

Abstract
Protein–protein interactions stabilized by multiple separate hot spots are highly challenging targets for synthetic scaffolds. Surface‐mimetic foldamers bearing multiple recognition segments are promising candidate inhibitors. In this work, a modular bottom‐up approach is implemented by identifying short foldameric recognition segments that interact with the independent hot spots, and connecting them through dynamic covalent library (DCL) optimization. The independent hot spots of a model target (calmodulin) are mapped with hexameric β‐peptide helices using a pull‐down assay. Recognition segment hits are subjected to a target‐templated DCL ligation through thiol–disulfide exchange. The most potent derivative displays low nanomolar affinity towards calmodulin and effectively inhibits the calmodulin–TRPV1 interaction. The DCL assembly of the folded segments offers an efficient approach towards the de novo development of a high‐affinity inhibitor of protein–protein interactions.
Keywords: dynamic covalent chemistry, foldamers, molecular recognition, peptidomimetics, protein–protein interactions
Finding synthetic means of targeting protein–protein interactions (PPIs) is a major challenge in chemistry. The class of PPIs in which a “hot spot pocket” or a contiguous system of conserved clefts is responsible for binding of the clustered “hot spot residues” projected from a secondary structure interface can be inhibited by secondary structure mimetics1 and small molecules.2 It is however inherently difficult to access two or more separated hot spots that accept residues from a non‐continuous peptide epitope or a flat surface of a globular protein.3 PPIs stabilized by symmetrically distributed anchor points have been targeted by multivalent surface mimetics.4 The structurally and enzymatically stable biomimetic foldamers are among the most promising scaffolds with which to generate tailor‐made protein recognition surfaces and PPI inhibitors.5 It has been shown for PPIs with single‐helix interfaces that β‐ and α/β‐peptidic foldamers produce excellent structural mimetics to decouple these interactions. Contiguous hot spot clusters with structurally complex or uncharacterized interacting partners have also been targeted in top‐down backbone homologation6 and bottom‐up de novo design7 approaches. Although the chemical accessibility and the programmable structure of peptidic foldamers are attractive features, these scaffolds have not been systematically tested on PPIs with two or more spatially non‐contiguous hot spot pockets. Such a surface‐mimetic foldamer can be constructed de novo in a modular way by finding the secondary structure elements that recognize the independent hot spots, and by connecting the binder segments in an optimized combination with a suitable flexible linker. Here, we set out to test this de novo bottom‐up approach on a protein that displays two separate hot spots. As a model protein, calmodulin (CaM) was selected, for which N‐ and C‐terminal EF‐hand motifs form the methionine‐rich hot spots that are connected by a flexible region. CaM is a well‐known model for protein recognition and inhibition studies.8 In our modular development workflow, we attempted to capture the canonical protein binding mode of calmodulin with high affinity, which recognizes a discontinuous epitope, displaying the hydrophobic anchor residues on the opposite faces of a helix within a distance of 3–5 helical turns (1.5–2.5 nm).9
Our hypothesis was that the target hot spot pockets could be mapped using short foldameric segments mimicking the local environment of the hot spot residues in terms of side‐chain presentation and solvent shielding (Figure 1 a). In order to address the problem of simultaneous optimization of the recognition segments and the linkage, a dynamic covalent library (DCL) method was deployed (Figure 1 b).10
Figure 1.
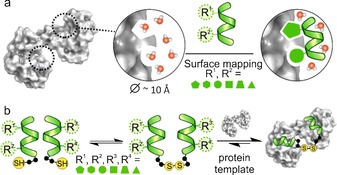
The concept of dynamically assembled folded segments. a) Mapping of the protein surface by a short folded segment library with protruding proteinogenic side chains and local solvent shielding. b) Self‐sorting of the folded segments in the presence of the protein target using a dynamic combinatorial library based on a disulfide‐exchange reaction, and selection of the highest‐affinity ligand.
