

Abstract
Caged organic fluorophores are established tools for localization-based super-resolution imaging. Their use relies on reversible deactivation of standard organic fluorophores by chemical reduction or commercially available caged dyes with ON switching of the fluorescent signal by ultraviolet (UV) light. Here, we establish caging of cyanine fluorophores and caged rhodamine dyes, i.e., chemical deactivation of fluorescence, for single-molecule Förster resonance energy transfer (smFRET) experiments with freely diffusing molecules. They allow temporal separation and sorting of multiple intramolecular donor–acceptor pairs during solution-based smFRET. We use this “caged FRET” methodology for the study of complex biochemical species such as multisubunit proteins or nucleic acids containing more than two fluorescent labels. Proof-of-principle experiments and a characterization of the uncaging process in the confocal volume are presented. These reveal that chemical caging and UV reactivation allow temporal uncoupling of convoluted fluorescence signals from, e.g., multiple spectrally similar donor or acceptor molecules on nucleic acids. We also use caging without UV reactivation to remove unwanted overlabeled species in experiments with the homotrimeric membrane transporter BetP. We finally outline further possible applications of the caged FRET methodology, such as the study of weak biochemical interactions, which are otherwise impossible with diffusion-based smFRET techniques because of the required low concentrations of fluorescently labeled biomolecules.
Förster resonance energy transfer (FRET) has become a complementary tool in structural biology.1−7 FRET can act as a molecular ruler based on a nonradiative energy transfer between two fluorescent probes, a donor and an acceptor, with distinct spectral properties. When designed properly, i.e., the orientation of fluorophore dipole moments does not govern energy transfer,1 the FRET efficiency depends only on the distance between both fluorophores. In that situation, a direct link between FRET efficiency and biochemical structure can be made by strategic labeling with fluorescent probes.1 In an intramolecular assay, FRET is then indicative of conformational states or ligand-induced structural changes.8 It can also visualize mobile parts of proteins that do not crystallize,9,10 but most importantly, it provides access to structural dynamics.1,2,11−13 For the latter, FRET is combined with single-molecule detection to allow the observation of unsynchronized reactions. Single-molecule FRET (smFRET) has become the tool of choice for investigating structural dynamics with a spatial resolution of nanometers (dynamic range of 2–10 nm) and a subsecond time resolution.14 Alternative strategies, which are also compatible with single-molecule detection, provide different dynamic ranges and exploit other photophysical effects [photoinduced electron transfer (PET)15 or protein-induced fluorescence enhancement (PIFE)16] or molecular properties such as diffusion that can be determined simultaneously with FRET to obtain multidimensional synergetic assays.17,18
The design of a molecular ruler, which monitors conformational states, requires a structure-guided identification of fluorophore labeling sites.11 These labeling sites are chosen such that changes in biochemical state result in a measurable photophysical signal, i.e., for the FRET ruler in a change of transfer efficiency E. Second, the structure of interest is modified to allow incorporation of the labels at the desired locations. This typically happens via site-directed mutagenesis of single amino acids in proteins to cysteines (alternatively “clickable” amino acids19,20) or the use of modified nucleic acids that allow labeling with reactive synthetic organic fluorophores.21−24 Because labeling might interfere with biochemical function, assays are needed that directly compare the protein activity and its degree of labeling as control experiments toward a relevant biophysical study. Ultimately, the quality of the final FRET data is related to not only the functionality of the protein but also the degree of labeling and the percentage of molecules containing both the donor and the acceptor dye, because only those provide FRET information. Especially for smFRET studies, these two requirements, i.e., high labeling efficiency and retained biochemical functionality, are challenging hurdles. Unfortunately, no established quality criteria exist. Optimized labeling protocols25 and using a bias-free diffusion-based method can prevent “cherry picking” when individual immobilized molecules are being studied at later stages.
It becomes clear that labeling is a crucial step in biophysical smFRET studies and is inherently complex when oligomeric or multisubunit proteins are studied. In this paper, we exploit reductive caging of cyanine fluorophores and photoactivatable rhodamine fluorophores for smFRET studies of exactly such complex biochemical systems. We present proof-of-principle experiments and a characterization of the photochemical uncaging process of dye-labeled oligonucleotides and proteins during their transit through a confocal excitation volume. Using a method dubbed “caged FRET”, we show that chemical caging and ultraviolet (UV) reactivation allows temporal uncoupling of convoluted fluorescence signals from, e.g., multiple donor or acceptor molecules. We use fluorescently labeled oligonucleotides, i.e., ruler structures, and the trimeric membrane transporter BetP as examples, demonstrating how caged FRET removes unwanted molecular species with more than two identical labels and hence allows proper interpretation of solution-based smFRET data. We finally outline further potential applications of the “caged FRET” methodology for studying weak biochemical interactions that are yet impossible to study with diffusion-based smFRET because of requirements for low concentrations of fluorescently labeled molecules.
Materials and Methods
Preparation of Labeled Oligonucleotides and Reagents
Unless otherwise stated, reagents of luminescent grade were used as received. Chemical compounds such as 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox), dithiothreitol (DTT), bovine serum albumin (BSA), methylviologen (MV), and tris(2-carboxyethyl)phosphine (TCEP) were purchased from Sigma-Aldrich. Fluorescently labeled 45 bp oligonucleotides were used as received (IBA). Labels comprise tetramethylrhodamine (TMR, IBA), Cy5 (GE Healthcare), ATTO647N (Atto-Tec), and Cage552 (Abberior). DNA single strands were annealed17 and stored in 10 mM Tris-HCl-containing buffer at suitable salt concentrations. Four different double-stranded DNA (dsDNA) scaffolds were used (Figure 1). For experiments that examined a reduced level of caging by TCEP, three DNA scaffolds (ds1–3) are used carrying the donor TMR at position 17 of the top strand. The acceptor (Cy5) was attached at position 8 (ds1), at position 33 (ds2), and at both positions of the bottom strand (ds3). The last DNA scaffold (cds4) is labeled with two donor fluorophores (Cage552) at the 5′ end and position 27 on the top strand. The corresponding acceptor is positioned on the bottom strand at position 18.
Figure 1.

DNA oligonucleotide sequences and fluorophore labeling positions.
Bacterial Strains, Plasmids, and Growth Conditions
The pASK-IBA5betP vector was used for heterologous expression of Strep-BetP and transformed into competent DH5α-T1 cells (Invitrogen). Cells were grown at 37 °C in Luria-Bertani medium supplemented with carbenicillin (50 μg/mL). Induction was initiated with anhydrotetracycline (200 μg/L), and cells were harvested after they had reached the stationary phase. Membranes were isolated and solubilized using N-dodecyl β-dodecyl-maltoside (DDM), and after ultracentrifugation, the supernatant was supplemented with 1 mM DTT and loaded onto a StrepTactin column (IBA GmbH), which was washed with 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 8.6% glycerol, and 0.1% DDM. The protein was eluted with the same buffer containing 5 mM desthiobiotin and loaded into an equilibrated size exclusion column (Superose 6 10/300 GL) for further evaluation.
