

Abstract
Influenza C is not included in the annual seasonal influenza vaccine, and has historically been regarded as a minor respiratory pathogen. However, recent work has highlighted its potential role as a cause of pneumonia in infants. We performed nasopharyngeal or nasal swabbing and/or serum sampling (n = 148) in Lancaster, UK, over the winter of 2014–2015. Using enzyme-linked immunosorbent assay (ELISA), we obtain seropositivity of 77%. By contrast, only 2 individuals, both asymptomatic adults, were influenza C-positive by polymerase chain reaction (PCR). Deep sequencing of nasopharyngeal samples produced partial sequences for 4 genome segments in one of these patients. Bayesian phylogenetic analysis demonstrated that the influenza C genome from this individual is evolutionarily distant to those sampled in recent years and represents a novel genome constellation, indicating that it may be a product of a decades-old reassortment event. Although we find no evidence that influenza C was a significant respiratory pathogen during the winter of 2014–2015 in Lancaster, we confirm previous observations of seropositivity in the majority of the population. (170 words).
Introduction
Clinical presentation
Influenza C (family Orthomyxoviridae, genus Influenzavirus C, species Influenza C virus) produces malaise, coryza and fever when administered to susceptible adult volunteers1. Historically, influenza C has been regarded as the least serious of the three species of influenza infecting humans, and seasonal vaccination programmes have been confined to influenzas A and B. More recent studies confirmed influenza C’s production of a mild respiratory illness in healthy adults, with only occasional complications2.
However, in a paediatric context, acute respiratory illness and/or pneumonia have been reported as a consequence of influenza C infection3,4,5,6,7,8 especially in those under 2 years old9, as well as vomiting, diarrhoea, acute otitis media10, a high rate of hospitalization11 and even acute encephalopathy12. This growing awareness of the paediatric clinical importance of influenza C raises the issue of its inclusion in the annual seasonal influenza vaccine, or its position as a candidate for vaccine development specifically for infants.
Epidemiology
Nearly 40% of adult volunteers were susceptible to administered influenza C1. The 60% who did not develop disease is consonant with observation of seropositivity levels of 59% in Spain13, 61% in France14 and 57% in Brazil15, and suggests that seropositivity may possibly confer resistance. By contrast, other studies have suggested that antibodies against influenza C tend to be more universal: 100% in an isolated Philippine village16 and in the USA17, 90% in Czechoslovakia18, 86% in the Soviet Union18 and 70% in East Germany19. Some studies have also found age-structured variability: in California, seropositivity of 64% in children under 5 but 98% in adults20; in Japan, 40–50% in early childhood to nearly 100% in adulthood21; in Louisiana, 47% in children to 96% in younger adults, but then a decline to 18% in the over-65s22; in France, 46% seropositivity in children, 76% in younger adults, but only 44% in the over-50s14.
Influenza C does not appear to be seasonal, based on contemporaneous two-year surveys of its occurrence in Bucharest and Japan from 1988–19906,23. Using this observation together with the seropositivity data, it is possible to propose several epidemiological scenarios. The first of these is that influenza C is an endemic virus in human populations, with approximately lifelong immunity conferred by first exposure. The decline in seropositivity in later life14,22, potentially due to immunosenescence, would then provide the virus with opportunities to infect individuals for a second time. The second scenario is that the virus is only intermittently epidemic, with variation in seropositivity a reflection of previous epidemic history in different locations. The third scenario is that the virus is endemic but antigenically variable over time24. Seropositivity would therefore be an unreliable guide to the true immune status of any individual.
Phylogenetics and molecular evolution
The rate of nucleotide substitution is lower in influenza C than in A and B25,26,27,28, and reassortment has been detected3,6,25,28,29,30. There is also evidence of positive selection at two residues in the receptor-binding domain of the haemagglutinin-esterase (HE) protein, but the overall ratio of non-synonymous to synonymous substitutions (omega) across the genome is low, individual proteins ranging from 0.05 to 0.1328. The low levels of omega indicate a virus that is well adapted to its host, but the presence of positive selection in the HE receptor-binding domain also indicates selective pressure from the host immune system. This provides a molecular explanation for the observed antigenic drift24 and some evidence against the scenario that humans are likely to acquire lifelong immunity.
The issue of endemicity versus sporadic epidemics also remains unresolved. Only one candidate epidemic surge has been identified, in Japan in 20044. The existence of reassorted strains indicates that double infection with two or more strains cannot be very infrequent, implying that it ought to be possible to detect numerous (or at least >1) strains co-circulating both temporally and geographically, previously demonstrated in Japan4. Indeed, a continually shifting pattern of segment combinations, referred to as genome constellations28, is observed when full genomes are studied, a phenomenon also seen in influenza B31. Eight genome constellations circulating in the 1990s differed from the genome constellations present in a set of reference genomes from the 1940s to the 1980s28.
