results section). The additional features of SMARTmiR are literature-derived evidence reflecting the direct effect of each miRNA candidate in cancer aetiology, progression, spread and miRNA specific relationship to colorectal cancer. This third feature calculates the total amount of literature evidence (PubMed IDs) linking the jth miRNA to the four cellular processes relevant to tumour progression (proliferation and diffrentiation, apoptosis, metastasis, and angiogenesis), depicted as
, where Ti is the node degree, and Ri is rank of ith target based on node degree, targeted by jth miRNA. Additionally, betweenness is calculated considering couples of nodes (v1, v2) and by counting the no. of shortest paths linking v1 and v2 and passing through a node n. The betweenness of a node in a biological network, such as a protein-signalling network, could indicate the relevance of a protein as functionally capable of holding together communicating proteins. Betweeness centrality was applied to identify the importance of a node in each of the four pathways (see the Results section). The additional features of SMARTmiR are literature-derived evidence reflecting the direct effect of each miRNA candidate in cancer aetiology, progression, spread and miRNA specific relationship to colorectal cancer. This third feature calculates the total amount of literature evidence (PubMed IDs) linking the jth miRNA to the four cellular processes relevant to tumour progression (proliferation and diffrentiation, apoptosis, metastasis, and angiogenesis), depicted as 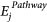 . Similarly, the fourth feature calculates the total amount of literature evidence (PubMed IDs) that reflects the association of the jth miRNA to colorectal cancer, depicted as
. Similarly, the fourth feature calculates the total amount of literature evidence (PubMed IDs) that reflects the association of the jth miRNA to colorectal cancer, depicted as 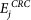 .
.
The ultimate goal of SMARTmiR is to prioritise the role of a candidate miRNA as a therapeutic biomarker for cetuximab treatment in CRC. Therefore, the last feature, 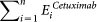 calculates the total amount of literature evidence (PubMed IDs) of the jth miRNA target's (ith) related to cetuximab.
calculates the total amount of literature evidence (PubMed IDs) of the jth miRNA target's (ith) related to cetuximab.
Seperately we have also calculated the miRNA scoring with betweeness centrality of the nodes as second feature; maintaing first, third, fourth and fifth features of SMARTmiR identical. The resulting scoring is provided in Supplementary Table S14.
Validation of predicted miRNA biomarkers
We used a list of differentially expressed miRNAs in cetuximab sensitive and resistant CRC patients (KRAS and VRAF wild type)31 to validate the miRNAs predicted and ranked by our workflow as significant therapeutic biomarker candidates for cetuximab treatment. To further substantiate the expression of targeted mRNAs by the most significant miRNA biomarkers, we also analysed the mRNA expression data58. The mRNA expression data analysis was performed using the appropriate R software packages. In particular, we used the MAS5 normalisation method59 and applied the SAM (Significance Analysis of Microarray) package to identify differentially expressed mRNAs60. All differentially expressed mRNA with its corresponding p-value has been marked in “red” in the fourth column of Table 2.
Supplementary Material
SI file
Table S001
Table S002
Table S003
Table S004
Table S005
Table S006
Table S007
Table S008
Table S009
Table S010
Table S011
Table S012
Table S013
Table S014
Cytoscape readable pathways representing 4 cellular processes and integrated map
Footnotes
The authors declare no competing financial interests.
Author Contributions A.D. and S.B. wrote the manuscript text. A.D. prepared figure 1, 3–5; S.B. prepared figure 2. A.D., P.S., M.H.A. and N.N. designed the research. A.D., S.B., P.S., M.H.A. and N.N. reviewed the manuscript.
References
- Jass J. R. Limitations of the adenoma-carcinoma sequence in colorectum. Clin. Cancer Res. 10, 5969–70; 5970 (2004). [DOI] [PubMed] [Google Scholar]
- Bradley C. J. et al. Productivity savings from colorectal cancer prevention and control strategies. Am. J. Prev. Med. 41, e5–e14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz L. B. et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 22, 1201–8 (2004). [DOI] [PubMed] [Google Scholar]
- Chung K. Y. et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol. 23, 1803–10 (2005). [DOI] [PubMed] [Google Scholar]
- Cunningham D. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 351, 337–45 (2004). [DOI] [PubMed] [Google Scholar]
- Frattini M. et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 97, 1139–45 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore-Bianchi A. et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 69, 1851–7 (2009). [DOI] [PubMed] [Google Scholar]
- Perrone F. et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann. Oncol. 20, 84–90 (2009). [DOI] [PubMed] [Google Scholar]
- Moroni M. et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 6, 279–86 (2005). [DOI] [PubMed] [Google Scholar]
- Prenen H. et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin. Cancer Res. 15, 3184–8 (2009). [DOI] [PubMed] [Google Scholar]
- Sartore-Bianchi A. et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One 4, e7287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R. A. Hallmarks of cancer: the next generation. Cell 144, 646–74 (2011). [DOI] [PubMed] [Google Scholar]
- Rukov J. L., Wilentzik R., Jaffe I., Vinther J. & Shomron N. Pharmaco-miR: linking microRNAs and drug effects. Brief. Bioinform. (2013) 10.1093/bib/bbs082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–97 (2004). [DOI] [PubMed] [Google Scholar]
- Lewis B. P., Shih I., Jones-Rhoades M. W., Bartel D. P. & Burge C. B. Prediction of mammalian microRNA targets. Cell 115, 787–98 (2003). [DOI] [PubMed] [Google Scholar]
- Tang F., Hajkova P., Barton S. C., Lao K. & Surani M. A. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 34, e9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L. P. et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–73 (2005). [DOI] [PubMed] [Google Scholar]
- Webster R. J. et al. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem. 284, 5731–41 (2009). [DOI] [PubMed] [Google Scholar]
- Lee K. M., Choi E. J. & Kim I. A. microRNA-7 increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Radiother. Oncol. 101, 171–6 (2011). [DOI] [PubMed] [Google Scholar]