Transport Measurements of BetP-Cysteine Mutants in Cells
The uptake of the 14C-labeled substrate by Escherichia coli cells was performed as described in ref (58). E. coli MKH13 cells expressing the strep-BetP mutant were cultivated at 37 °C in LB medium containing carbenicillin (50 μg/mL) and induced at an OD600 of 0.5 by adding anhydrotetracycline (200 μg/L) to the growth medium. After growing for an additional 2 h, the cells were harvested and washed with a buffer containing 25 mM KPi (pH 7.5) and 100 mM NaCl and then resuspended in the same buffer containing 20 mM glucose. For uptake measurements of radiolabeled substrates, the external osmolality was adjusted with KCl. Cells were incubated for 3 min at 37 °C before the addition of 250 μM 14C-labeled substrate for osmotic regulation profiles. Uptake was measured at various time intervals after the cell samples were passed through glass fiber filters and washed twice with 2.5 mL of 0.6 M KPi buffer. The radioactivity retained on the filters was quantified by liquid scintillation counting.
Labeling of BetP Derivatives with Thiol-Specific Reagents
BetP cysteine-containing derivatives were obtained as described previously30,59 and stored at −20 °C in 500 μL aliquots (1–6 mg/mL) in 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 8.6% glycerol, and 0.1% DDM. Stochastic labeling with maleimide derivatives of donor and acceptor fluorophores was performed on ∼5 nmol of protein. Proteins were labeled with Alexa 555-maleimide (donor) and Alexa647-maleimide (acceptor) in a protein:donor:acceptor ratio of 1:4:3. Briefly, purified proteins were diluted and treated with 10 mM DTT for 60 min in a deoxygenated buffer that consisted of 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 8.6% glycerol, and 0.1% DDM (buffer A), to fully reduce oxidized cysteines. The protein mix was further diluted to a DTT concentration of 1 mM and loaded into an equilibrated desalting column (ZEBA, 2 mL) with a 7 kDa molecular weight cut-off to remove the DTT from the protein solution. The protein was washed with deoxygenated buffer that consisted of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% DDM (buffer B). Simultaneously, the applied fluorophore stocks (50 nmol dissolved in 5 μL of anhydrous dimethyl sulfoxide) were added to appropriate amounts of buffer B, immediately applied to the protein solution, and incubated for 3 h at 4 °C (under mild agitation). After labeling, unreacted dyes was removed when the samples were sequentially washed with buffer B and a ZEBA desalting column. The protein was eluted in 500 μL of buffer B and analyzed with a size exclusion column (Superose 6 10/300 GL) equilibrated with 50 mM Tris-HCl (pH 7) and 200 mM NaCl with 0.1% DDM.
Steady-State Fluorescence Anisotropy
Free fluorophore rotation and hence the correlation between FRET efficiency and distance were validated by steady-state anisotropy measurements of BetP with Alexa dyes showing R values of ≤0.22 (Table 1). The values for the dyes on protein were even lower than those found on established double-stranded ruler DNAs with a known donor−acceptor separation of 13 bp, where we established before that FRET indeed serves as a molecular ruler.17 We used a published theory60 to estimate the relative error associated with distance determination in both dsDNA and BetP when erroneously assuming a fixed dye orientation (κ ∼ 2/3). Haas and co-workers provide this error as ratio r/r′ of true distance r to apparent distance r′; this ratio (=uncertainty) is moderate for anisotropies R < 0.3 of both dyes.60 We found r/r′ < 20% for dsDNA and BetP variants. The experimental procedure for determining anisotropy values R can be summarized as follows. Fluorescence spectra were derived on a standard scanning spectrofluorometer (Jasco FP-8300, 20 nm excitation and emission bandwidth, 8 s integration time) and calculated at the emission maxima of the fluorophores (for donor, λex = 535 nm and λem = 580 nm; for acceptor, λex = 635 nm and λem = 660 nm), according to the relationship R = (IVV – GIVH)/(IVV + 2GIVH), where IVV and IVH describe the emission components relative to the vertical (V) or horizontal (H) orientation of the excitation and emission polarizers, respectively. The sensitivity of the spectrometer to different polarizations was corrected using horizontal excitation to obtain G = IHV/IHH. Typical G values for donor and acceptor dyes were 0.47 and 0.49, respectively. We used 50 mM Tris-HCl, 200 mM NaCl, and 0.1% DDM (pH 7.5) as a buffer and analyzed the anisotropy of the labeled protein and DNA samples in a concentration range of 50–2000 nM. The determined anisotropy values are summarized in Table 1.
Table 1. Anisotropies (R) Derived from Ensemble Measurements.
| anisotropy R |
||
|---|---|---|
| compound | Alexa555 | Alexa647 |
| free dye | 0.19 ± 0.01 | 0.16 ± 0.01 |
| Ds42 (dsDNA/donor-13 bp-acceptor) | 0.22 ± 0.02 | 0.22 ± 0.03 |
| BetPC252T/S516C | 0.19 ± 0.02 | 0.19 ± 0.01 |
Sample Preparation for Single-Molecule Experiments
ALEX experiments were performed at room temperature with a 25–50 pM solution of protein and DNA samples. For the DNA sample, we used imaging buffer phosphate-buffered saline (PBS) at pH 9.0, containing 2 mM Trolox and 2 mM MV with varying concentrations of TCEP; the pH value of the respective buffer was adjusted after addition of TCEP. Protein samples were also analyzed at 25–50 pM in an imaging buffer containing 50 mM Tris, 150 mM NaCl, 2 mM Trolox, 2 mM MV, 0.1% DDM, and varying concentrations of TCEP; the pH value was either 7.4 or 9 (see the text for details). In typical single-molecule experiments, sample solutions were transferred to coverslips that were previously incubated with 1 mg/mL BSA for 5 min for surface passivation.
Single-Molecule FRET and ALEX Spectroscopy
We used a custom-built confocal microscope for μs-ALEX, which we described in detail previously.17,61 In brief, the setup was extended by a single-line 375 nm UV laser (Coherent, Obis) that was employed at power densities of ≤500 kW/cm2 at the confocal volume. A 60× oil-immersion objective with NA = 1.35 (Olympus, UPLSAPO 60XO) or a water-immersion objective with NA = 1.2 was used to generate a diffraction-limited excitation spot. The excitation intensity was typically set to 30–60 μW at 532 nm and 15–25 μW at 640 nm with an alternation period of 50 μs. Fluorescence emission was collected in epi-fluorescence mode, spatially filtered by a 50 μm pinhole, matching bandpass filters, and registered by two avalanche photodiode detectors (τ-Spad, Picoquant).
In this mode, three photon streams were extracted from the data corresponding to donor-based donor emission DD, donor-based acceptor emission DA, and acceptor-based acceptor emission AA. S and apparent FRET efficiencies E* were calculated for each fluorescent burst during their diffusion time trough confocal spot above a certain threshold, yielding a two-dimensional (2D) histogram. Uncorrected FRET efficiency E* is calculated according to the equation E* = DA/[DD + DD]. Stoichiometry S is defined as the ratio between the overall green fluorescence intensity to the total green and red fluorescence intensity during the green excitation period and describes the ratio of donor to acceptor fluorophores in the sample {S = DA + DD/[DD + DA + AA]}.