Results
Participants
Of the 148 participants, 69 were male and 79 female. 71 were symptomatic and 77 asymptomatic. Distribution of male and female participants within symptomatic and asymptomatic groups was assessed by a 2 × 2 chi-square test and was not statistically significant. Except for a relative excess of age group 20–29 participants (mostly from the university), age approximated a normal distribution.
Influenza C seropositivity
Of the 148 participants, 129 consented to donate serum. Of these 99 were seropositive and 30 negative, giving a figure of 77% seropositivity. Figure 1 shows the anti-influenza C IgG concentration by age. Gender differences in seropositivity were also nearly absent (male mean 2.5 mg/dl, female mean 2.3 mg/dl) with no statistical significance on t-test, but symptomatic individuals had slightly more IgG (symptomatic mean 2.6 mg/dl, asymptomatic mean 2.2 mg/dl), significant on a t-test at p < 0.05. A Mann Whitney U-test was performed on the distribution of seropositive individuals between each age group, and was not statistically significant.
Figure 1. Anti-influenza C IgG concentration (mg/dl), plotted for each individual against age.
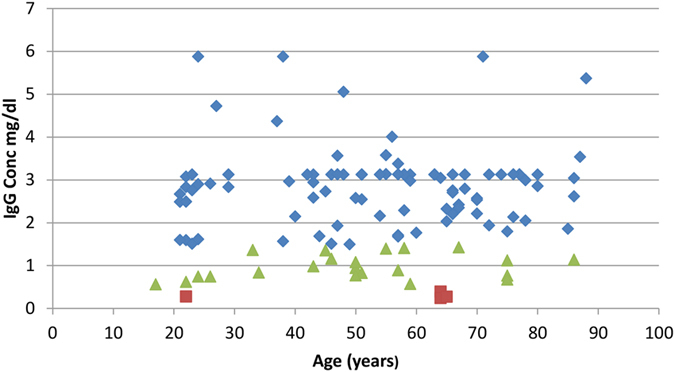
Blue: >2 standard deviations above negative control; green: 1–2 standard deviations above negative control; red: <1 standard deviation above negative control.
Detection of viral RNA
Two participants out of 148 (1.4%), aged 51 years and 70 years, both asymptomatic, were detected as positive for influenza C using quantitative PCR, at 135 ng and 160 ng total viral RNA respectively, corresponding to 1.9 × 1010 and 2.2 × 1010 genome copies. Since influenza viruses are believed to have a single genome copy per virion32, this indicates approximately 2 × 1010 virions per individual nasopharyngeal swab. On deep sequencing (accessions SRR4733498 and SRR4733494), only one patient showed sufficient levels of influenza C reads for genome assembly to be attempted (SRR4733498).
Genetic relationships of isolated influenza C genome segments
Partial genome segment sequences were obtained from deep sequencing for segments 1, 5, 6 and 7, encoding PB2, NP, M1/CM2 and NS1/NS2 respectively. Those greater than 200 bases are deposited in GenBank, accession numbers KY075640 - KY075642. Insufficient reads were available to assemble the other segments. Although breadth of coverage across segments is low (ranging from 22% in segment 5 to 32% in segment 6), there is sufficient genetic information to assign each fragment to a clade as defined previously28, using Bayesian phylogenetics. Plotting of the root-to-tip genetic distance on a neighbour-joining tree using TempEst showed that molecular clocks apply best to segments 2 and 7 (PB2 and NS1/NS2), but that both segments 5 and 6 (NP and M1/CM2) have lower root-to-tip distances for C/Lancaster/1/2015 than expected. Figures 2 and 3 shows the TempEst plots for segments 1 and 6 (PB2 and M1/CM2), giving examples of clock-like and non-clock-like behaviour, respectively. The TempEst plots for segments 5 and 7 (NP and NS1/NS2) are Supplementary Figures 3 and 4 respectively. The new strain of influenza C identified was designated C/Lancaster/1/2015.
Figure 2. Root-to-tip distance in a neighbour joining tree for segment 1 (encoding PB2) of the influenza C genome.