The scaffold for the folded segments was chosen to match the geometrical requirements of a hot spot pocket in general. It was equipped with protruding proteinogenic side chains and designed to be sufficiently rigid and bulky to locally exclude the solvent from the binding cleft. A hexameric 14‐helical β‐peptide scaffold11 with a diameter of 10 Å fulfilled these requirements. The structure was stabilized by trans‐1,2‐aminocyclohexane amino acids and projected two proteinogenic side chains of β3‐amino acids from the same face (positions 2 and 5) of the folded helix (Figure 2 a). To cover the diverse chemical characteristics, a 256‐membered fragment library was designed using 16 different β3‐amino acids in both positions. The suitability of β‐peptides for constructing structural mimetics for protein recognition has been established,5a including libraries generated by using one‐bead‐one‐compound12 or parallel synthesis13 methods. Here, we utilized a mixture‐based library approach.14 To simplify the synthesis and analysis, a total of 256 fragments was divided into four 64‐membered sublibraries (L1–L4, Figure 2 a).
Figure 2.
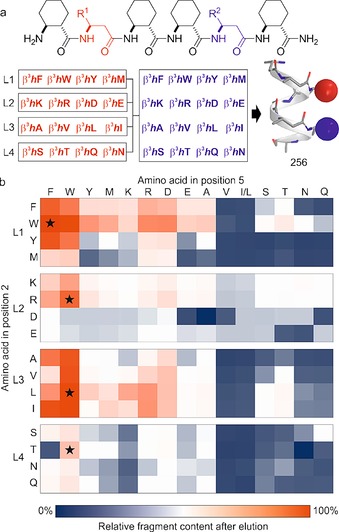
Library of folded segments and mapping of the protein surface with a pull‐down assay. a) Design principle and the sequences of the hexameric foldamer library with 16 different amino acids coupled in positions 2 and 5, which resulted in a side‐chain display on one face of the 14‐helical structure. A total of 256 compounds were synthesized in four sublibraries (L1–L4) based on the chemical characteristics of the amino acids in position 2. b) Results of the pull‐down assay expressed in percentages relative to the control experiment. Based on HPLC–MS peak integration, relative fragment content was calculated for each library member using the following formula: (AUCeluted/AUCcontrol)×100, where AUCeluted is the AUC (area under the curve) of a specific fragment in the eluted fraction and AUCcontrol is the AUC of the same fragment in the control sample. The color gradient scale indicates the differences in CaM binding affinity; coloring corresponds to low (blue) and high (red) abundance of the fragment after elution of the protein–foldamer complex. Asterisks indicate the segments that exhibited the highest concentration due to binding in the specific sublibrary. The side chains of β3‐amino acids in positions 2 and 5 are indicated by the standard α‐amino acid one‐letter codes. The pull‐down assay was repeated three times, and we did not observe deviation in the results above the experimental error associated with the HPLC–MS measurements. Here, we show representative data from one experiment.
It has been noted that careful selection of the DCL components is necessary because the correlation between binding efficiency and amplification might break down in large libraries if weak binders significantly outnumber the few high‐affinity ligands.15 We therefore prefiltered the library members through an affinity‐based pull‐down assay, in which the library members were incubated with immobilized CaM, and after washing away the unbound fragments, the CaM–foldamer complexes were eluted and quantified using HPLC–MS (Figure 2 b). Fragments highlighted in bound to CaM, and were almost quantitatively recovered after elution of the protein—foldamer complex. Due to their low affinities, fragments highlighted in blue were removed by the washing procedure, therefore a small number of these helices were identified after the final elution. Foldamers that contained aromatic residues (R1, R2: W, F; Figure 2 a) in combination with aliphatic side chains were the best binders, which is consistent with the observation that CaM has a preference for tryptophan, leucine, and isoleucine residues.9, 16 On the basis of relative fragment content (Figure 2 b and Figure S2 in the Supporting Information), one fragment was selected from each sublibrary for characterization.
The binding of the selected foldameric recognition segment candidates 1 (R1: W, R2: F), 2 (R1: R, R2: W), 3 (R1: L, R2: W), and 4 (R1: T, R2: W) (Figure S3) were validated and characterized quantitatively. Isothermal titration calorimetry (ITC) experiments were performed to identify the thermodynamic parameters of the binding. The measured dissociation constants (K d) were 0.076, 0.706, 0.139 and 17.1 μm for 1–4, respectively (Figure S4). The titration curves could be fitted with a two independent sites model and indicated that the target protein binds uniformly two foldamer segments in these interactions. The negative ΔH values for most of the fragments (Figure S4b) suggested a noncovalent bond complementarity between the foldameric fragments and the protein,17 except for 3, for which a positive enthalpy change was found. For 1, the interaction is enthalpy‐driven, suggesting that the binding is not dominated by the hydrophobic interactions, which is an advantageous characteristic for drug design.