Using published procedures to identify fluorescent bursts corresponding to single molecules, we obtained bursts characterized by three parameters (M, T, and L).17 A fluorescent signal is considered a burst provided it meets the following criteria: a total of L photons having M neighboring photons within a time interval of T microseconds. For data shown in Figures 2 and 7, an all-photon burst search with parameters of M = 15, T = 500 μs, and L = 50 was applied; for data shown in Figures 4–6, a dual-color burst search using parameters of M = 15, T = 500 μs, and L = 25 was applied. Additional thresholding removed spurious changes in fluorescence intensity and selected for intense single-molecule bursts (all photons >100/150 photons unless otherwise mentioned). The detected bursts were binned into a 2D E*/S histogram in which subpopulations are separated according to their S values. E* and S distributions were fitted using a Gaussian function, yielding mean values μi of the distribution and an associated standard deviations wi. Experimental values for E* and S were corrected for background (Figures 2 and 7) and additionally for spectral crosstalk (Figures 4–6) according to published procedures.17
Figure 2.

Structure and FRET properties of the homotrimeric C252T/S516C BetP mutant. (A) Side and top views of the crystal structure of the mutant marking the three label positions and related distances. Protein Data Bank entry 4AIN. (B) Normalized uptake rate of Cys-less BetP (wt) and BetP cysteine mutant C252T/S516C in E. coli cells depending on osmotic stress. The relative rate of uptake of 14C-labeled betaine by E. coli cells for wild-type protein (green) is comparable to that of the mutant protein (red, C252T/S516C), which exhibits one-third of the total wild-type activity. (C) Cartoon of different labeling possibilities, including their degeneracy. (D) 2D ALEX histogram of Alexa555/647-labeled BetPS516C showing the convolution of FRET interactions and difficulties in using these data for structural analysis. (E) Photon count rate of single-molecule bursts from different subpopulations in the S region between 0.2 and 0.8. (F) Related one-dimensional E* histograms of the different species.
Figure 7.

Caged FRET investigations of BetP(C252T/S516C) with a periplasmic label position. (A) ALEX histogram of labeled BetP using an excess of acceptor dye to remove donor–donor–acceptor species. (B) BetP ALEX data set with one relevant donor–acceptor population via use of 1.5 mM TCEP-containing buffer. (C) Frequency of the photon count rate of acceptor emission signals as a function of TCEP concentration. (D) Relative numbers of molecules in the different populations as a function of TCEP concentration. Molecules were assigned by use of stoichiometry threshold values indicated in panels A and B.
Figure 4.

Caged FRET methodology implemented in μs-ALEX. (A) Confocal ALEX microscope extended by an additional UV laser in continuous-wave mode. (B) Cartoon view of the excitation volume where diffusing species produce only green signals (top, caged acceptor) and both green and red signals (bottom, UV-activated acceptor) with the corresponding photon stream shown in panel C for an applied UV power of ∼100 kW/cm2. (D) 2D ALEX histograms of dsDNA in PBS (pH 9) under different buffer conditions: active acceptor (PBS), caged/inactive acceptor (PBS with 50 mM TCEP), and photoactivated acceptor (PBS with 50 mM TCEP at ∼100 kW of UV power/cm2) illustrating the caged FRET methodology. (E) Associated frequency histograms of photon count rates in different detection channels: donor-based donor emission (DD), donor-based acceptor emission (DA), and acceptor-based acceptor emission (AA). Distributions were obtained after applying a standard burst search (see Materials and Methods) and subsequent normalization of fluorescence signals in each burst to its respective duration to obtain normalized count rates in kHz.
Figure 6.

Caged FRET allows the determination of two distances. (A) TCEP caging of dsDNA containing TMR donor and two Cy5 acceptors. (B) DNA with two Cage552 donors and one ATTO647N acceptor. The figure shows that under conditions with more than two labels the FRET information is ambiguous because of fluorophore interactions that afterward cannot be disentangled. The desired information can be seen in FRET efficiency histograms in panels 1 and 2 (A) and 1–3 (B). The convoluted FRET histogram is shown in panel 3 (A); caged conditions are shown in panel 4. The desired information can be restored with caged FRET as seen in panel 5 (A and B).
Results
Various methods and approaches that allow specific incorporation of fluorescent labels into nucleic acids and proteins for in vitro biophysical studies exist.19,21,26 While synthetic oligonucleotides with labels or reactive groups can be purchased, proteins are a more challenging target. The most straightforward approach uses incorporation of cysteines at strategic positions, which allows stochastic labeling with two distinct fluorophores, e.g., for a FRET assay. In multimeric proteins or multisubunit proteins, however, this approach is complicated by ambiguous interactions of the fluorescent labels. Although site-specific labeling using unnatural amino acids20 allows for selective tagging of more than two positions in protein complexes, labeling of multimeric proteins for smFRET studies remains challenging. Our group has recently started to explore the structure–function relationship and molecular mechanisms of active membrane transporters with single-molecule FRET.8 In this study, we describe the first smFRET studies on the homotrimeric osmoregulated transporter BetP (Figure 2A) that serves as a good example of complications encountered when labeling multimeric proteins.
The sodium-coupled betaine symporter BetP from Corynebacterium glutamicum is a well-characterized member of the betaine–choline–carnitine transporter (BCCT) family.27 Several crystal structures show BetP as an asymmetric trimer, in which each protomer can adopt distinct conformations. These were assigned as individual transport states in the alternating access cycle28 that allow uphill substrate transport driven by the electrochemical Na+ potential, i.e., accumulation of betaine, which is the exclusive substrate for BetP, to molar amounts in the cytosol under hyperosmotic conditions.28 Thus, BetP has two major biochemical functions: sensing of osmotic stress and regulated transport of betaine. The 45-amino acid α-helical C-terminal domain of BetP binds cytoplasmic K+, which is a prerequisite for activation of BetP during hyperosmotic stress.29 The catalytic domain of BetP consists of 12 transmembrane helices (TMs) and is divided into a transporter core of two inverted five-helix repeats (TM3–TM12) and the two N-terminal helices, TM1 and TM2, which contribute to the trimer contacts. The symmetry between the two repeats (TM3–TM7 and TM8–TM12) is a key to the alternating access mechanism in BetP. For this study, cysteine mutants of BetP were designed to establish a dynamic picture of its structure–function relationship. Mutants were constructed on a cysteine-less BetP (C252T, TM5) containing an engineered cysteine at periplasmic position 516 in transmembrane domain 12, TM12 (Figure 2A). This position is part of the periplasmic gate and undergoes subtle conformational changes in the range of 3 Å during the isomerization from outward- to inward-facing states. Thus we expect only small changes in BetP structure that can be read out with this mutant via FRET. It serves, however, as a relevant example of the type of problems encountered with labeling during smFRET studies of multimeric proteins. The mutant protein was purified and solubilized in a detergent solution according to published procedures30 as described in Materials and Methods. It shows slightly reduced uptake activity but an activation profile and potassium dependence comparable to those of wild-type BetP31 (Figure 2B).
The top view of the BetP crystal structure (Figure 2A) reveals the problems of the FRET approach of a multimeric protein. Because the protein is expressed and purified as a stable homotrimer, the cysteine residue appears in each subunit. Stochastic labeling with the donor and acceptor fluorophore results in a mixture of different subpopulations, comprising various donor-only, acceptor-only, and donor–acceptor species with distinct degeneracy (Figure 2C).
We used μs-ALEX (microsecond alternating-laser excitation32), in which fluorescently labeled biomolecules diffuse through the excitation volume of a confocal microscope, for smFRET studies of BetP. During its diffusional transit, the labeled protein produces fluorescent bursts in two distinct detection channels that are chosen to selectively monitor donor and acceptor emission. In ALEX, green excitation of the sample generates fluorescent signals that allow calculation of apparent FRET E* and Stoichiometry S. While E* is indicative of the donor–acceptor separation, S distinguishes molecular species by their relative labeling ratio of green to red fluorophores. A low S of <0.2 is indicative of acceptor-only labeled protein, while a high S of >0.8 corresponds to a donor-only species. Macromolecules containing both dyes are found at S values between these two boundaries (0.2 > S > 0.8) (see Figure 2D).
A two-dimensional ALEX histogram of BetP reveals five different subpopulations, which cannot be used for further structural analysis of BetP without additional information and refinement of the experimental conditions (Figure 2D; labels donor Alexa555 and acceptor Alexa647). While donor- and acceptor-only species can be excluded from the analysis easily by considering only bursts within the S range of 0.2–0.8, Figure 2C suggests the existence of three possible species that contain both fluorophores: donor–donor–acceptor, donor–acceptor, and donor–acceptor–acceptor. To establish a direct link between S range and molecular composition, we analyzed the frequency distribution of photon count rates within single-molecule bursts. For this analysis of green DD and red AA emission channels, we separated the data set into three regions: (i) 0.2 > S > 0.4 (low S), (ii) 0.4 > S > 0.55 (intermediate S), and (iii) 0.55 > S > 0.68 (high S). The analysis shown in Figure 2E clearly reveals that the low-S regime corresponds to a donor–acceptor–acceptor species, the high-S regime is related to donor–donor–acceptor molecules, and only intermediate S values contain donor–acceptor molecules.
To understand which populations contain meaningful structural information in the form of E* distributions that are related to the donor–acceptor distances, we compared the E* histograms in the three regions with that of a double-stranded dsDNA with a 13 bp fluorophore separation, i.e., a distance similar to that of S516C label positions. We found that only the intermediate-S population provides the correct FRET measure while low-S and high-S populations show unexpected E* values. In species with more than two fluorophores, the relation of FRET efficiency and interprobe distance R seems to be lost because of the ambiguous interaction of, e.g., multiple donor with multiple acceptor fluorophores or signal loss via homo-FRET and energy dissipation. A change in the labeling ratio of donor to acceptor allows the relative abundance of the populations (data not shown) to shift; it remains, however, difficult to isolate a single donor–acceptor species.
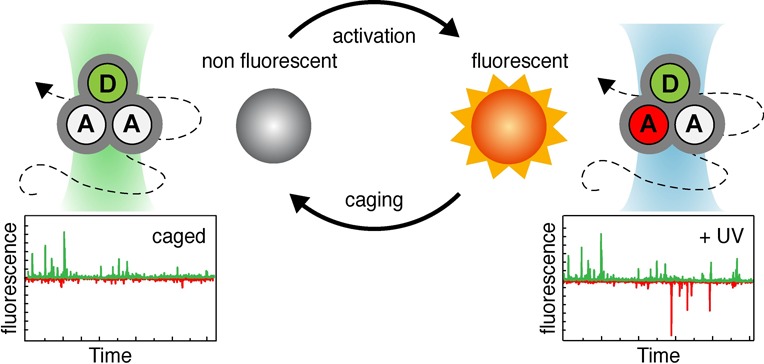
To solve these problems and to allow smFRET studies of BetP and other complex protein systems, where subpopulations can be assigned clearly, we developed a novel experimental concept that we dub caged FRET. Here, unwanted fluorophore interactions are prohibited via use of reductive caging of synthetic organic fluorophores. This approach is so far typically used in localization-based super-resolution microscopy33,34 and for FRET studies of surface-immobilized molecules using stochastic photoswitching.35,36 In caged FRET, a fluorescent dye is treated with reducing chemicals to disable fluorescence; the photoactivation and hence recovery of the fluorescent signal are achieved with UV light (Figure 3A). Cyanine dyes such as Cy5 are ideal for this because they undergo caging even with mild reducing agents such as TCEP (Figure 3B). As an example of the caging process, the concentration-dependent reaction of Cy5 was monitored via changes in the UV/vis absorption spectrum of the fluorophore (Figure 3C). The spectra also reveal that photoactivation (“uncaging”) by UV light is achieved efficiently for wavelengths of <375 nm. Both the efficiency of caging and photoactivation heavily depend on fluorophore structure, redox potential, reducing agent, and the presence of oxidizing compounds in the imaging buffer as described in the literature.33,34
Figure 3.

Caging of fluorescent dyes by reducing agents. (A) Principle. (B) Reductive caging of Cy5 by TCEP to a nonfluorescent from of Cy5 as described in ref (33). The fluorescent state can be recovered by absorption of UV light and subsequent photochemical uncaging. (C) Absorbance of a 5 μM solution of Cy5-NHS in PBS in the presence of varying concentrations of TCEP. Similar effects of ON/OFF-switching can be achieved using other reducing agents (e.g., NaBH4)34 or using synthetic caged fluorophores.37−39
Caged FRET is implemented in this study using μs-ALEX-based smFRET32 (Figure 4A) with diffusing biomolecules. We tested the concept with donor–acceptor-labeled double-stranded DNA (donor fluorophore TMR, acceptor Cy5). In a reducing buffer with 50 mM TCEP (pH 9), only “green” DD signals are observed at ∼50 pM dsDNA. As soon as an additional continuous-wave UV laser (375 nm) illuminates the sample also the sensitized acceptor signal via FRET can be observed (Figure 4B/C). Doubly labeled FRET species can hence be “switched” off by TCEP and activated with UV light as seen in the corresponding ALEX histograms in Figure 4D. The data shows a reduction of FRET bursts to less than 20% (Figure 4D, PBS vs PBS + TCEP). Caged molecules can be reactivated with an efficiency of 83%, a value that is close to the original level (Figure 4D, PBS + TCEP + UV). The achievable photon counts of both donor and acceptor are altered in systematic fashion when TCEP is added or UV illumination is applied (Figure 4E).
The analysis of photon count rates reveals that mostly the number of fluorescent molecules is decreased in all three channels when TCEP is added (Figure 4E), but both a high number of fluorescent molecules and the average brightness of donor and acceptor are restored after UV activation (Figure 4E). As seen in both Figure 4D/E the quality of the FRET histograms and the statistics are reduced insignificantly in caged FRET. The same results as presented for a high FRET sample with 8 bp separation between donor and acceptor fluorophore (Figure 4D/E) are also observed for intermediate or low FRET samples with 18 and 33 bp separation (data not shown).
It should be noted that the concrete distributions of photon count rates and ALEX histograms depend on the (subjective) choice of burst search parameters and per-bin thresholds applied, which has to be done in a consistent fashion for a set of data. Weighing algorithms that consider the statistical significance of a burst from obtained photon counts or other burst-related parameters40−44 would be preferential for data analysis instead of plotting each burst with a unity signal in the plot. While the data presented here show the working principle of caged FRET for caging of Cy5 with TCEP, it raises the question of how the quality of the data, the dye photophysics, and reactivation properties depend on the settings of the ALEX (green/red excitation power) and UV laser. An excellent analysis of caging and photoactivation properties of various fluorophores in TIRF-based super-resolution microscopy is given in refs (37), (39), and (45).
To understand the interrelation of setup parameters and fluorophore properties in caged FRET, we studied a DNA labeled with caged rhodamine Abberior Cage552. This nonfluorescent chromophore efficiently photoconverts into a structural analogue of the fluorophore TMR upon UV absorption.37−39 As seen in Figure 5A, Cage552 is nonfluorescent before UV activation, as indicated by the small amount of coincidence between the red and green signal (Figure 5A, DA species).
Figure 5.

Caged rhodamine fluorophores in smFRET. Fluorophore Cage552 can be activated by 375 nm excitation and serves as a FRET donor molecule after photochemical conversion. (A) 2D histogram of dsDNA labeled with ATTO647N and two donor molecules at distances of 17 and 9 bp. Upon 375 kW/cm2 UV radiation, the FRET population at EPr values of 0.5 and 0.9 is enhanced. (B) Corresponding frequency histogram of photon count rates of the donor species in the absence and presence of 375 kW of UV power/cm2. (C) Number of active donor molecules of the FRET species for 0 and 500 kW/cm2 UV excitation for 60/15 and 30/15 green/red power ALEX lasers. (D) Absolute ratio of DD, DA, and AA molecules to the total number of detected bursts as a function of applied UV laser power for a 60/15 green/red power of ALEX lasers.
The successful activation of Cage552 is also seen in a frequency histogram of photon count rates of DD of single-stranded DNA containing two Cage552 fluorophores (Figure 5B); we note that the bright fraction of molecules before photoactivation seen in Figure 5A–C could not be determined accurately because we also found that non-photoinduced uncaging occurs slowly on a time scale of weeks. We hence performed the set of experiments presented in Figure 5 within a short time interval. Thus, the calculated contrast between nonfluorescent and UV-induced bright molecules for Cage552 (Figure 5C) represents only a lower threshold. It can be improved by use of fresh Cage552 and, e.g., protein/DNA labeling only just before the respective experiment. The experiments reveal, however, that contrast values of >10 can be achieved with caged FRET using the two approaches with a caged donor or acceptor fluorophore (Figures 4 and 5). As seen by a comparison of relative acceptor– to donor–acceptor-containing molecules [via inspection of S distributions as a function of UV activation intensity (Figure 5D)], a linear dependence is observed for increasing levels of UV activation. Contrast and activation efficiency depend on UV laser power and on applied green/red excitation intensity and choice of burst search parameters (Figure 5C). At UV powers of >0.4 mW (500 kW/cm2), both elevated background signals in the green detection channel and increased acceptor photophysics were observed and are not recommended for caged FRET experiments.
As a next step, we tested caged FRET in DNA constructs with two acceptor (Figure 6A) or donor fluorophores (Figure 6B). Here, information about FRET processes is typically convoluted as for BetP and does not permit extraction of the desired information, i.e., donor–acceptor separation. This is shown in Figure 6A, where three different labeled DNAs are compared to each other. Two dsDNA with a TMR–Cy5 donor–acceptor pair show intermediate and high FRET according to interprobe separations of 17 and 9 bp, respectively (Figure 6A, panels 1 and 2). As soon as two acceptor dyes are simultaneously adjacent to the donor fluorophore, only a single FRET distribution with a high mean value is observed (Figure 6A, panel 3). This distribution does not contain information about the two molecular distances. Instead, the convoluted signal does not even allow the proper determination of one of the two distances. Upon application of reductive caging of the acceptor fluorophores by TCEP, the convoluted population is reduced (Figure 6A, panel 4). Subsequent UV activation leads to a stochastic mixture of uncaged molecules with one donor and one acceptor, where the latter has two distinct distances to the donor fluorophore (Figure 6A, panel 5). While the efficiency of the uncaging process is imbalanced, the information about the two donor–acceptor distances can be restored. Such behavior with more efficient activation of high-FRET species was also described previously.36 Although different high- and low-FRET samples could be uncaged with similar efficiency in the presence of only one donor and acceptor molecule (see Figure 4 for the high-FRET sample; low- and intermediate-FRET data not shown), the interactions are apparently more complex for the combined construct where two acceptor dyes are present.
To optimize the photoactivation process, we performed a similar experiment with two Cage552 donor fluorophores in combination with one ATTO647N acceptor fluorophore. As a reference, we analyzed a DNA-based ladder with 8, 13, and 18 bp separations for TMR-ATTO647N (Figure 6B, panels 1–3) showing the FRET ruler character. The silent as well as photoactivated DNA with a Cage552 donor show two different FRET species that can be distinguished clearly. However, much better statistics are obtained with additional UV illumination. As indicated above, the contrast with and without UV can be optimized further by fresh labeling. The results presented in Figure 6 suggest that donor-based activation with Cage552 is a practically more relevant method compared to use of TCEP caging with Cy5, because the activation efficiency of the Cage552 donor does not depend on FRET interactions with the acceptor fluorophore.
Finally, we tested caged FRET on the S516C mutant with the goal of fully isolating a donor–acceptor species. First, we used an excess of acceptor dye for labeling to bias the formation of donor–acceptor and donor–acceptor–acceptor species (Figure 7A). Under these conditions, we obtain only species in the low- and intermediate-S regime (<0.65), in agreement with data shown in Figure 2. Next, we applied caged FRET to remove the unwanted donor–acceptor–acceptor population at low S values via simple addition of low concentrations of TCEP to the buffer. This stochastically reduces the size of the active acceptor population (Figure 7B) and thereby allows us to obtain histograms with only one DA species related to a single (and relevant) distance between both probes.
We found that for this specific BetP mutant, pH 9 and 1.5 mM TCEP resulted in a significant decrease in the amount of unwanted DAA species (Figure 7C,D). When the number of molecules in D-only, A-only, and donor–acceptor-containing fractions (including both DA and DAA) is plotted as a function of TCEP concentration, it is apparent that caged FRET without photoactivation allows improvement of the clarity of the histogram. The latter of course has to be balanced with measurement time and overall data quality because the available mean photon counts (Figure 7D) of both the donor and the acceptor fluorophore are also reduced when TCEP is applied at increasing concentrations.
Discussion and Conclusion
We herein establish the use of caged fluorophores for smFRET studies of diffusing biomolecules. For the “caged FRET” methodology with photoactivation, we suggest the simple addition of an UV laser to a confocal microscope for photoactivation during diffusion. The applied laser wavelength needs to be chosen according to the absorbance properties of the caged species that is often found in a range below 400 nm.33,34,37 Using this approach, we could remove ambiguous interactions of fluorophores that appear in FRET assays of oligonucleotides and multisubunit proteins with more than two fluorescent labels. For this, we used caged rhodamines and reductive caging of cyanines with subsequent photoactivation. In an even simpler approach, reductive caging can be used to remove overlabeled protein (more than one donor or acceptor) without any UV activation as demonstrated in detergent-solubilized membrane transporter BetP. Caged FRET is also distinct from other established approaches such as photoswitchable FRET,35,36 which relies on surface-immobilized molecules and stochastic activation of, e.g., acceptor dyes. Stochastic switching would be compatible with caged FRET only if the photoswitching could be made substantially faster as is now to allow photoactivation during diffusion through the confocal volume.46,47
In the future, we envision that caged FRET not only will be useful for improvement of labeling properties but also might allow solution-based smFRET at elevated concentrations.48 This would allow studies of two interacting biomolecules with nanomolar to micromolar affinity.49 For such experiments, the respective biochemical partners would be labeled with a caged donor and caged acceptor. To allow smFRET observation, simultaneous photoactivation of both labels is needed during diffusion through the confocal volume. A strict requirement for such an assay is that both donor and acceptor fluorophore be caged and activated similarly well in the same buffer and for the same UV intensity. In this respect, a combination of caged FRET with local activation of dye,50 where a FRET acceptor is photoactivated (more) efficiently whenever it is close to the donor fluorophore, could be useful. Out of curiosity, we explored the practical limits of the general idea. When incubating a 1 μM solution of Cy5-COOH with 100 mM NaBH4 for 48 h, we observed <1 burst/s under standard ALEX conditions (data not shown), indicating that micromolar concentrations are indeed accessible. While the Cy5 fluorophore was caged effectively, the photoactivation reaction was extremely inefficient and has to be optimized for practical future use. The low activation efficiency of Cy5 in the presence of strong reducing agents is in accordance with published studies and relates to the need for strong oxidants. Ultimately, the achievable concentration of caged smFRET will be a compromise of different factors because effective caging is often linked to inefficient photoactivation. Thus, a potent donor and acceptor pair must be identified, where the two requirements, i.e., efficient caging and photoactivation, are fulfilled.
While the experiments presented here are of proof-of-principle character, they demonstrate the possibilities of temporal separation of fluorescent signals for FRET-based assays. Such a strategy is already widely used in localization-based super-resolution microscopy (PALM,51 STORM,52 and PAINT53). We consequently think that the caged FRET methodology relates to other multiruler techniques in a manner like stochastic super-resolution techniques (STORM and PALM) compare to targeted nanoscopy (STED and RESOLFT54,55). This idea might be useful for distinguishing multidimensional smFRET-based approaches such as photoswitchable FRET and caged FRET (temporal signal separation) from combinations of different rulers, e.g., PIFE-FRET,17,18 PET-FRET,56 or farFRET57 (spatial signal separation).
Acknowledgments
We thank G. Gkouridis for useful advice, J. Oelerich for experimental help with labeling of oligonucleotides, and J. Hohlbein for fruitful discussions regarding the systematic categorization of FRET techniques.
Author Present Address
∥ E.P.: Department of Chemistry and CeNS, Ludwig-Maximilians-Universität München, 81377 Munich, Germany.
Author Contributions
A.A.J. and E.P. contributed equally to this work.
This work was financed by the Zernike Institute for Advanced Materials, the Centre for Synthetic Biology (start-up grant to T.C.), and an ERC Starting Grant (ERC-STG 638536-SM-IMPORT to T.C.). A.A.J. was supported by Ubbo Emmius funding (University of Groningen) and by the DFG in the frame of SFB699. E.P. was supported by a DFG fellowship (PL696/2-1).
The authors declare no competing financial interest.
References
- Medintz I., and Hildebrandt N. (2013) FRET-Förster resonance energy transfer: From theory to applications, John Wiley & Sons, New York. [Google Scholar]
- Widom J. R.; Dhakal S.; Heinicke L. A.; Walter N. G. (2014) Single-molecule tools for enzymology, structural biology, systems biology and nanotechnology: an update. Arch. Toxicol. 88, 1965–1985. 10.1007/s00204-014-1357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschielok A.; Andrecka J.; Jawhari A.; Bruckner F.; Cramer P.; Michaelis J. (2008) A nano-positioning system for macromolecular structural analysis. Nat. Methods 5, 965–971. 10.1038/nmeth.1259. [DOI] [PubMed] [Google Scholar]
- Sisamakis E.; Valeri A.; Kalinin S.; Rothwell P. J.; Seidel C. A. (2010) Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 475, 455–514. 10.1016/S0076-6879(10)75018-7. [DOI] [PubMed] [Google Scholar]
- Craggs T. D.; Kapanidis A. N. (2012) Six steps closer to FRET-driven structural biology. Nat. Methods 9, 1157–1158. 10.1038/nmeth.2257. [DOI] [PubMed] [Google Scholar]
- Choi U. B.; Strop P.; Vrljic M.; Chu S.; Brunger A. T.; Weninger K. R. (2010) Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 17, 318–324. 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindbert S.; Kalinin S.; Nguyen H.; Kienzler A.; Clima L.; Bannwarth W.; Appel B.; Müller S.; Seidel C. A. M. (2011) Accurate Distance Determination of Nucleic Acids via Förster Resonance Energy Transfer: Implications of Dye Linker Length and Rigidity. J. Am. Chem. Soc. 133, 2463–2480. 10.1021/ja105725e. [DOI] [PubMed] [Google Scholar]
- Gouridis G.; Schuurman-Wolters G. K.; Ploetz E.; Husada F.; Vietrov R.; de Boer M.; Cordes T.; Poolman B. (2015) Conformational dynamics in substrate-binding domains influences transport in the ABC importer GlnPQ. Nat. Struct. Mol. Biol. 22, 57–64. 10.1038/nsmb.2929. [DOI] [PubMed] [Google Scholar]
- Nagy J.; Grohmann D.; Cheung A. C. M.; Schulz S.; Smollett K.; Werner F.; Michaelis J. (2015) Complete architecture of the archaeal RNA polymerase open complex from single-molecule FRET and NPS. Nat. Commun. 6, 6161. 10.1038/ncomms7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb N. C.; Cordes T.; Hwang L. C.; Gryte K.; Duchi D.; Craggs T. D.; Santoso Y.; Weiss S.; Ebright R. H.; Kapanidis A. N. (2013) The transcription bubble of the RNA polymerase-promoter open complex exhibits conformational heterogeneity and millisecond-scale dynamics: implications for transcription start-site selection. J. Mol. Biol. 425, 875–885. 10.1016/j.jmb.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. (2008) A practical guide to single-molecule FRET. Nat. Methods 5, 507–516. 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T.; Zhuang X.; Kim H. D.; Orr J. W.; Williamson J. R.; Chu S. (1999) Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. U. S. A. 96, 9077–9082. 10.1073/pnas.96.16.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T.; Ting A. Y.; Liang J.; Caldwell W. B.; Deniz A. A.; Chemla D. S.; Schultz P. G.; Weiss S. (1999) Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc. Natl. Acad. Sci. U. S. A. 96, 893–898. 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohlbein J.; Craggs T. D.; Cordes T. (2014) Alternating-laser excitation: single-molecule FRET and beyond. Chem. Soc. Rev. 43, 1156–1171. 10.1039/C3CS60233H. [DOI] [PubMed] [Google Scholar]
- Doose S.; Neuweiler H.; Sauer M. (2009) Fluorescence quenching by photoinduced electron transfer: a reporter for conformational dynamics of macromolecules. ChemPhysChem 10, 1389–1398. 10.1002/cphc.200900238. [DOI] [PubMed] [Google Scholar]
- Hwang H.; Kim H.; Myong S. (2011) Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc. Natl. Acad. Sci. U. S. A. 108, 7414–7418. 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploetz E.; Lerner E.; Husada F.; Roelfs M.; Chung S.; Hohlbein J.; Weiss S.; Cordes T. (2016) Förster resonance energy transfer and protein-induced fluorescence enhancement as synergetic multi-scale molecular rulers. Sci. Rep. 6, 33257. 10.1038/srep33257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner E.; Ploetz E.; Hohlbein J.; Cordes T.; Weiss S. (2016) A Quantitative Theoretical Framework For PIFE-FRET. J. Phys. Chem. B 120, 6401–6410. 10.1021/acs.jpcb.6b03692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S.; Lemke E. A. (2015) Single-molecule FRET and crosslinking studies in structural biology enabled by noncanonical amino acids. Curr. Opin. Struct. Biol. 32, 66–73. 10.1016/j.sbi.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Brustad E. M.; Lemke E. A.; Schultz P. G.; Deniz A. A. (2008) A General and Efficient Method for the Site-Specific Dual-Labeling of Proteins for Single Molecule Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 130, 17664–17665. 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis A. N.; Ebright Y. W.; Ebright R. H. (2001) Site-specific incorporation of fluorescent probes into proteins: hexahistidine-tag-mediated fluorescent labeling with (Ni(2+):nitrilotriacetic Acid (n)-fluorochrome conjugates. J. Am. Chem. Soc. 123, 12123–12125. 10.1021/ja017074a. [DOI] [PubMed] [Google Scholar]
- Seo Y. J.; Malyshev D. A.; Lavergne T.; Ordoukhanian P.; Romesberg F. E. (2011) Site-specific labeling of DNA and RNA using an efficiently replicated and transcribed class of unnatural base pairs. J. Am. Chem. Soc. 133, 19878–19888. 10.1021/ja207907d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod S. H.; Baccaro A.; Marx A. (2011) Site-specific DNA labeling by Staudinger ligation. Methods Mol. Biol. 751, 195–207. 10.1007/978-1-61779-151-2_12. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Ho S. O.; Gassman N. R.; Korlann Y.; Landorf E. V.; Collart F. R.; Weiss S. (2008) Efficient Site-Specific Labeling of Proteins via Cysteines. Bioconjugate Chem. 19, 786–791. 10.1021/bc7002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husada F.; Gouridis G.; Vietrov R.; Schuurman-Wolters G. K.; Ploetz E.; de Boer M.; Poolman B.; Cordes T. (2015) Watching conformational dynamics of ABC transporters with single-molecule tools. Biochem. Soc. Trans. 43, 1041–1047. 10.1042/BST20150140. [DOI] [PubMed] [Google Scholar]
- Chakraborty A.; Wang D.; Ebright Y. W.; Ebright R. H. (2010) Azide-specific labeling of biomolecules by Staudinger-Bertozzi ligation phosphine derivatives of fluorescent probes suitable for single-molecule fluorescence spectroscopy. Methods Enzymol. 472, 19–30. 10.1016/S0076-6879(10)72018-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler C.; Bremer E.; Kramer R. (2010) The BCCT family of carriers: from physiology to crystal structure. Mol. Microbiol. 78, 13–34. 10.1111/j.1365-2958.2010.07332.x. [DOI] [PubMed] [Google Scholar]
- Perez C.; Koshy C.; Yildiz O.; Ziegler C. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature 490, 126–130. 10.1038/nature11403. [DOI] [PubMed] [Google Scholar]
- Kappes R. M.; Kempf B.; Bremer E. (1996) Three transport systems for the osmoprotectant glycine betaine operate in Bacillus subtilis: characterization of OpuD. J. Bacteriol. 178, 5071–5079. 10.1128/jb.178.17.5071-5079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenhagen R.; Morbach S.; Kramer R. (2001) The osmoreactive betaine carrier BetP from Corynebacterium glutamicum is a sensor for cytoplasmic K+. EMBO J. 20, 5412–5420. 10.1093/emboj/20.19.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclawska I. J. (2015) Dynamics in Stress-regulated Betain trasnport and role of pathogen-relevant choline transport. Ph.D. Thesis, University of Regensburg, Regensburg, Germany.
- Kapanidis A. N.; Laurence T. A.; Lee N. K.; Margeat E.; Kong X.; Weiss S. (2005) Alternating-laser excitation of single molecules. Acc. Chem. Res. 38, 523–533. 10.1021/ar0401348. [DOI] [PubMed] [Google Scholar]
- Vaughan J. C.; Dempsey G. T.; Sun E.; Zhuang X. (2013) Phosphine quenching of cyanine dyes as a versatile tool for fluorescence microscopy. J. Am. Chem. Soc. 135, 1197–1200. 10.1021/ja3105279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan J. C.; Jia S.; Zhuang X. (2012) Ultrabright photoactivatable fluorophores created by reductive caging. Nat. Methods 9, 1181–1184. 10.1038/nmeth.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsang J.; Cordes T.; Tinnefeld P. (2009) Single-molecule photophysics of oxazines on DNA and its application in a FRET switch. Photochemical & Photobiological Sciences 8, 486–496. 10.1039/b822318c. [DOI] [PubMed] [Google Scholar]
- Uphoff S.; Holden S. J.; Le Reste L.; Periz J.; van de Linde S.; Heilemann M.; Kapanidis A. N. (2010) Monitoring multiple distances within a single molecule using switchable FRET. Nat. Methods 7, 831–836. 10.1038/nmeth.1502. [DOI] [PubMed] [Google Scholar]
- Belov V. N.; Mitronova G. Y.; Bossi M. L.; Boyarskiy V. P.; Hebisch E.; Geisler C.; Kolmakov K.; Wurm C. A.; Willig K. I.; Hell S. W. (2014) Masked Rhodamine Dyes of Five Principal Colors Revealed by Photolysis of a 2-Diazo-1-Indanone Caging Group: Synthesis, Photophysics, and Light Microscopy Applications. Chem. - Eur. J. 20, 13162–13173. 10.1002/chem.201403316. [DOI] [PubMed] [Google Scholar]
- Aloi A.; Vargas Jentzsch A.; Vilanova N.; Albertazzi L.; Meijer E. W.; Voets I. K. (2016) Imaging Nanostructures by Single-Molecule Localization Microscopy in Organic Solvents. J. Am. Chem. Soc. 138, 2953–2956. 10.1021/jacs.5b13585. [DOI] [PubMed] [Google Scholar]
- Belov V. N.; Wurm C. A.; Boyarskiy V. P.; Jakobs S.; Hell S. W. (2010) Rhodamines NN: A Novel Class of Caged Fluorescent Dyes. Angew. Chem., Int. Ed. 49, 3520–3523. 10.1002/anie.201000150. [DOI] [PubMed] [Google Scholar]
- Enderlein J.; Robbins D. L.; Ambrose W. P.; Goodwin P. M.; Keller R. A. (1997) The statistics of single-molecule detection: an overview. Bioimaging 5, 88–98. . [DOI] [Google Scholar]
- Ingargiola A.; Lerner E.; Chung S.; Weiss S.; Michalet X. (2016) FRETBursts: Open Source Burst Analysis Toolkit for Confocal Single-Molecule FRET. PLoS One 11 (8), e0160716. 10.1371/journal.pone.0160716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingargiola A.; Laurence T.; Boutelle R.; Weiss S.; Michalet X. (2016) Photon-HDF5: An Open File Format for Timestamp-Based Single-Molecule Fluorescence Experiments. Biophys. J. 110, 26–33. 10.1016/j.bpj.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingargiola A.; Laurence T.; Boutelle R.; Weiss S.; Michalet X. (2016) Open Computational Tools for Freely Diffusing Single-Molecule Fluorescence Analysis. Biophys. J. 110, 634a. 10.1016/j.bpj.2015.11.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfs M. (2014) A novel algorithm for reduced data acquisition times in diffusion-based single-molecule FRET microscopy. Bsc Thesis, University of Groningen, Groningen, The Netherlands. [Google Scholar]
- Wysocki L. M.; Grimm J. B.; Tkachuk A. N.; Brown T. A.; Betzig E.; Lavis L. D. (2011) Facile and general synthesis of photoactivatable xanthene dyes. Angew. Chem., Int. Ed. 50, 11206–11209. 10.1002/anie.201104571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilemann M.; Margeat E.; Kasper R.; Sauer M.; Tinnefeld P. (2005) Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 127, 3801–3806. 10.1021/ja044686x. [DOI] [PubMed] [Google Scholar]
- Vogelsang J.; Cordes T.; Forthmann C.; Steinhauer C.; Tinnefeld P. (2009) Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc. Natl. Acad. Sci. U. S. A. 106, 8107–8112. 10.1073/pnas.0811875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling C.; Hilbert M.; Bock H.; Ringemann C.; Hofmann M.; Stiel A. C.; Andresen M.; Jakobs S.; Egner A.; Schonle A.; Hell S. W. (2007) Reversible photoswitching enables single-molecule fluorescence fluctuation spectroscopy at high molecular concentration. Microsc. Res. Tech. 70, 1003–1009. 10.1002/jemt.20505. [DOI] [PubMed] [Google Scholar]
- Holzmeister P.; Acuna G. P.; Grohmann D.; Tinnefeld P. (2014) Breaking the concentration limit of optical single-molecule detection. Chem. Soc. Rev. 43, 1014–1028. 10.1039/C3CS60207A. [DOI] [PubMed] [Google Scholar]
- Geertsema H. J.; Schulte A. C.; Spenkelink L. M.; McGrath W. J.; Morrone S. R.; Sohn J.; Mangel W. F.; Robinson A.; van Oijen A. M. (2015) Single-Molecule Imaging at High Fluorophore Concentrations by Local Activation of Dye. Biophys. J. 108, 949–956. 10.1016/j.bpj.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Rust M. J.; Bates M.; Zhuang X. (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (storm). Nat. Methods 3, 793–796. 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharonov A.; Hochstrasser R. M. (2006) Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. U. S. A. 103, 18911–18916. 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S. W. (2007) Far-field optical nanoscopy. Science 316, 1153–1158. 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- Hell S. W.; Sahl S. J.; Bates M.; Zhuang X.; Heintzmann R.; Booth M. J.; Bewersdorf J.; Shtengel G.; Hess H.; Tinnefeld P.; Honigmann A.; Jakobs S.; Testa I.; Cognet L.; Lounis B.; Ewers H.; Davis S. J.; Eggeling C.; Klenerman D.; Willig K. I.; Vicidomini G.; Castello M.; Diaspro A.; Cordes T. (2015) The 2015 super-resolution microscopy roadmap. J. Phys. D: Appl. Phys. 48, 443001. 10.1088/0022-3727/48/44/443001. [DOI] [Google Scholar]
- Haenni D.; Zosel F.; Reymond L.; Nettels D.; Schuler B. (2013) Intramolecular Distances and Dynamics from the Combined Photon Statistics of Single-Molecule FRET and Photoinduced Electron Transfer. J. Phys. Chem. B 117, 13015–13028. 10.1021/jp402352s. [DOI] [PubMed] [Google Scholar]
- Krainer G.; Hartmann A.; Schlierf M. (2015) farFRET: Extending the Range in Single-Molecule FRET Experiments beyond 10 nm. Nano Lett. 15, 5826–5829. 10.1021/acs.nanolett.5b01878. [DOI] [PubMed] [Google Scholar]
- Ott V.; Koch J.; Spate K.; Morbach S.; Kramer R. (2008) Regulatory properties and interaction of the C- and N-terminal domains of BetP, an osmoregulated betaine transporter from Corynebacterium glutamicum. Biochemistry 47, 12208–12218. 10.1021/bi801325r. [DOI] [PubMed] [Google Scholar]
- Peter H.; Burkovski A.; Kramer R. (1998) Osmo-sensing by N- and C-terminal extensions of the glycine betaine uptake system BetP of Corynebacterium glutamicum. J. Biol. Chem. 273, 2567–2574. 10.1074/jbc.273.5.2567. [DOI] [PubMed] [Google Scholar]
- Haas E.; Katchalski-Katzir E.; Steinberg I. Z. (1978) Effect of the orientation of donor and acceptor on the probability of energy transfer involving electronic transitions of mixed polarization. Biochemistry 17, 5064–5070. 10.1021/bi00616a032. [DOI] [PubMed] [Google Scholar]
- van der Velde J. H. M.; Ploetz E.; Hiermaier M.; Oelerich J.; de Vries J. W.; Roelfes G.; Cordes T. (2013) Mechanism of Intramolecular Photostabilization in Self-Healing Cyanine Fluorophores. ChemPhysChem 14, 4084–4093. 10.1002/cphc.201300785. [DOI] [PubMed] [Google Scholar]