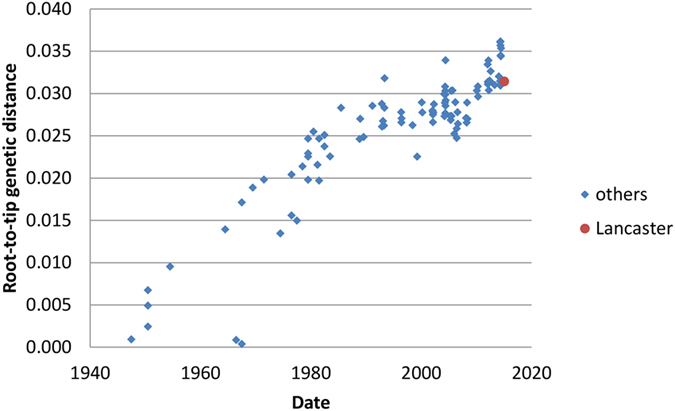
100 full-length or near full-length genome segments (2365 bases) are used plus the 724 discontinuous bases of segment 1 derived from deep sequencing. C/Lancaster/1/2015 has a degree of divergence from the root consistent with molecular clock-like behaviour in its lineage.
Figure 3. Root-to-tip distance in a neighbour joining tree for segment 6 (encoding M1/CM2) segment of the influenza C genome.
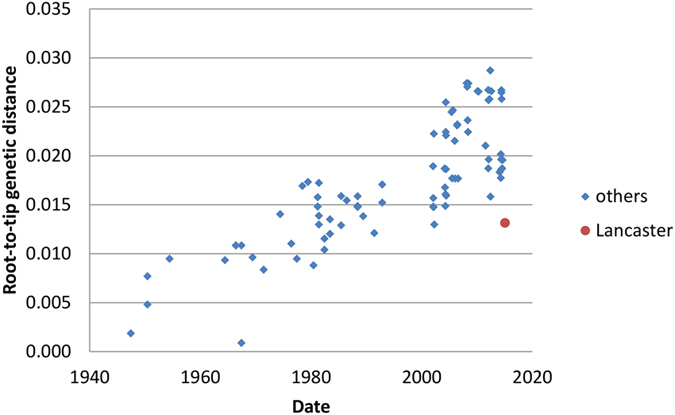
86 full-length or near full-length genome segments (1180 bases) are used plus the 380 discontinuous bases of segment 6 derived from deep sequencing. C/Lancaster/1/2015 is less divergent from the root than it should be given its known sampling date, consistent with a perturbation of molecular clock-like behaviour in its lineage.
Clade memberships were determined by examination of Bayesian phylogenetic trees produced in BEAST, as previously28 and then annotated onto the neighbour-joining trees used for the molecular clock analysis. Figure 4 shows the tree for segment 5 (encoding NP), demonstrating that C/Lancaster/1/2015 belongs to the C/Miyagi/1/93 clade, and not to the C/Greece/79 and C/pig/Beijing/81 clades circulating in recent isolates. Figure 5 shows the tree for segment 7 (encoding NS1/NS2) has an even more distant relationship to recent genomes, being part of the C/Sapporo/71 clade last seen in 1979. The phylogenetic trees for PB2 and MP are given in Supplementary Figures 1 and 2, and further confirm the genetic distance between C/Lancaster/1/2015 and other recently sequenced genomes. Clade memberships are then synthesised to derive the relationship between C/Lancaster/1/2015 and defined genome constellations (Table 1).
Figure 4. Neighbour joining tree rooted on C/Taylor/1233/1947 for segment 5 (NP), annotated with clades as previously derived28, demonstrating the closer relationship of C/Lancaster/1/2015 (red) to NP segments of the C/Miyagi/1/93 clade than to recent isolates.

Scale: substitutions per site.
Figure 5. Neighbour joining tree rooted on C/Taylor/1233/1947 for segment 7 (NS1/NS2), annotated with clades as previously derived28, demonstrating the closer relationship of C/Lancaster/1/2015 (red) to NS1/NS2 segments of the C/Sapporo/71 clade from the 1970s than to recent isolates.

Scale: substitutions per site.
Table 1. Clade membership of segments of C/Lancaster/1/2015 and the prior clade and genome constellation classifications as previously derived28.
| Genome segment (encoded protein) | Clade of segment in Lancaster consensus, as defined by Gatherer28 | Genome constellation in which that clade is present | Clade(s) of other post 2009 genomes |
|---|---|---|---|
| 1 (PB2) | C/Sapporo/71 | All, except 5 | C/Greece/79; C/Sapporo/71 |
| 5 (NP) | C/Miyagi/1/93 | 4a | C/pig/115/Beijing/81; C/Greece/79 |
| 6 (M1/CM2) | C/Sapporo/71 | All, except 2 & 3 | C/Sapporo/71 |
| 7 (NS1/NS2) | C/Sapporo/71 | None: clade not seen since 1970s | C/Shizuoka/79 |
The rightmost column lists those clades found in other segments sequenced from 2010 onwards. Segments 1 and 6 of C/Lancaster/1/2015 are outliers within clades found in other recent genomes, but segments 5 and 7 are not.