All selected fragments contained a β3‐hW residue, which afforded the opportunity to measure the blueshift of its side‐chain fluorescence emission (from 350 to 330 nm) upon transfer from a solvent‐exposed environment to the hydrophobic clefts of CaM.18 The blueshift was observed for 1, 2, 3, and 4 (Figure S5). The phenomenon was not detected in the absence of Ca2+ (removed with EDTA), which confirmed that only the Ca2+‐bound tertiary structure of the protein generates the hot spot sockets that were recognized by the foldamers. Fluorescence titration experiments displayed the same trend of the binding affinities as those observed by ITC (Figure S6).
The propensity to fold into an H14‐helix in aqueous buffer was confirmed by ROESY experiments on 1–4 (Figure S7); the long‐range i–i+3 inter‐residue interactions were detected. To test the effects of the folding on binding, a non‐helical control derivative of 1 was designed by preventing helix formation with a non‐matching backbone stereochemistry (1R,2R‐ACHC) at position 4 (5, Figure S8).19 Sequence 5 did not show any sign of binding and exhibited disorder in water thus supporting the necessity of the compact and bulky structure. The affinities in the micromolar region for 2 and 4, with their fast exchange, afforded transferred NOESY (tr‐NOESY) measurements, which confirmed helical conformations also in the bound state. (Figure S9) These binding phenomena were not detected in the absence of Ca2+ (removed from CaM with EDTA, Figure S5), which confirmed that only the Ca2+‐bound tertiary structure of the protein generates the hot spot pockets that were recognized by the foldamers. We tested the foldamer–CaM interactions with 15N‐HSQC NMR spectroscopic titrations, which were conclusive for 2 and 3 due to sufficient affinity and signal‐to‐noise ratio (limited line broadening). Significant chemical shift perturbation and/or resonance broadening were observed for target residues L39, M36, M71, M72, M109, M144, and M145 (Figure S10), which are key residues in the CaM–protein contacts and line the hot spot pockets in the N‐ and C‐terminal EF‐hand motifs.20
DCLs are attractive tools for the discovery of new ligands for biomolecules,10b and they rely on the reversible generation of compound mixtures under thermodynamic control. Assembling the fragments with the template in the mixture shifts the dynamic equilibrium towards the tight binders, thereby increasing the concentration of the high‐affinity ligands.21 The three best recognition segment candidates were selected from each sublibrary and synthesized individually with a Gly–Gly–Cys tag at the C termini (6–17, Figure 3 a) to generate the DCL through a disulfide‐exchange reaction.22 DCLs were prepared in a glutathione redox buffer in the presence and in the absence (as a control) of the template. The concentration was 10 μm for each library member. CaM was used as a template at three concentrations (1, 6 and 30 μm). On the basis of quantitative evaluation with HPLC–MS chromatograms, amplification factors were determined relative to the control. The DCL mixture reached equilibrium within a reasonably short time21 (96 h), and the final reaction mixture contained 12 monomers, their 12 glutathione adducts and 78 different dimers of the folded segments (Table S5). The same product distribution was obtained from different starting mixture compositions (Figure S11), demonstrating that thermodynamic equilibrium had been reached. The most amplified dimers contained foldamers 9–11 in combination with sequences 6–8 or 12–14 (Figure 3 b). We found that the presence of positively charged side chains together with aromatic or aliphatic residues were essential for the amplification. Despite the quasi‐symmetry of CaM, the homodimers of the best binder fragments were not identified, which points to an emergent feature originating from the systems chemistry approach. The use of elevated template concentrations resulted in increased amplification factors (Figure 3 c and d), but the higher number of enriched heterodimers led to a lower selectivity.15a, 23 The selectivity pattern differences can be explained by the global behavior of the equilibrated DCL in response to the multiple molecular‐recognition events facilitated by the higher concentration of the template.24 For the series of ligands in which the dissociation constants of binding to the template are similar, better selectivity was found at a lower template concentration due to the competition between the building blocks. Accordingly, the DCL with 1 μm CaM concentration was used for the selection of the most amplified heterodimer (9‐SS‐12).