- Xu X. T. et al. MicroRNA expression profiling identifies miR-328 regulates cancer stem cell-like SP cells in colorectal cancer. Br. J. Cancer 106, 1320–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. et al. Increased expression of microRNA-21and its association with chemotherapeutic response in human colorectal cancer. J. Int. Med. Res. 39, 2288–95 (2011). [DOI] [PubMed] [Google Scholar]
- Li J.-M. et al. Down-regulation of fecal miR-143 and miR-145 as potential markers for colorectal cancer. Saudi Med. J. 33, 24–9 (2012). [PubMed] [Google Scholar]
- Zhu H. et al. EGFR signals downregulate tumor suppressors miR-143 and miR-145 in Western diet-promoted murine colon cancer: role of G1 regulators. Mol. Cancer Res. 9, 960–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nautiyal J. et al. EGFR regulation of colon cancer stem-like cells during aging and in response to the colonic carcinogen dimethylhydrazine. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G655–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. M., Iida M. & Wheeler D. L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 11, 777–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T. & Kudo M. Signaling pathways governing tumor angiogenesis. Oncology 81 Suppl 124–9 (2011). [DOI] [PubMed] [Google Scholar]
- Wittekind C. & Neid M. Cancer invasion and metastasis. Oncology 69 Suppl 114–6 (2005). [DOI] [PubMed] [Google Scholar]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovskikh V. A. & Herceg Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. Bioessays 32, 894–904 (2010). [DOI] [PubMed] [Google Scholar]
- Witkos T. M., Koscianska E. & Krzyzosiak W. J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 11, 93–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosakhani N. et al. MicroRNA profiling predicts survival in anti-EGFR treated chemorefractory metastatic colorectal cancer patients with wild-type KRAS and BRAF. Cancer Genet. 205, 545–51 (2012). [DOI] [PubMed] [Google Scholar]
- Maragkakis M. et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics 10, 295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand T. M., Iida M. & Wheeler D. L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 11, 777–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardiello F. et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 10, 784–93 (2004). [DOI] [PubMed] [Google Scholar]
- Sabatel C. et al. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One 6, e16979 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Rieu M. C. et al. MicroRNA-21 is induced early in pancreatic ductal adenocarcinoma precursor lesions. Clin. Chem. 56, 603–12 (2010). [DOI] [PubMed] [Google Scholar]
- Lu Y. et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 67, 8240–7 (2007). [DOI] [PubMed] [Google Scholar]
- Wheeler D. L. et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol. Ther. 8, 696–703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn E. F. et al. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 30, 561–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov E., Tang Z., Aifantis I. & Statnikov A. Assessing quality and completeness of human transcriptional regulatory pathways on a genome-wide scale. Biol. Direct 6, 15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi A. & Lehner B. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 5, 260 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman Y. Y., Tuller T., Shlomi T., Sharan R. & Ruppin E. Translation efficiency in humans: tissue specificity, global optimization and differences between developmental stages. Nucleic Acids Res. 38, 2964–74 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes T. J. S. et al. Tissue-specific subnetworks and characteristics of publicly available human protein interaction databases. Bioinformatics 27, 2414–21 (2011). [DOI] [PubMed] [Google Scholar]
- Witkos T. M., Koscianska E. & Krzyzosiak W. J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 11, 93–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann S. et al. Global microRNA level regulation of EGFR-driven cell-cycle protein network in breast cancer. Mol. Syst. Biol. 8, 570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M. miRNAs versus oncogenes: the power of social networking. Mol. Syst. Biol. 8, 569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q., Yu Z., Pan Y., Purisima E. O. & Wang E. MicroRNAs preferentially target the genes with high transcriptional regulation complexity. Biochem. Biophys. Res. Commun. 352, 733–8 (2007). [DOI] [PubMed] [Google Scholar]
- Cui Q., Yu Z., Purisima E. O. & Wang E. MicroRNA regulation and interspecific variation of gene expression. Trends Genet. 23, 372–5 (2007). [DOI] [PubMed] [Google Scholar]
- Ekins S., Nikolsky Y., Bugrim A., Kirillov E. & Nikolskaya T. Pathway mapping tools for analysis of high content data. Methods Mol. Biol. 356, 319–50 (2007). [DOI] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M. et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–31 (2003). [DOI] [PubMed] [Google Scholar]
- Nikitin A., Egorov S., Daraselia N. & Mazo I. Pathway studio--the analysis and navigation of molecular networks. Bioinformatics 19, 2155–7 (2003). [DOI] [PubMed] [Google Scholar]
- Vergoulis T. et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 40, D222–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F. et al. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 37, D105–10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.-D. et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 39, D163–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q. et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 37, D98–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scardoni G., Petterlini M. & Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 25, 2857–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khambata-Ford S. et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 25, 3230–7 (2007). [DOI] [PubMed] [Google Scholar]
- Irizarry R. A. et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V. G., Tibshirani R. & Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98, 5116–21 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI file
Table S001
Table S002
Table S003
Table S004
Table S005
Table S006
Table S007
Table S008
Table S009
Table S010
Table S011
Table S012
Table S013
Table S014
Cytoscape readable pathways representing 4 cellular processes and integrated map