Discussion
Our participant group were 77% seropositive to influenza C. This is slightly higher than the 57–61% levels from studies in western Europe and Brazil13,14,15, within the range of the 70–90% found in eastern Europe18,19 but still considerably short of those studies reporting universal seropositivity in the USA and east Asia16,17. As in previous studies, our antibody titre levels were widely variable among those classed as seropositive, and our choice of threshold is purely statistical. However, we also found no statistically significant age-structured or gender-structured variability in seropositivity (Fig. 1). This is at variance with some previous studies in the USA, Japan and Europe14,20,21,22. It should also be noted that many serological studies on influenza C are now some decades old and techniques have varied over the years, so individual studies are not necessarily directly comparable. We also cannot exclude the possibility of some cross-reactivity of our influenza C antigen with antibodies to other influenza viruses, but this is also an issue in all previous studies.
Neither of the two participants who were identified as influenza C-positive by PCR generated sufficient deep sequencing reads for complete genomes to be assembled. Our deep sequencing of the nasopharyngeal swabs of both of our PCR-positive participants, produced much fuller genome sequence results for other RNA viruses apart from influenza C, as well as sequences from a range of bacterial species (Atkinson et al. in preparation). We therefore do not think that the difficulty in detecting influenza C, or in generating complete genomes, is due to RNA degradation or other technical failure. The 4 segments partially assembled are the least variable segments, but only segment 4, encoding HE, is an outlier in terms of its variability, at 0.042 substitutions per site since 1947, compared to a range of 0.017 to 0.027 for the other segments28. Even within HE there are relatively conserved regions within the stalk domain, so we do not believe that failure to assemble HE or other segments is an artefact of excessive stringency in our assembly process.
In the individual with the 4 partial genome segment sequences, it is evident that C/Lancaster/1/2015 is a reassortant that does not fall into any of the genome constellations previously classified28 (see also Table 1). It contains a rare NS1/NS2 segment of the C/Sapporo/71 clade, related to sequences that were last observed in the late 1970s. Influenza C genomes sequenced since 2010 all have the C/Shizuoka/79 clade in the NS1/NS2 segment (Fig. 5). C/Lancaster/1/2015 also has a rare NP segment of the C/Miyagi/1/93 clade, related to sequences that were last observed around 2000 (Fig. 4) and typical of genome constellation 4a (Table 1). The other segments are within clades found more recently, although C/Lancaster/1/2015’s position within these clades is never close to any of the recent genome sequences (Supplementary Figures 1 and 2). The exact position of C/Lancaster/1/2015 on each segment’s phylogenetic tree is rarely well supported by Bayesian phylogenetics posterior probability density, but its location within each of the broader clades is well supported. We therefore conclude that its apparent reassortant nature is unlikely to be simply an artefact of partial sequence information.
Tentative reconstruction of the reassortment event may be attempted. Previous work28 defines genome constellation 4a as consisting of C/Sapporo/71, C/Miyagi/1/93, C/Sapporo/71 and C/Shizuoka/79 in segments 1, 5, 6 and 7 respectively. The corresponding clades for C/Lancaster/1/2015 are C/Sapporo/71, C/Miyagi/1/93, C/Sapporo/71 and C/Sapporo/71 respectively (Table 1), suggesting that a strain of constellation 4a reassorted with one containing a C/Sapporo/71-clade segment 7. Since no strain containing a segment 7 of this clade has been seen since the 1970s and constellation 4a was only seen in the 1990s, it seems likely that the reassortment event occurred in the 1990s. This would also explain the dissimilarity of C/Lancaster/1/2015 in all of its segments, to other recently sequenced strains. We are tempted to speculate that this reassortant occurred locally in Lancaster, but in the absence of any other British genomes since C/England/892/198333, which is itself incomplete, it is impossible to come a conclusion.
If this scenario is common in small isolated populations, influenza C diversity in terms of shifting genome constellations may be even greater than suggested from the available genomes. The M1/CM2 (Fig. 3) and NP segments (Supplementary Figure 3) for C/Lancaster/1/2015 have lower root-to-tip distances than expected under the assumption of molecular clock-like evolution. When this method is used on database-derived sequences, it is often taken as indicative of incorrect dating. However, given that we know precisely when our samples were collected, it is more likely to reflect a genuinely slower rate of evolution in these samples. The M1/CM2 segment of C/Lancaster/1/2015 is positioned in the phylogenetic tree near segments from the 1980s (Supplementary Figure 1) and the NP segment near segments from the 1990s and 2000 (Fig. 4). This same phenomenon of slowed molecular clock, and aberrant positioning with the phylogenetic tree, has been seen in some strains of Zaire ebolavirus34 and also in the 1977 “Russian Flu” H1N1 outbreak35, and is thought be a consequence of the virus entering a host population where the serial interval – the time between infection of one host and the next in a transmission chain – is reduced and the virus therefore spends longer in a non-replicative state. For ebolavirus, this is assumed to be a non-typical animal reservoir host, and for Russian Flu possibly a laboratory freezer. Neither of these options would seem to be possible for influenza C, so it may simply be a cumulative result of low transmission rates within relatively small populations slightly delaying the average serial interval, conditions which could apply in Lancaster.