Figure 3.
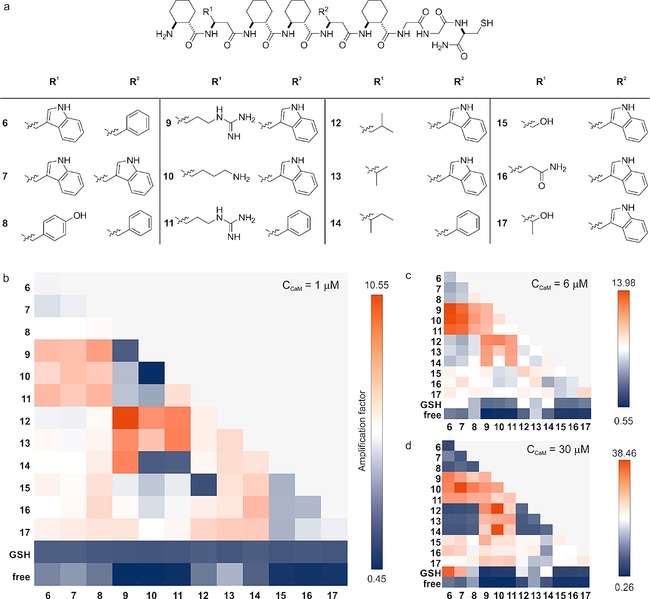
Results of the dynamic combinatorial experiments with three different template concentrations. a) Structures of the DCL building blocks. The amplification factors measured at thermodynamic equilibrium in the presence of CaM at b) 1 μm, c) 6 μm, and d) 30 μm. The amplification factor is defined as the ratio of AUCs measured in the presence and the absence of the template. The color scheme indicates the lowest and the highest amplification factors with blue and red, respectively. “GSH” refers to glutathione adducts.
To enhance the synthetic efficiency and avoid a possible instability caused by the disulfide bond in further investigations, a chemically stable thioether linkage25 was used to couple the individual helical segments of 9‐SS‐12 (18; Figure 4 a and Figure S12). ITC characterization revealed a two‐step process for the binding of 18 with the protein (Figure 4 b). First, a high‐affinity binding step was found with a K d value of 1.54±0.16 nm (n=1.04), which is a dissociation constant two orders of magnitude lower than that of the monomeric fragments. The 1:1 stoichiometry strongly suggested that we had successfully targeted the separate hot spots on the protein surface with a single ligand assembled from two folded fragments. The thermodynamic driving forces for the strong binding were found to be balanced. The binding enthalpies (ΔH) were −2.5 and −4.8 kcal mol−1, whereas the entropic contributions (−TΔS) were −9.1 and −7.2 kcal mol−1 at 25 and 35 °C, respectively. Second, a lower‐affinity step with a fractional stoichiometry was detected (Figure 4 b). This pointed towards the capability of the helical segments of 18 to interact separately with both lobes of CaM, which led to crosslinking of the protein by the ligand at micromolar concentrations. This result was confirmed by native gel electrophoresis, indicating multiple types of complexes after the provision of more than one equivalent of the foldamer to CaM (Figure S13). Fluorescence titration experiments with 18 revealed a Ca2+‐dependent binding to CaM with a K d value of 30 nm (Figure S14), which represents an averaged affinity of the two binding modes.
Figure 4.
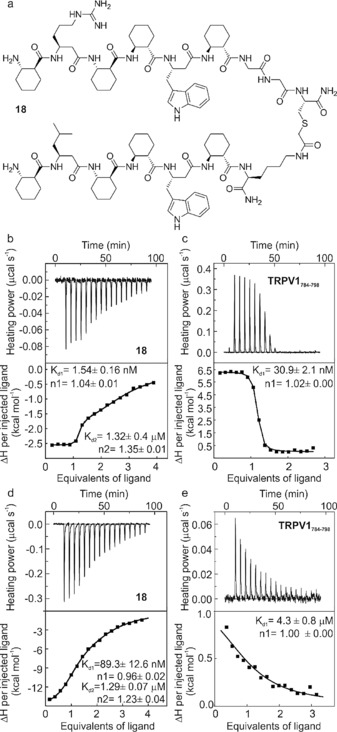
Binding and inhibition properties of 18. a) The chemical structure of 18. ITC titrations showing raw data (upper) and integrated and fitted curves (lower) with the fitted K d and stoichiometry (n) for b) 18 titrated with 3 μm CaM at 25 °C, and c) TRPV‐CT15 titrated with 7 μm CaM at 25 °C. ITC competition experiments for d) 18 titrated with a mixture of 3 μm CaM and 6 μm TRPV1‐CT15, and e) TRPV1‐CT15 titrated with a mixture of 7 μm CaM and 18.
CaM exerts its Ca2+ sensing function through a number of PPIs. We selected the CaM–TRPV1 (vanilloid receptor)26 interaction as a model system to test the inhibitory potential of 18. It has been shown that a 15‐mer fragment of the TRPV1 C terminus (TRPV1‐CT15) binds CaM with high affinity,27 and the X‐ray structure of the complex has been reported.26 Our ITC measurements confirmed a K d value of 30.9±2.1 nm (n=1.02) (Figure 4 c). After saturation of CaM with 2 equivalents of TRPV1‐CT15 in the cell, the titration with 18 resulted in an apparent K d value of 89.3±12.6 nm for the first step and 1.29±0.07 μm for the second (Figure 4 d). This suggested that the foldameric ligand can replace TRPV1‐CT15 in the first, high‐affinity step of the interaction. In a reverse experimental setup, pre‐binding of 18 to CaM resulted in the blockade of the TRPV1‐CT15–CaM interaction, as indicated by the micromolar apparent affinity (Figure 4 e). ITC experiments were repeated at 35 °C with similar results (Figure S15). Competitive inhibition was further supported by a pull‐down assay, in which immobilized CaM was saturated with TRPV1‐CT15 and titrated with 18 at different concentrations, which resulted in elution of TRPV1‐CT15 from CaM in a concentration‐dependent manner (Figure S16).
In summary, we have shown that a high‐affinity synthetic PPI inhibitor can be developed de novo through the combination of local surface mimetic foldamer segments and their dynamic covalent coupling. This strategy is a synthetically efficient optimization pathway, which could be extended to other targets, and suggests it is a feasible approach for high‐affinity PPI inhibitor synthesis.
Experimental Section
Pull‐Down Assay
Filtering of the folded fragment library with CaM was performed by pull‐down assay. Cobalt affinity resin suspension (50 % w/v, 100 μL; TALON, Takara Bio USA, Inc., Mountain View, CA) was pipetted into a paper filter spin cup (Thermo Scientific), centrifuged at 1000 rpm for 2 min and washed three times with HEPES buffer (20 mm, pH 7.4, 300 μL) containing NaCl (150 mm) and CaCl2 (1 mm). Polyhistidine‐tagged CaM was conjugated to the resin at a concentration of 2 mg mL−1, and the mixture was shaken at 100 rpm at room temperature for 30 min. After the conjugation, the resin was washed three times as described previously to remove the excess of the protein, and then it was incubated with the foldamer sublibrary, in which each library member was used at 10 μm concentration. The library with the immobilized CaM was also shaken at 100 rpm at room temperature for 30 min, and then it was centrifuged at 1000 rpm for 2 min at room temperature. The resin was washed three more times following the same procedure to remove unbound fragments, and freshly prepared imidazole (200 mm, 200 μL) was added to the sample to elute the polyhistidine‐tagged CaM and bound library members from the resin at room temperature for 5 min. Then, the resin was centrifuged as described previously. Negative control experiments were performed using the same procedure in the absence of polyhistidine‐tagged CaM to measure the nonspecific binding between the resin and the foldamer library. Eluted fractions and control samples were measured using HPLC–MS and Thermo Xcalibur 2.2 software was used for peak identification and integration.
Isothermal Titration Calorimetry Experiments
ITC experiments were performed with a MicroCal VP‐ITC microcalorimeter. A buffer of HEPES (20 mm, pH 7.0) containing CaCl2 (30 mm) was used. Peptide solutions were sonicated 20 min before titration to avoid aggregation. Foldamer solution (10 or 15 μL) was injected from the computer‐controlled microsyringe into the CaM solution at intervals of 240 s. The CaM concentration in the cell was between 3 and 7 μm, and the concentration of foldamers in the syringe was 85–200 μm. The temperature was adjusted to 25 or 35 °C. The control experiments were performed by injecting foldamers into the cell containing buffer but no target. Experiments were repeated twice. The experimental data were fitted to the one binding site or two independent sites model (adjustable parameters: ΔH b1, K d1, n 1 and ΔH b2, K d2, n 2) using a nonlinear least‐squares procedure. Errors were calculated by jackknife resampling.