We began this study with the premise that influenza C might be a candidate for inclusion in the seasonal influenza vaccine. Our results do not provide any support for the proposition that vaccination of adults is appropriate, consistent with the conclusion of one other recent report36. Although we recruited 71 symptomatic individuals with a range of cold/flu-like symptoms, none of these was influenza C-positive, and none of the respiratory disease burden in Lancaster during our study period can be attributed to influenza C.
There may still be a case for vaccination of children in the light of published reports of serious respiratory disease caused by influenza C in that age group3,4,5,6,7,8,9,10,11,12,37. We recruited 6 participants in the <9 years age group but none were consented to allow serum sampling. In the single participant in the 10-19 year age group, anti-influenza C IgG levels were at <1 mg/dl and this individual is classified as seronegative (Fig. 1). Haemagglutinin-inhibition assay (HI) would potentially clarify this issue, but in its absence we can only draw limited conclusions at best concerning the clinical implications of seropositivity, as quantified by ELISA.
Methods
Ethics
Ethical approval was granted by the UK National Research Ethics Service (NRES), reference 14/LO/1634, Integrated Research Application System (IRAS) Project 147631. The project was registered with the National Institute of Health Research (NIHR), UK as part of the NIHR Clinical Research Network (UKCRN) Portfolio, ID 17799. All methods were carried out in accordance with the relevant guidelines and regulations. Informed consent was obtained from all volunteers of 18 years and older. For those under 18 years, informed parental consent was obtained, together with supervised assent of the volunteer.
Patient recruitment
Lancaster (54.05°N 2.80°W) is a small city with a population of 45,000 (141,000 including surrounding towns and villages). The permanent resident population is >95% white and 18% are over age 65. Participants were approached in 3 locations from November 2014 to May 2015: 1) Lancaster University, 2) a general practice (GP), 3) hospital clinics. After informed consent was given, patients with coryza and/or other symptoms consistent with respiratory infection, were classified as the symptomatic group (n = 71) and the remainder as asymptomatic (n = 77). The latter were included to investigate if influenza C could be detected in patients without coryza. Nasopharyngeal (or nasal) swabbing, blood sampling, or both, were performed on the patients, according to consent.
Sample processing
Nasopharyngeal swabs (MW951SENT, Medical Wire) were used on the rear wall of the nasopharynx or nose (according to consent) of patients, and the tips then snapped off directly into Sigma Virocult® medium.
Blood was drawn or taken from a finger prick, according to consent, using Beckton Dickinson Serum Separator® tubes (SST™). Serum was separated at 1000–2000 g for 10 minutes (for arm samples) or at 6000–15000 g for 90 s (for finger-prick samples) and then stored at −80 °C.
RNA was extracted from the nasopharyngeal swabs using a MagMAX™ Viral RNA Isolation Kit (Ambion). The quality and quantity of RNA extracted from samples was assessed by spectrophotometry using the NanoDrop® 1000 Spectrophotometer V3.3.0 (Thermo Fisher Scientific). cDNA was prepared using a High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems®, Life Technologies™) and a Veriti® Thermal Cycler (Applied Biosystems®, Life Technologies™). The samples were incubated at 37 °C for 60 minutes, before stopping the reaction at 95 °C for 5 minutes and then holding at 4 °C. Once completed, the plates were stored at −20 °C.
Polymerase chain reaction (PCR) was then performed using a 7500 FAST Real-Time PCR system (Applied Biosystems®, Life Technologies™) with thermo-cycling carried out as follows: one cycle of 95 °C for 10 min and 45 cycles of 95 °C for 15 s and 60 °C for 1 min. PCR primers for influenza C were as used previously38, and quantification was performed by reference to a positive control sample at 32 ng/μl. Concentrations were converted into genome copy numbers using http://scienceprimer.com/nucleotide-molecular-weight-calculator. Samples judged positive after quantitative PCR were processed using the Illumina Nextera XT library kit and deep sequenced in 2 × 126 bp format using an Illumina HiSeq2500 system.
Enzyme-linked immunosorbent assay (ELISA) was performed on the serum samples using influenza C antigen as previously described, and using the same antigen preparation38, with goat anti-human HRP-conjugated secondary antibody (ab6858, Abcam®) and SureBlue™ TMB Microwell Peroxidase Substrate solution. Absorbance was measured at 450 nM using a Wallac Victor2™ (Perkin Elmer) plate reader. Anti-influenza C IgG was quantified by calibration of the peroxidase reaction against a standard dilution series of IgG concentrations. The threshold for seropositivity was placed at 2 standard deviations above the mean level of the negative control serum.
Genome segment sequence assembly
Illumina reads were trimmed of adapters and other non-genomic elements using CutAdapt 1.139 (https://pypi.python.org/pypi/cutadapt), fastq-mcf 0.11.340 (https://expressionanalysis.github.io/ea-utils), and trim_galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), within the Read_cleaner pipeline (Gatherer, unpublished). Ethical approval required that no genetic material remain within the samples which could enable identification of patients. Therefore, human genome and transcriptome sequences were removed by iterative alignment onto the NCBI, Ensembl and UCSC human iGenomes (http://support.illumina.com/sequencing/sequencing_software/igenome.html), first using bowtie 1.1.141 (http://bowtie-bio.sourceforge.net/index.shtml), then BWA 0.7.12-r103942 (http://bio-bwa.sourceforge.net) within the Valet pipeline (Gatherer, unpublished). Following each alignment, extraction of unaligned reads was achieved using samtools 0.1.1943 (http://samtools.sourceforge.net) and the next alignment commenced. Bowtie, BWA and samtools were co-ordinated using the Vanator pipeline44 (https://sourceforge.net/projects/vanator-cvr). The resulting trimmed and cleaned reads are available from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/: BioSamples SAMN05954290 and SAMN05954291, Runs SRR4733498 and SRR4733494).
Influenza C genome C/Victoria/2/2012 (Genbank ref. KM504282) was selected as a representative of recently circulating influenza C and alignment of cleaned reads carried out using bowtie within the Valet pipeline. Consensus sequences were constructed using samtools 0.1.19 (bcftools and vcfutils functions). C/Victoria/2/2012 was used to fill gaps in the consensi and the bowtie alignment repeated. This cycle was performed until a stable consensus was obtained for each genome segment. The same process was repeated using BWA and combined consensi obtained. Alignment of reads to the final consensi was examined with Tablet45 (https://ics.hutton.ac.uk/tablet). Resulting assemblies of more than 200 bases were submitted to GenBank (references KY075640 - KY075642). The new strain of influenza C identified was designated C/Lancaster/1/2015.
Phylogenetics and genome constellations
Sequence alignments of composite partial segments with full influenza C genomes from GenBank, were performed using Muscle46 in MEGA47 (http://www.megasoftware.net) and neighbour joining trees48 constructed. Clock-like behaviour in sequence evolution on those trees was checked using TempEst49 (http://tree.bio.ed.ac.uk/software/tempest). Bayesian phylogenetic analysis was performed in BEAST v.1.8.350 (http://tree.bio.ed.ac.uk/software/beast/). A Tamura 3-parameter (T93 + G) substitution model51, coalescent constant size tree prior and relaxed lognormal clock were run for 100 million iterations in BEAST, with a burn-in of 25%. Genome constellations were determined by establishing the clade, as previously defined28, in which each genome segment was located.
Statistical analyses
on volunteers and ELISAs, BAM files and reference genomes for genome assemblies, genome fragments too short for inclusion in GenBank, BEAST inputs and outputs, TempEst inputs and outputs and pipeline Perl scripts, are available from: doi://10.17635/lancaster/researchdata/111.
Additional Information
How to cite this article: Atkinson, K. V. et al. Influenza C in Lancaster, UK, in the winter of 2014-2015. Sci. Rep. 7, 46578; doi: 10.1038/srep46578 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
KVA received a Service Increment from Teaching (SIFT) studentship from University Hospitals of Morecambe Bay (UHMB) National Health Service (NHS) Foundation Trust, UK and performed this work as part of the requirements for the degree of Master of Science (MSc). Rosetrees Trust, UK, provided additional funding for deep sequencing (grant M395).
Footnotes
The authors declare no competing financial interests.
Author Contributions K.V.A. collected the samples and performed all laboratory work except deep sequencing. LAB supervised laboratory work on ELISAs. G.R. supervised laboratory work on PCR. N.S. prepared influenza C antigens, PCR primers and positive and negative control samples for PCR. N.R.M. performed deep sequencing. M.J.H. performed deep sequencing. J.R. assisted in sample collection. N.H. assisted in sample collection. S.W. provided clinical supervision in the general practice surgery. R.M.L. provided executive laboratory supervision on immunology. R.W.P. provided executive laboratory supervision on molecular biology and was a named co-supervisor for the MSc degree of KVA. M.W. provided clinical supervision in the hospital and was a named co-supervisor for the MSc degree of KVA D.G. wrote the grant and ethics applications, drafted and corrected the paper, and was principal supervisor for the MSc degree of KVA.
References
- Joosting A. C., Head B., Bynoe M. L. & Tyrrell D. A. Production of common colds in human volunteers by influenza C virus. British medical journal 4, 153–154 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila J. et al. Influenza C virus infection in military recruits-symptoms and clinical manifestation. J Med Virol, doi: 10.1002/jmv.23756 (2013). [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y. et al. Frequent reassortment among influenza C viruses. J Virol 77, 871–881 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y. et al. A nationwide epidemic of influenza C virus infection in Japan in 2004. Journal of clinical microbiology 45, 783–788, doi: 10.1128/JCM.01555-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Principi N., Scala A., Daleno C. & Esposito S. Influenza C virus-associated community-acquired pneumonia in children. Influenza and other respiratory viruses, doi: 10.1111/irv.12062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriuchi H., Katsushima N., Nishimura H., Nakamura K. & Numazaki Y. Community-acquired influenza C virus infection in children. The Journal of pediatrics 118, 235–238 (1991). [DOI] [PubMed] [Google Scholar]
- Calvo C., Garcia-Garcia M. L., Centeno M., Perez-Brena P. & Casas I. Influenza C virus infection in children, Spain. Emerging infectious diseases 12, 1621–1622, doi: 10.3201/eid1210.051170 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y., Abiko C., Ikeda T., Mizuta K. & Matsuzaki Y. Influenza C Virus and Human Metapneumovirus Infections in Hospitalized Children With Lower Respiratory Tract Illness. The Pediatric infectious disease journal 34, 1273–1275, doi: 10.1097/INF.0000000000000863 (2015). [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y. et al. Clinical features of influenza C virus infection in children. The Journal of infectious diseases 193, 1229–1235, doi: 10.1086/502973 (2006). [DOI] [PubMed] [Google Scholar]
- Laxdal O. E., Blake R. M., Cartmill T. & Robertson H. E. Etiology of acute otitis media in infants and children. Canadian Medical Association journal 94, 159–163 (1966). [PMC free article] [PubMed] [Google Scholar]
- Gouarin S. et al. Study of influenza C virus infection in France. J Med Virol 80, 1441–1446, doi: 10.1002/jmv.21218 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi M. et al. Acute encephalopathy associated with influenza C virus infection. The Pediatric infectious disease journal 28, 554, doi: 10.1097/INF.0b013e3181a064b2 (2009). [DOI] [PubMed] [Google Scholar]
- Manuguerra J. C., Hannoun C., Saenz Mdel C., Villar E. & Cabezas J. A. Sero-epidemiological survey of influenza C virus infection in Spain. European journal of epidemiology 10, 91–94 (1994). [DOI] [PubMed] [Google Scholar]
- Manuguerra J. C., Hannoun C. & Aymard M. Influenza C virus infection in France. The Journal of infection 24, 91–99 (1992). [DOI] [PubMed] [Google Scholar]
- Motta F. C., Luiz M. O. & Couceiro J. N. Serological analysis reveals circulation of influenza C viruses, Brazil. Revista de saude publica 34, 204–205 (2000). [DOI] [PubMed] [Google Scholar]
- Nishimura H., Sugawara K., Kitame F., Nakamura K. & Sasaki H. Prevalence of the antibody to influenza C virus in a northern Luzon Highland Village, Philippines. Microbiology and immunology 31, 1137–1143 (1987). [DOI] [PubMed] [Google Scholar]
- Hilleman M. R., Werner J. H. & Gauld R. L. Influenza antibodies in the population of the USA; an epidemiological investigation. Bulletin of the World Health Organization 8, 613–631 (1953). [PMC free article] [PubMed] [Google Scholar]
- Vasil’eva V. I., Zakstel’skaia L., Govorkova E. A., Rusakova E. V. & Alekseenkova L. I. [Immunostructure of the population to the influenza C virus]. Voprosy virusologii 30, 661–664 (1985). [PubMed] [Google Scholar]
- Tumova B., Scharfenorth H. & Adamczyk G. Incidence of influenza C virus in Czechoslovakia and German Democratic Republic. Acta virologica 27, 502–510 (1983). [PubMed] [Google Scholar]
- Dykes A. C., Cherry J. D. & Nolan C. E. A clinical, epidemiologic, serologic, and virologic study of influenza C virus infection. Archives of internal medicine 140, 1295–1298 (1980). [PubMed] [Google Scholar]
- Kaji M. et al. Distribution of antibodies to influenza C virus. The Kurume medical journal 30, 121–123 (1983). [DOI] [PubMed] [Google Scholar]
- O’Callaghan R. J., Gohd R. S. & Labat D. D. Human antibody to influenza C virus: its age-related distribution and distinction from receptor analogs. Infection and immunity 30, 500–505 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita E., Lupulescu E., Alexandrescu V., Matepiuc M. & Tecu C. Seroepidemiological study of the circulation of influenza C virus in man. Roumanian archives of microbiology and immunology 51, 263–269 (1992). [PubMed] [Google Scholar]
- Chakraverty P. Antigenic relationship between influenza C viruses. Archives of virology 58, 341–348 (1978). [DOI] [PubMed] [Google Scholar]
- Buonagurio D. A., Nakada S., Fitch W. M. & Palese P. Epidemiology of influenza C virus in man: multiple evolutionary lineages and low rate of change. Virology 153, 12–21 (1986). [DOI] [PubMed] [Google Scholar]
- Yamashita M., Krystal M., Fitch W. M. & Palese P. Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology 163, 112–122 (1988). [DOI] [PubMed] [Google Scholar]
- Muraki Y., Hongo S., Sugawara K., Kitame F & Nakamura K. Evolution of the haemagglutinin-esterase gene of influenza C virus. J Gen Virol 77 (Pt 4), 673–679 (1996). [DOI] [PubMed] [Google Scholar]
- Gatherer D. Tempo and mode in the molecular evolution of influenza C. PLoS currents 2, RRN1199, doi: 10.1371/currents.RRN1199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V. R. & Palese P. Isolation of influenza C virus recombinants. J Virol 32, 1006–1014 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G. et al. Genetic Reassortment of Influenza-C Viruses in Man. Journal of General Virology 75, 3619–3622, doi: 10.1099/0022-1317-75-12-3619 (1994). [DOI] [PubMed] [Google Scholar]
- Chen R. & Holmes E. C. The evolutionary dynamics of human influenza B virus. J Mol Evol 66, 655–663, doi: 10.1007/s00239-008-9119-z (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. Y. et al. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proceedings of the National Academy of Sciences of the United States of America 109, 9101–9106, doi: 10.1073/pnas.1206069109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y. et al. Genetic Lineage and Reassortment of Influenza C Viruses Circulating between 1947 and 2014. J Virol 90, 8251–8265, doi: 10.1128/JVI.00969-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T. T., Zhu H., Chong Y. L., Holmes E. C. & Guan Y. Puzzling Origins of the Ebola Outbreak in the Democratic Republic of the Congo, 2014. J Virol 89, 10130–10132, doi: 10.1128/JVI.01226-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim J. O. The re-emergence of H1N1 influenza virus in 1977: a cautionary tale for estimating divergence times using biologically unrealistic sampling dates. PloS one 5, e11184, doi: 10.1371/journal.pone.0011184 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. B., Gaunt E. R., Digard P., Templeton K. & Simmonds P. Detection of influenza C virus but not influenza D virus in Scottish respiratory samples. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology 74, 50–53, doi: 10.1016/j.jcv.2015.11.036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G. et al. Frequent occurrence of genetic reassortment between influenza C virus strains in nature. J Gen Virol 77 (Pt 7), 1489–1492 (1996). [DOI] [PubMed] [Google Scholar]
- Salez N. et al. Influenza C virus high seroprevalence rates observed in 3 different population groups. The Journal of infection 69, 182–189, doi: 10.1016/j.jinf.2014.03.016 (2014). [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal 17, 10–12, http://dx.doi.org/10.14806/ej.14817.14801.14200 (2011). [Google Scholar]
- Aronesty E. Comparison of sequencing utility programs. The Open Bioinformatics Journal 7, doi: 10.2174/1875036201307010001 (2013). [DOI] [Google Scholar]
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25, doi: 10.1186/gb-2009-10-3-r25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, doi: 10.1093/bioinformatics/btp698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, doi: 10.1093/bioinformatics/btp352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett R. F., Gallagher A. & Gatherer D. Molecular methods of virus detection in lymphoma. Methods Mol Biol 971, 277–293, doi: 10.1007/978-1-62703-269-8_16 (2013). [DOI] [PubMed] [Google Scholar]
- Milne I. et al. Using Tablet for visual exploration of second-generation sequencing data. Briefings in bioinformatics 14, 193–202, doi: 10.1093/bib/bbs012 (2013). [DOI] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797, doi: 10.1093/nar/gkh340 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Nei M., Dudley J & Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in bioinformatics 9, 299–306, doi: 10.1093/bib/bbn017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N. & Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution 4, 406–425 (1987). [DOI] [PubMed] [Google Scholar]
- Rambaut A., Lam T. T., Carvalho L. M. & Pybus O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evolution 2, doi: 10.1093/ve/vew1007 doi: 10.1093/ve/vew007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. & Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC evolutionary biology 7, 214, doi: 10.1186/1471-2148-7-214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. The rate and pattern of nucleotide substitution in Drosophila mitochondrial DNA. Molecular biology and evolution 9, 814–825 (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.