Generation and Analysis of the Dynamic Combinatorial Library
DCLs were prepared from Gly–Gly–Cys‐functionalized building blocks (6–17) at a concentration of 10 μm in a redox buffer [pH 7.4, HEPES (20 mm), NaCl (150 mm), CaCl2 ( 1 mm), NaN3 (3 mm) reduced glutathione (GSH, 500 μm) and oxidized glutathione (GSSG, 125 μm)]. CaM was used as a template at 1, 6, and 30 μm, and a control DCL was started in parallel in the absence of template protein. Libraries were shaken (250 rpm, 37 °C) for five days in Eppendorf LoBind microcentrifuge tubes. At the beginning and every 24 h, reaction mixtures (100 μL) were removed for analysis and quenched with 10 % TFA in water. All quenched reaction mixtures were analyzed using HPLC–MS, and library members were identified according to their mass and hydrophobic characteristics. Amplification factors were determined as the component concentration ratio relative to the control experiment.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Hungarian Academy of Sciences, Momentum Programme (LP‐2011‐009) and the Ministry of National Economy, National Research Development and Innovation Office (GINOP‐232‐15‐2016‐00014), Gedeon Richter Plc. (TP7‐017) and Gedeon Richter's Talentum Foundation (Ph.D. Scholarship to É.B.). E.W. thanks the Postdoctoral Fellowship Program 2014 of the Hungarian Academy of Sciences. Lívia Fülöp and Zsolt Bozsó are gratefully acknowledged for discussions on synthesizing compound 18.
É. Bartus, Z. Hegedüs, E. Wéber, B. Csipak, G. Szakonyi, T. A. Martinek, ChemistryOpen 2017, 6, 236.
References
- 1. Pelay‐Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9022–9054. [Google Scholar]
- 2. Scott D. E., Bayly A. R., Abell C., Skidmore J., Nat. Rev. Drug Discovery 2016, 15, 533. [DOI] [PubMed] [Google Scholar]
- 3. Jubb H., Blundell T. L., Ascher D. B., Prog. Biophys. Mol. Biol. 2015, 119, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hewitt S. H., Wilson A. J., Chem. Commun. 2016, 52, 9745–9756. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Checco J. W., Gellman S. H., Curr. Opin. Struct. Biol. 2016, 39, 96–105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Guichard G., Huc I., Chem. Commun. 2011, 47, 5933–5941; [DOI] [PubMed] [Google Scholar]
- 5c. Cabrele C., Martinek T. S. A., Reiser O., Berlicki Ł., J. Med. Chem. 2014, 57, 9718–9739. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Horne W. S., Johnson L. M., Ketas T. J., Klasse P. J., Lu M., Moore J. P., Gellman S. H., Proc. Natl. Acad. Sci. USA 2009, 106, 14751–14756; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Checco J. W., Kreitler D. F., Thomas N. C., Belair D. G., Rettko N. J., Murphy W. L., Forest K. T., Gellman S. H., Proc. Natl. Acad. Sci. USA 2015, 112, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fülöp L., Mándity I. M., Juhász G., Szegedi V., Hetényi A., Wéber E., Bozsó Z., Simon D., Benkő M., Király Z., PLoS One 2012, 7, e39485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Orner B. P., Ernst J. T., Hamilton A. D., J. Am. Chem. Soc. 2001, 123, 5382–5383; [DOI] [PubMed] [Google Scholar]
- 8b. Milanesi L., Hunter C. A., Sedelnikova S. E., Waltho J. P., Chem. Eur. J. 2006, 12, 1081–1087. [DOI] [PubMed] [Google Scholar]
- 9. Tidow H., Nissen P., FEBS J. 2013, 280, 5551–5565. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Rowan S. J., Cantrill S. J., Cousins G. R., Sanders J. K., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993; [Google Scholar]
- 10b. Herrmann A., Chem. Soc. Rev. 2014, 43, 1899–1933; [DOI] [PubMed] [Google Scholar]
- 10c. Mondal M., Hirsch A. K., Chem. Soc. Rev. 2015, 44, 2455–2488; [DOI] [PubMed] [Google Scholar]
- 10d. Huang R., Leung I. K., Molecules 2016, 21, 910; [Google Scholar]
- 10e. Eisenberg M., Shumacher I., Cohen-Luria R., Ashkenasy G., Bioorg. Med. Chem. Bioorg. Med. Chem. 2013, 21, 3450–3457. [DOI] [PubMed] [Google Scholar]
- 11. Seebach D., Matthews J. L., Chem. Commun. 1997, 2015–2022. [Google Scholar]
- 12.
- 12a. Kritzer J. A., Luedtke N. W., Harker E. A., Schepartz A., J. Am. Chem. Soc. 2005, 127, 14584–14585; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Murray J. K., Farooqi B., Sadowsky J. D., Scalf M., Freund W. A., Smith L. M., Chen J., Gellman S. H., J. Am. Chem. Soc. 2005, 127, 13271–13280. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Murray J. K., Gellman S. H., Nat. Protoc. 2007, 2, 624–631; [DOI] [PubMed] [Google Scholar]
- 13b. Sadowsky J. D., Murray J. K., Tomita Y., Gellman S. H., ChemBioChem 2007, 8, 903–916. [DOI] [PubMed] [Google Scholar]
- 14. Pinilla C., Appel J. R., Borràs E., Houghten R. A., Nat. Med. 2003, 9, 118–122. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Corbett P. T., Sanders J. K., Otto S., J. Am. Chem. Soc. 2005, 127, 9390–9392; [DOI] [PubMed] [Google Scholar]
- 15b. Ramström O., Lehn J.-M., Nat. Rev. Drug Discovery 2002, 1, 26–36. [DOI] [PubMed] [Google Scholar]
- 16. Rhoads A. R., Friedberg F., FASEB J. 1997, 11, 331–340. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Leavitt S., Freire E., Curr. Opin. Struct. Biol. 2001, 11, 560–566; [DOI] [PubMed] [Google Scholar]
- 17b. Ladbury J. E., Klebe G., Freire E., Nat. Rev. Drug Discovery 2010, 9, 23–27. [DOI] [PubMed] [Google Scholar]
- 18. Gomes A. V., Barnes J. A., Vogel H. J., Arch. Biochem. Biophys. 2000, 379, 28–36. [DOI] [PubMed] [Google Scholar]
- 19. Mándity I. M., Wéber E., Martinek T. A., Olajos G., Tóth G. K., Vass E., Fülöp F., Angew. Chem. Angew. Chem. 2009, 121, 2205–2209. [DOI] [PubMed] [Google Scholar]
- 20. Villarroel A., Taglialatela M., Bernardo-Seisdedos G., Alaimo A., Agirre J., Alberdi A., Gomis-Perez C., Soldovieri M. V., Ambrosino P., Malo C., J. Mol. Biol. 2014, 426, 2717–2735. [DOI] [PubMed] [Google Scholar]
- 21. Corbett P. T., Leclaire J., Vial L., West K. R., Wietor J.-L., Sanders J. K., Otto S., Chem. Rev. 2006, 106, 3652–3711. [DOI] [PubMed] [Google Scholar]
- 22. Black S. P., Sanders J. K., Stefankiewicz A. R., Chem. Soc. Rev. 2014, 43, 1861–1872. [DOI] [PubMed] [Google Scholar]
- 23. Li J., Nowak P., Otto S., J. Am. Chem. Soc. 2013, 135, 9222–9239. [DOI] [PubMed] [Google Scholar]
- 24. Corbett P. T., Sanders J. K., Otto S., Angew. Chem. Int. Ed. 2007, 46, 8858–8861. [DOI] [PubMed] [Google Scholar]
- 25. Robey F. A., Fields R. L., Anal. Biochem. 1989, 177, 373–377. [DOI] [PubMed] [Google Scholar]
- 26. Lau S.-Y., Procko E., Gaudet R., J. Gen. Physiol. 2012, 140, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hetényi A., Németh L., Wéber E., Szakonyi G., Winter Z., Jósvay K., Bartus É., Oláh Z., Martinek T. A., FEBS Lett. 2016, 590, 2768–2775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary