

Abstract
Tetrahydropyranyl (Thp) is recognized as a useful protecting group for alcohols in organic synthesis. It has several advantages, including low cost, ease of introduction, general stability to most non‐acidic reagents, it confers good solubility, and the ease with which it can be removed if the functional group it protects requires manipulation. However, little attention has been paid to Thp in peptide chemistry. Provided here is a concise analysis of the Thp protection of various amino acid functionalities (OH, SH, NH and COOH) and its application to peptide synthesis. Thp is a useful moiety for the side‐chain protection of serine, threonine and cysteine and is suitable for the Fmoc/tBu solid‐phase peptide synthesis strategy. The immobilized version of 3,4‐dihydro‐2H‐pyran, the so‐called Ellman resin, is also discussed as a useful solid support for anchoring the side chains of serine, threonine and tryptophan residues.
Keywords: 3,4-dihydro-2H-pyran; hydroxyl groups; protecting groups; tetrahydropyranyl group; thiols
1. Introduction
The formation of a peptide bond requires the reaction between an activated carboxylic acid of an amino acid—typically an active ester—and the amino function of another amino acid.1, 2 To avoid side reactivity, the remaining functional groups of each amino acid have to be conveniently protected.3 In solid‐phase peptide synthesis (SPPS), the carboxylic acid of the reacting amine is usually attached to the solid support. Furthermore, it is notable that protection of amino acid side chains is often required in SPPS due to the presence of a wide range of reactive functional groups along a peptide sequence. Thus, during peptide elongation, various types of protecting groups might co‐exist.4
In the area of peptide synthesis, protecting groups5 are divided into three main classes: 1) permanent protecting groups, which are removed at the end of the synthetic process. These comprise mainly side‐chain protecting groups and the C‐terminal protecting group, which, in the case of SPPS, is effectively the linker to the solid support; 2) temporary protecting groups, which are removed after each synthetic step and include mainly those protecting the α‐amino groups; and 3) semi‐permanent protecting groups, which are removed in the middle of the synthetic process to perform a reaction that requires the presence of other protecting groups, such as for the formation of branched6, 7 or cyclic peptides.8, 9
For SPPS, amino acid protecting groups should be: 1) easily introduced, 2) stable during peptide sequence elongation, and 3) easily and orthogonally removed. To measure the balance between stability and instability of the protecting groups, orthogonal strategies represent the best choice in the sense that two protecting groups belong to independent chemical classes and consequently they can be removed by different chemistry.10 Furthermore, the two groups can be removed in any order. Orthogonal protection schemes are milder because the selective deprotection is governed by alternative cleavage mechanisms rather than by reaction rates. This is perfectly implemented if N α‐(9‐fluorenylmethyloxycarbonyl) (Fmoc) amino acids are used in combination with acid‐labile side‐chain protecting groups, as well as a C‐terminal amino acid that is anchored to a resin. Whereas the Fmoc group is removed after each coupling step by repetitive treatment with piperidine/DMF (1:4), the side‐chain protecting groups and C‐terminal peptide cleavage is achieved using acid treatment, typically with trifluoroacetic acid (TFA). These acid‐labile protecting groups are based on benzyl‐derived electron‐donating groups or derivatives of tertiary alcohols. In this regard, tert‐butyl (tBu) and trityl (Trt) are the most commonly used protecting groups. In order to trap the carbocation formed, these derivatives are removed in acidic conditions in the presence of scavengers.4
In peptide chemistry, little attention has been paid to the tetrahydropyranyl (Thp) group, which has otherwise been recognized as a useful protecting group for alcohols in organic synthesis.11 Historically, the extensive contribution of Paul to the chemistry of pyran and furan rings attributes him with the discovery of Thp as a hydroxyl protecting group (Paul et al., in 1934, were credited for the discovery of pyran and furan rings, and Thp was not used for −OH protection until then; however, Anderson et al., in 1948, used them for hydroxyl protection). In 1934, Paul observed that 2‐methoxytetrahydropyran was obtained after adding methanol to 3,4‐dihydro‐2H‐pyran (DHP) in the presence of HCl.12 Later, Woods and Kramer studied the addition of various alcohols to the dihydropyran ring in greater depth and also postulated the stability of these tetrahydropyran acetals under basic conditions.13 However, the description of Thp as an alcohol‐protecting group was not investigated until 1948, when Parham and Anderson reported the conversion of alcohols to acetals and their easy cleavage in mildly acidic aqueous conditions with the regeneration of the hydroxyl group.14
The Thp group offers versatile protection of hydroxyl functional groups (alcohols and sometimes phenols) during multistep organic synthesis. Thp ethers are attractive because of the low cost of DHP, its easy incorporation, the stability of the resulting Thp derivatives in various reaction conditions, and ease of removal under mildly acidic conditions (Scheme 1). Thp has the advantage over benzyl‐based protecting groups, such as Trt, diphenylmethyl (Dpm), (4‐methoxyphenyl)diphenylmethyl (methoxytrityl, Mmt) or benzyloxymethyl (Bom), because it lacks aromaticity and offers better solubility. In addition to producing more protected hydrophobic peptides, the use of bulky aromatic protecting groups in SPPS affects the inter‐/intrachain interactions during peptide elongation, which might compromise the purity of the final product.3, 15
Scheme 1.
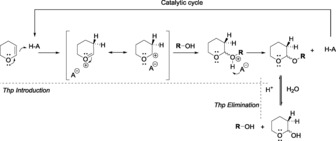
Protection of an alcohol by Thp and its elimination mechanism.
In view of the aforementioned observations, here we provide a concise analysis of the Thp protection of various amino acids functionalities (NH, SH, OH and COOH) and its application to peptide synthesis (Figure 1). This Review is organized according to the functional group protected. For each case, methods for the introduction and removal of the protecting groups, as well as the scope and limitations, are discussed.
Figure 1.
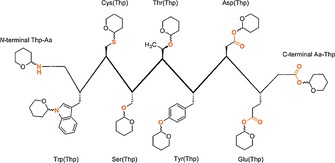
Thp protection of amino acids.
2. Thp Protection of the NH Group (Tryptophan)
2.1. General
The dihydropyranylation of nitrogen‐based functional groups in SPPS has not been extensively studied. The reversible binding of purines16, 17 and benzimidazoles18 through one of the nitrogen atoms of imidazole has been reported, as well as through the indolic nitrogen, for the synthesis of 2,3‐disubstituted indoles.19 Additionally, Cheng and Hii20 reported the solution‐phase synthesis of α‐alkylaminotetrahydrofurans, as well as α‐alkylamino‐ and α‐arylaminotetrahydropyrans under neutral pH conditions by a hydroamination reaction catalyzed by palladium thiocyanate complexes. The authors concluded that the α‐aminotetrahydropyran ring underwent rapid reversible ring‐opening in aqueous solution, as revealed in studies on N‐glycosidic bond lability in aqueous media with N‐glucosamines.21 More recently, Nicolás et al.22 used the dihydropyranyl linker to anchor the indolic nitrogen of the amino acid Trp to a solid support for the synthesis of Trp‐containing diketopiperazines. Furthermore, they determined the conditions required for Thp removal, which is achieved using a strong acid (TFA) in the presence of a carbocation scavenger, as discussed below.22
2.2. Introduction of the Protecting Group
The protocol for alkylamine tetrahydropyranylation described by Cheng and Hii20 considered only the use of dialkylamines and aniline for the hydroamination reaction catalyzed by palladium thiocyanate complexes over long reaction times (>12 h) and high temperature (70–100 °C).
The introduction of a tetrahydropyranyl moiety into primary amines is challenging, and our lab attempted the protection of Phe with Thp. For this purpose, we used various commercially available Pd catalysts and also prepared the catalyst that the aforementioned authors demonstrated to be the most efficient for the N‐Thp linkage, that is, K2Pd(SCN)4. However, we were unable to introduce an N‐Thp group into Phe using PdII chemistry under these conditions.
Interestingly, the introduction of the Thp moiety at the indolic nitrogen of Trp was more successful and has a greater impact on SPPS. The protocol described by Nicolás et al.22 involved the reaction of Fmoc‐Trp‐OR (R=Allyl or Me) with DHP using pyridinium p‐toluenesulfonate (PPTS) as catalyst and 1,2‐dichloroethane (DCE) as solvent at 70 °C for 8 h. A yield of 80 % was achieved for Fmoc‐Trp(Thp)‐OAllyl and 69 % for Fmoc‐Trp(Thp)‐OMe. Furthermore, they also anchored a Trp derivative on Ellman resin through its side chain using PPTS as a catalyst in anhydrous DCE under microwave conditions for 2 h at 120 °C. The authors realized that, in order to attach Trp to a tetrahydropyran‐functionalized resin through its indole ring, the α‐amino and carboxyl functions had to be protected with groups compatible with the conditions necessary for the formation of the hemiaminal linkage and peptide chain elongation.22
In order to amplify the use of Thp as a protecting group for SPPS, and taking into account the side‐chain anchoring of Trp, we attempted to protect the indole side chain of Fmoc‐Trp‐OH with Thp (see Scheme 1). The protocol followed involved reaction of Fmoc‐Trp‐OH with DHP using p‐toluenesulfonic acid (PTSA) as catalyst. The mixture was left to react at room temperature for 2.5 h and then washed with water and brine. Purification by column chromatography on silica gel using CH2Cl2/MeOH (96:4) as eluent yielded the protected Fmoc‐Trp(Thp)‐OH (82 % yield). Although the carboxylic acid reacts with DHP in a similar way, the corresponding hemiacetal ester was not stable to the aqueous work‐up, so Thp was cleaved from the carboxyl group. Thus, it can be concluded that Fmoc‐Trp‐OH can be side‐chain protected with Thp without prior protection of the carboxyl group (Scheme 2).
Scheme 2.
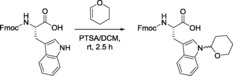
Thp protection of the side‐chain indole NH of Trp.
2.3. Removal of the Protecting Group
The reported conditions for the cleavage of the hemiaminal bond between the indolic nitrogen and the Thp moiety of Ellman resin involve a mixture of TFA/1,3‐dimethoxybenzene/CH2Cl2 (5:10:85) at room temperature for 1 h.22 In addition, we have studied the conditions of cleavage of Thp from Fmoc‐Trp(Thp)‐OH using various concentrations of TFA in the presence of scavengers (see Table 1). A comparison was made between Fmoc‐Trp(Boc)‐OH and Fmoc‐Trp(Thp)‐OH to test lability under different acidic conditions (Table 1). In this regard, the cleavage cocktail containing TFA/H2O/CH2Cl2 (10:2:88) was the best choice, achieving nearly 90 % deprotection within 1 h of treatment for Fmoc‐Trp(Thp)‐OH (Table 1, entries 11–13). In contrast, only 69 % deprotection was achieved for Fmoc‐Trp(Boc)‐OH (Table 1, entries 4–6). However, even at a higher concentration of TFA (60 %), complete cleavage of Thp could not be achieved.
Table 1.
Acid lability studies of Fmoc‐Trp(Boc)‐OH and Fmoc‐Trp(Thp)‐OH.[a]
| Entry | Compound | Cocktail | Reaction time [min] | Deprotected amino acid [%] |
|---|---|---|---|---|
| 1 | Fmoc‐Trp(Boc)‐OH | TFA/CH2Cl2 (10:90) | 10 | 28 |
| 2 | 30 | 59 | ||
| 3 | 60 | 82 | ||
| 4 | TFA/H2O/CH2Cl2 (10:2:88) | 10 | 0 | |
| 5 | 30 | 42 | ||
| 6 | 60 | 69 | ||
| 7 | TFA/CH2Cl2 (60:40) | 10 | >99 | |
| 8 | Fmoc‐Trp(Thp)‐OH | TFA/CH2Cl2 (10:90) | 10 | 63 |
| 9 | 30 | 68 | ||
| 10 | 60 | 77 | ||
| 11 | TFA/H2O/CH2Cl2 (10:2:88) | 10 | 72 | |
| 12 | 30 | 79 | ||
| 13 | 60 | 90 | ||
| 14 | TFA/CH2Cl2 (60:40) | 10 | 59 | |
| 15 | 30 | 73 | ||
| 16 | 60 | 90 | ||
| 17 | TFA/H2O/CH2Cl2 (60:10:30) | 10 | 92 | |
| 18 | 30 | 94 | ||
| 19 | 60 | 96 |
[a] Unpublished data from our group.
2.4. Peptide Elongation
As mentioned above, Nicolás et al. used a tetrahydropyran moiety to perform the synthesis of diketopiperazines using SPPS. They demonstrated that the N‐Thp bond is compatible with the steps of SPPS: protecting group introduction and elimination, amino acid coupling, and peptide cleavage.22 In order to expand the scope of this work, we introduced Fmoc‐Trp(Thp)‐OH smoothly into several model peptides. However, the removal of Thp from Thp‐protected Trp‐containing peptides did not take place quantitatively under the cleavage conditions tested. After several trials, we concluded that Thp does not prevent alkylation of the indole during TFA cleavage, because this is the main side reaction to be avoided in peptide synthesis.23, 24
3. Thp Protection of the Thiol Group (Cysteine)
3.1. General
Protection of the thiol group is crucial for many fields of organic chemistry research, particularly in peptide and protein syntheses, which often involve the use of Cys.25 Thp has also been found to be efficient and stable for the protection of thiol groups. Parham and DeLaitsch studied and compared the pyranylation of hydroxyl and thiol groups, observing higher reactivity for pyranylation of hydroxyl groups and less stability for the resulting tetrahydropyran acetal than for thioacetals.14, 26 In 1958, Holland and Cohen addressed the introduction of DHP into Cys for the protection of the thiol group for the synthesis of insulin peptides in solution.27
Although thiol groups are protected most widely as thioethers, especially Trt‐, Dpm‐, and benzyl‐like groups, developed in the labs of Yajima and Nishiuchi, S,O‐acetal groups, such as Bom for Boc chemistry28 and more recently methoxybenzyloxymethyl (MBom) for Fmoc chemistry,29 have also been used. However, these derivatives can result in a very low level of racemization, hinder Bom and MBom synthesis, and hamper the quality of the final product because formaldehyde is formed as a side product during cleavage and is accompanied by concomitant hydroxymethylation.30 With the same idea of exploiting the S,O‐acetal protecting group concept, we introduced Thp as a Cys‐protecting group for SPPS.31 We demonstrated that the use of Thp resulted in lower racemization levels than the conventional protecting groups for Cys (Trt, Dpm and S‐tBu), either if using Thp in the peptide sequence or as a C‐terminal Cys‐protecting group.31
Additionally, the solubility of Thp‐protected peptides was improved with respect to strategies involving the commonly used hindered aromatic protecting groups. Furthermore, Thp is suitable for protecting N‐terminal Cys residues, and, in comparison to other protecting groups, it does not lead to formylation upon removal of the protecting group.32 Our results revealed that Thp is a useful protecting group for Cys if applied to the Fmoc/tBu strategy for SPPS.
3.2. Introduction of the Protecting Group
Thp‐protected Cys can be prepared from Cys and DHP in the presence of BF3 ⋅Et2O using Et2O as the solvent at room temperature for 30 min33 or by reaction with DHP in an acidic medium.27 However, we have recently demonstrated that the most convenient method for protecting Fmoc‐Cys‐OH with Thp is in the presence of PTSA in CH2Cl2 for 60 min at room temperature,31 as shown in Scheme 3.
Scheme 3.
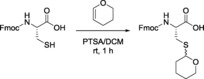
Thp protection of the thiol group of Cys.
3.3. Removal of the Protecting Group
The methods reported in the early literature described the removal of Thp based on the use of H2SO4, Lewis acids, aqueous acetic acid, Selectfluor, or PPTS in alcoholic solvent.34, 35, 36, 37 We demonstrated that a convenient method for Thp removal involves the use of >10 % TFA in the presence of scavengers such as water and triisopropylsilane (TIS). Therefore, the conventional cleavage conditions used in SPPS, such as TFA/TIS/CH2Cl2 (10:2.5:87.5), TFA/TIS/H2O (95:2.5:2.5), and 0.1 n HCl in HFIP–TIS (99:1) will ensure complete elimination of Thp in short treatments.38
A comparison was made between the protected amino acids Fmoc‐Cys(Thp)‐OH and Fmoc‐Ser(Thp)‐OH to test elimination conditions in acidic media. Table 2 shows a comparison of the lability of Fmoc‐Ser(Thp)‐OH and Fmoc‐Cys(Thp)‐OH in aqueous solutions. It was concluded from the data that Fmoc‐Cys(Thp)‐OH is more stable compared to Fmoc‐Ser(Thp)‐OH under moderately acidic aqueous conditions (Table 2, entries 1–5).
Table 2.
Study of Thp removal from Fmoc‐Cys(Thp)‐OH and Fmoc‐Ser(Thp)‐OH.31
| Entry | Compound | Cocktail composition | Reaction time [h] | Deprotected amino acid [%] |
|---|---|---|---|---|
| 1 | Fmoc‐Cys(Thp)‐OH | 100 mm MES[a] (pH 4.8) | 48 | 0 |
| 2 | Fmoc‐Ser(Thp)‐OH | 40 | 50 | |
| 3 | Fmoc‐Cys(Thp)‐OH | 100 mm MES, 137 mm NaCl, 2.7 mm KCl (pH 4.8) | 48 | 0 |
| 4 | Fmoc‐Ser(Thp)‐OH | 20 | 25 | |
| 5 | Fmoc‐Cys(Thp)‐OH | 0.1 m MES, 1.5 % TIS (pH 4.8) | 48 | 0 |
| 6 | Fmoc‐Cys(Thp)‐OH | PBS: 10 mm PO4 3−, 2.7 mm KCl, 137 mm NaCl (pH 7.4) | 120 | 0 |
| 7 | Fmoc‐Ser(Thp)‐OH | 0 |
[a] 2‐(N‐Morpholino)ethanesulfonic acid.
During studies on the removal of Thp, we observed that the thioacetal in Fmoc‐Cys(Thp)‐OH remained stable to mildly acidic conditions, whereas the acetal in Fmoc‐Ser(Thp)‐OH underwent smooth cleavage. Accordingly, a hydrolysis study at pH 4.8 was carried out by HPLC over 3 days (Figure 2).
Figure 2.
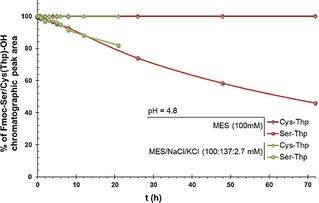
HPLC study of the kinetics of Fmoc‐Ser(Thp)‐OH and Fmoc‐Cys(Thp)‐OH acidolysis.
4. Thp Protection of the Hydroxy Group (Serine and Threonine)
4.1. General
The use of Thp as a protecting group for Ser and Thr was tentatively addressed due to the generation of a new stereocenter that complicated the isolation of intermediates. Nevertheless, some successful examples of Thp protection in the solution phase have been reported, including the preparation of the iron‐chelating siderophores enterobactin39, 40 and salmochelin,41 a cyclic trimer of l‐Ser, as well as for the syntheses of cyclic depsipeptides,42, 43 oxazoles,44 oxazolidin‐5‐ones45 and analogues of 7‐keto‐8‐aminopelargonic acid vitamers.46 Furthermore, the O‐Thp, N‐Fmoc protected version of Thr was used as part of interesting SPPS strategies described by Krishnamoorthy et al.47 and Iijima et al.48 for the total syntheses of callipeltin B and (+)‐antimycin A3b, respectively.
In addition to the abovementioned use as a protecting group, the Thp moiety was also studied as a cleavable linker for SPPS. Thompson and Ellman introduced the use of a DHP‐functionalized support.49 The use of Ellman resin provides a general and straightforward method for attachment of primary and secondary alcohols through the base‐stable tetrahydropyranyl ether linkage. Furthermore, this resin has proved convenient for the synthesis of a wide range of hydroxyl‐containing organic compounds.49 Accordingly, Ellman resin has found application in the synthesis of alcohol‐containing peptides.
4.2. Introduction of the Protecting Group
The common method for preparing Thp ethers is by reaction of the hydroxyl group with DHP in the presence of appropriate catalysts. The examples described for hydroxyl group protection include the use of hydrochloric acid,50 PTSA,51 BF3 ⋅Et2O,52 PPTS,34 electrogenerated acid (EG acid),53 Amberlyst H‐15,54 Nafion‐H,55 montmorillonite clay K‐10,56 H‐Y zeolite,57 Spanish sepiolite clay,58 and dicyanoketene ethylene.59 However, only a few studies have addressed the use of Thp as a protecting group of Ser and Thr for SPPS. Few examples can be found in the literature describing the introduction of Thp as a protecting group for these amino acids. The protocols most widely used involve the introduction of DHP either in the presence of PPTS at 60 °C for 16 h or PTSA at room temperature for 30–60 min using CH2Cl2 or DCE as the solvent.11 For instance, Fmoc‐Thr(Thp)‐OAllyl was synthesized by adding DHP followed by PPTS (2.5 mol %) in DCE. The reaction was then stirred for 12 h at 60 °C.47 Finally, removal of the allyl group afforded Fmoc‐Thr(Thp)‐OH as the key intermediate for the synthesis of callipeltin B.
Nevertheless, we have demonstrated that the protection of the carboxylic acid group is not necessary for the preparation of Fmoc‐Thr/Ser(Thp)‐OH. Although the carboxylic acid reacts with DHP similarly to hydroxyl groups, the corresponding hemiacetal ester was found to be unstable to aqueous work‐up, generating the free carboxylic acid.60 Thus it can be concluded that Fmoc‐Thr/Ser‐OH can be side‐chain protected without extra protection of the carboxyl group (Scheme 4). This conclusion is highly relevant for the protection of unnatural ω‐hydroxy amino acids, which are difficult to synthesize, such as the allo‐Thr derivatives and other β‐hydroxy amino acids that naturally occur as cyclodepsipeptides.61, 62
Scheme 4.
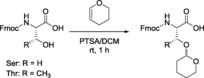
Thp protection of the hydroxyl groups of Ser and Thr.
Furthermore, we have also demonstrated that the use of PTSA as a catalyst is more convenient and efficient than PPTS. Accordingly, PPTS has been reported as a catalyst for pyranylation, a process that involves heating the reaction mixture to almost 60 °C for 10–12 h. However, compared to PPTS, PTSA is more efficient as the rate of the reaction increases.63 The side‐chain anchoring of protected Thr and Ser and amino alcohols to Ellman resin has been achieved using PPTS as catalyst in DCE for 16 h at 60–80 °C.64, 65 Furthermore, we have demonstrated that the hydroxyl group can also be introduced onto Ellman resin using PTSA as catalyst in THF for 30 min at room temperature.63
4.3. Removal of the Protecting Group
It has been reported that Ser and Thr hydroxyl side chains protected with Thp are deprotected under acidic conditions. In this regard, HCl,41, 46 TFA‐containing cocktails (1, 2 or 95 % TFA),47, 48, 49 TsOH42 or PPTS39, 40, 43, 49 in alcoholic solvents have been reported to cleave the Thp group. Accordingly, we have recently confirmed that Thp removal can be achieved using 2 % TFA in CH2Cl2 in the presence of scavengers.63 Unexpectedly, it was found that Thp protection of the Thr hydroxyl side chain is slightly more labile than in the case of Ser. We also showed that the Thp protecting group of the Ser hydroxyl can be partially removed by acidic aqueous solutions (see Table 2). This latter observation might be interesting for the application of this protecting group for the preparation of bioconjugates. It was also found that cleavage of a peptide from Ellman resin can be accomplished using the conditions mentioned above.
5. Thp Protection of a Phenol (Tyrosine)
5.1. General
Although successful protection of different kinds of phenols with the Thp group has been described in acidic media,13, 14, 54, 56, 57, 58, 66 the O‐Thp side‐chain protection of Tyr has not been studied extensively, and, to the best of our knowledge, Thp‐protected Tyr has not been used in SPPS. The introduction of Thp into the side chain of Tyr was first described by Iselin and Schwyzer62 using a trace of HCl as a catalyst. Shortly after, Thp protection of Tyr was attempted for the controlled synthesis of peptides in aqueous media using α‐amino acid N‐carboxyanhydrides,67 and also for the preparation of water‐soluble random l‐Tyr‐containing copolymers with N 5‐(3‐hydroxypropyl)‐l‐glutamine.68 Moreover, the Tyr phenolic hydroxyl was protected with Thp for the synthesis of Tyr‐containing compounds or analogues philanthotoxin,69 a potent inhibitor of l‐glutamate and nicotinic acetylcholine receptors, and related macrocycles of tirobifan, an anti‐platelet aggregation agent.70 As Thp protection had not been addressed in detail for SPPS, we decided to initiate a study to investigate the scope and limitations of the reaction.
5.2. Introduction of the Protecting Group
Similar to protection of the alcohol, and as discussed above, Thp ether formation at the phenolic group in Tyr is achieved through the reaction of the phenolic hydroxyl group with DHP in the presence of acid catalysts. Accordingly, Thp‐protected Tyr has been reported with N‐carboxybenzyl (N‐Cbz)60, 69 or N‐(para‐toluenesulfonyl) (N‐Ts)70 groups but not in the N‐Fmoc form. In this regard, our lab attempted to introduce Thp as a protecting group into the side chain of Tyr through the reaction of Fmoc‐Tyr‐OH with DHP using PTSA as catalyst and CH2Cl2 as solvent at room temperature for 35 min (Scheme 5). After completion of the reaction, followed by aqueous work‐up and silica gel purification, Fmoc‐Tyr(Thp)‐OH was obtained in 63 % yield. It was difficult to monitor the purity using HPLC and also molecular mass through LC–MS as cleavage of Thp (>60 % yield) occurred due to the presence of 0.1 % TFA in the mobile phase. This demonstrates the lability of Thp if it is used as a protecting group for the phenolic side chain of Tyr.
Scheme 5.
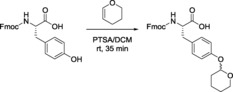
Thp protection of the hydroxyl group of Tyr.
5.3. Removal of the Protecting Group
Similar to Ser and Thr, Thp in Fmoc‐Tyr(Thp)‐OH is removed by acid treatment. The conditions described in the literature involve the use of TFA/CHCl3 (1:2) at room temperature for 5 h69 or PPTS in CH2Cl2/MeOH (1:1).70 In our experiments, Thp removal from the phenolic side chain of Fmoc‐protected Tyr was observed under HPLC and LC–MS conditions, thereby clearly indicating the high acid sensitivity of the phenolic Thp ether.
6. Thp Protection of the Carboxyl Group
6.1. General
Initially, the Thp group was used to activate carboxylic acids in order to achieve amide bonds. However, compared with other active esters, Thp esters are not highly suited for synthetic purposes because they are easily hydrolyzed.60 Reports on Thp protection for carboxyl protection are rare. In 1958, Holland and Cohen reported that the use of one equivalent of DHP in acidic medium resulted in blocking the carboxyl group in preference to the SH group. They reported that DHP, used in excess, reacted with N‐protected Cys to protect both SH and COOH functional groups, and the resulting compound was resistant to mild alkaline saponification.27 Additionally, Pappo71 and Bernady72 exploited the protection of the carboxyl function of prostaglandins with Thp. Using a peptidic scaffold, Wipf and Kim73 protected the C‐terminal Phe with Thp for the convergent total synthesis of cyclotheonamide A. However, in 2001, Holden and co‐workers74 attempted to protect carboxylic acids with Thp as an intermediate for the preparation of pilocarpine derivatives. They explained that use of Thp to protect the carboxyl group was problematic due to its instability at room temperature and its decomposition on silica during purification.
6.2. Introduction of the Protecting Group
As described for other functional groups, the introduction of Thp is achieved by treating the corresponding carboxylic acid with DHP in the presence of a catalytic amount of acid, such as PTSA⋅H2O,72 methanesulfonic acid,64, 74 HCl,27, 60 or PPTS.73 We recently protected the carboxylic acid group of several N α‐protected amino acids with Thp (Table 3). The amino acids studied were treated with DHP in the presence of a catalytic amount of PTSA at room temperature. The reaction time varied from 10 to 30 min depending on the amino acids (Scheme 6 and Table 3). Fmoc‐Gly‐OH was successfully protected using Thp. The crude product was purified by silica gel column chromatography after aqueous work‐up. However, undesired removal of Thp from other amino acids was observed during aqueous work‐up. This could be attributed to the high lability of the hemiacetal esters, as previously described.60, 74, 75 Therefore, after monitoring the reaction to completion by using thin‐layer chromatography, N,N‐diisopropylethylamine (DIEA) was added as a part of the work‐up to neutralize the PTSA used during the reaction. The crude product was then purified directly on basic silica using n‐hexane and ethyl acetate (1:1) as a solvent system—an approach that afforded the pure compounds, which were then characterized by NMR spectroscopy.
Table 3.
Thp protection of the carboxyl group of Fmoc‐protected amino acids.[a]
| Entry | Starting material | Product | Reaction time [min] | Yield [%] |
|---|---|---|---|---|
| 1 | Fmoc‐Gly‐OH | Fmoc‐Gly‐OThp | 30 | 80 |
| 2 | Fmoc‐Ala‐OH | Fmoc‐Ala‐OThp | 10 | 74 |
| 3 | Fmoc‐Val‐OH | Fmoc‐Val‐OThp | 10 | 76 |
| 4 | Fmoc‐Asp‐OH | Fmoc‐Asp(Thp)‐OThp | 25 | 79 |
[a] Unpublished data from our group.
Scheme 6.
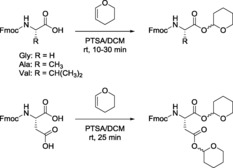
Thp protection of the carboxyl groups of different amino acids.
Additionally, Ellman resin was studied for the immobilization of Fmoc‐protected amino acids, with the goal of using it for the preparation of peptide acids under very mildly acidic conditions. Thus, DHP resin was added to a solution of Fmoc‐Gly‐OH (4 equiv.) and PTSA (0.025 equiv.) in THF and left to react for 30 min at room temperature. The stability of Fmoc‐Gly‐O–Ellman‐resin was studied using DMF, 1 % ethyl (hydroxyimino)cyanoacetate (Oxyma Pure) in DMF and 1 % hydroxybenzotriazole (HOBt) in DMF. After monitoring stability by HPLC, we concluded that Fmoc‐Gly‐OH was unstable on DHP resin under the above‐mentioned conditions and therefore not suitable for SPPS.
6.3. Removal of the Protecting Group
Although it is described that Thp esters are easily hydrolyzed,60, 74 some authors report that they are easily cleaved in mildly acidic conditions such as by treatment with a mixture of acetic acid/THF/H2O (4:2:1) at 45 °C.71, 72 According to our results, complete removal of Thp from Fmoc‐Gly‐OThp requires the use of TFA at greater than 10 % concentration. The need for such a high concentration of TFA can be explained by the fact that the lability of hemiacetal esters is a reversible reaction. In contrast, for other Fmoc‐protected amino acids, Thp was removed at low TFA concentrations (<1 %).
7. Conclusions
Here we have reviewed the use of Thp as a protecting group in SPPS. Although the early literature describes several methods based on rather strong conditions for the introduction of the Thp group, there is now consensus that the use of PTSA as catalyst is more amenable in terms of reaction rate and purity of the final product. Thus, the reaction time using PTSA varies from 10 min to 3 h, depending upon the functional group protected, whereas that of PPTS involves longer heating times (nearly 12 h). Furthermore, it has been demonstrated that Thp is a useful group for protecting the side‐chain functions of Ser, Thr and Cys and thus is compatible with the Fmoc/tBu SPPS strategy. However, Thp has been demonstrated to be useful for the protection of neither amines, due to difficulty of introduction, nor for the carboxyl group, due to the extremely high acid lability of the hemiacetal ester. Due to the high acid lability of Thp‐protected carboxylic acids, hydroxyl‐ and thiol‐containing amino acids can be conveniently protected leaving the carboxyl group free. This observation reflects the superiority of Thp over other acid‐labile protecting groups such as tBu and Trt, the introduction of which into the side‐chain of hydroxyl‐bearing amino acids first requires protection of the carboxyl group.
The solid‐phase‐supported version of Thp—Ellman resin—has proved to be suitable for side‐anchoring of Ser/Thr and Trp. However, the use of Thp for protecting the indole moiety of Trp is not optimal. It is envisaged that Ellman resin will also be useful for thiol anchoring.
Due to its low cost, ease of installation, general stability to most non‐acidic reagents, lower tendency for side reactions, better solubility and the ease with which it is removed, we conclude that Thp will find application for the synthesis of Ser‐, Thr‐ or Cys‐containing peptides.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Anamika Sharma obtained her M.Sc. in General Chemistry and Ph.D. in Organic Chemistry at University of Mysore (Mysore, India). She worked in the area of synthetic chemistry as a Research Associate at Syngene International (Bangalore, India). Her Ph.D. work involved classical solution‐phase peptide synthesis and bioconjugation. Afterwards, she worked as Assistant Professor at Techno India (Kolkata, India). She is currently working as a postdoctoral fellow at the University of KwaZulu Natal (Durban, South Africa). Her research includes the investigation of protecting groups for amino acids in solid‐phase peptide synthesis and also broad areas in medicinal chemistry.

Biographical Information
Dr. Iván Ramos‐Tomillero obtained his B.Sc., M.Sc. and Ph.D. in Organic Chemistry at the University of Barcelona and the Institute for Research of Biomedicine (Barcelona, Spain). Iván has been working on methodological improvements for organic chemistry and peptide science. During his Ph.D. he worked on the development of novel linkers for bioconjugation, specifically for antibody–drug conjugates, in collaboration with the company PharmaMar. At present, Iván is undertaking his first postdoctoral research at ChemBioLab, located at the Barcelona Science Park, studying the preparation of complex cell‐penetrating bicyclic peptides.

Biographical Information
Professor Ayman El‐Faham received his B.Sc. and M.Sc. in Physical Organic Chemistry from the University of Alexandria (Egypt). In 1991 he received his Ph.D. in organic chemistry through a joint project between the University of Alexandria and the University of Massachusetts. From 1992 to 1999, he worked on new coupling reagents in Professor Louis Carpino's laboratory. Following a position as Head of the Chemistry Department at Beirut Arab University (2000–2004), and as Direct Manager of both the NMR laboratory and the Central Lab at Alexandria University (2004–2008), he worked at King Saud University (Riyadh, Saudi Arabia) as a Professor of Organic Chemistry (2008–2010). Currently, he is again working at King Saud University. His research interests include the synthesis of peptides, natural products, heterocycles, and biologically active targets.

Biographical Information
Dr. Ernesto Nicolas obtained his Ph.D. in Chemical Science at the University of Barcelona (Spain), working on solid‐phase peptide synthesis. He completed postdoctoral research in the Department of Chemistry at the University of Arizona, working on the asymmetric synthesis of β‐branched amino acids. In 1992 he joined the Department of Chemistry at the University of Barcelona as a Professor. His research is focused on the field of bioorganic chemistry, specifically the development of new methods for solid‐phase peptide synthesis and amino acid chemistry.

Biographical Information
Dr. Hortensia Rodríguez obtained a B.Sc. in Radiochemistry from the Superior Institute of Science and Nuclear Technology (ISCTN, Cuba) and his M.Sc. and Ph.D. at the University of Havana (Cuba). Her thesis was focused on the development of nonconventional methods for the synthesis of bioactive heterocyclic compounds. Afterwards, she served as chair of the Laboratory of Organic Synthesis (2006–2009) and of the Department of Organic Chemistry (2009–2011) in the Chemistry Faculty of the University of Havana. Between 2011 and 2014 she worked as a Research Associate at the Institute for Research in Biomedicine in the group of Professor Fernando Albericio. At present, she is a Research Professor at Yachay Tech University (Urcuquí, Ecuador). Her research covers a broad range of topics in synthetic bioorganic chemistry and nanobioscience.

Biographical Information
Professor Beatriz G. de la Torre obtained her Ph.D. from the University of Barcelona (Spain). At present, she is Professor at the School of Laboratory of Medicine and Medical Sciences at the University of KwaZulu‐Natal (Durban, South Africa). She has worked extensively on glyco‐, nucleo‐, and lipopeptides. Her scientific interests are focused on the discovery of new antimicrobial peptides including those against tuberculosis, peptide‐based vaccines, and peptide‐based drug delivery systems.

Biographical Information
Professor Fernando Albericio developed his academic career in Europe, USA, Latin America, and presently in Africa. His major research interests cover practically all aspects of peptide synthesis and combinatorial chemistry methodologies, as well as the synthesis of peptides and small molecules with therapeutic activities, especially against tuberculosis, infectious diseases, and cancer. His research group is also involved in the development of new strategies for drug delivery. He is deeply involved in the development of the third mission of the University: the transference of knowledge and technology to society.

Acknowledgements
We thank Dr. Thomas Bruckdorfer from Iris Biotech (Germany) for supporting this work and providing free samples of protected amino acids. The work in the author's laboratories was funded in part by the following: National Research Foundation (NRF) and the University of KwaZulu‐Natal (South Africa); the International Scientific Partnership Program (ISPP #0061) at King Saud University (Saudi Arabia); and MEC (CTQ2015‐67870‐P) and Generalitat de Catalunya (2014 SGR 137; Spain).
A. Sharma, I. Ramos-Tomillero, A. El-Faham, E. Nicolas, H. Rodriguez, B. G. de la Torre, F. Albericio, ChemistryOpen 2017, 6, 168.
References
- 1. Montalbetti C. A., Falque V., Tetrahedron 2005, 61, 10827–10852. [Google Scholar]
- 2. El-Faham A., Albericio F., Chem. Rev. 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- 3. Isidro-Llobet A., Alvarez M., Albericio F., Chem. Rev. 2009, 109, 2455–2504. [DOI] [PubMed] [Google Scholar]
- 4. Albericio F., Biopolymers 2000, 55, 123–139. [DOI] [PubMed] [Google Scholar]
- 5. Schelhaas M., Waldmann H., Angew. Chem. Int. Ed. Engl. 1996, 35, 2056–2083; [Google Scholar]; Angew. Chem. 1996, 108, 2192–2219. [Google Scholar]
- 6. Sheridan J. M., Hayes G. M., Austen B. M., J. Pept. Sci. 1999, 5, 555–562. [DOI] [PubMed] [Google Scholar]
- 7. Li D., Elbert D., J. Pept. Res. 2002, 60, 300–303. [DOI] [PubMed] [Google Scholar]
- 8. Kates S. A., Solé N. A., Johnson C. R., Hudson D., Barany G., Albericio F., Tetrahedron Lett. 1993, 34, 1549–1552. [Google Scholar]
- 9. White C. J., Yudin A. K., Nat. Chem. 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- 10. Royo M., Alsina J., Giralt E., Slomcyznska U., Albericio F., J. Chem. Soc. Perkin Trans. 1 1995, 1095–1102. [Google Scholar]
- 11. Wuts P. G., Greene T. W., Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Hoboken, NJ, 2006. [Google Scholar]
- 12. Paul R., Bull. Soc. Chim. 1934, 5, 973. [Google Scholar]
- 13. Woods G. F., Kramer D. N., J. Am. Chem. Soc. 1947, 69, 2246–2246. [Google Scholar]
- 14. Parham W. E., Anderson E., J. Am. Chem. Soc. 1948, 70, 4187–4189. [DOI] [PubMed] [Google Scholar]
- 15. Postma T. M., Albericio F., Eur. J. Org. Chem. 2014, 3519–3530. [Google Scholar]
- 16. Nugiel D. A., Cornelius L., Corbett J. W., J. Org. Chem. 1997, 62, 201. [DOI] [PubMed] [Google Scholar]
- 17. Chang J., Dong C., Guo X., Hu W., Cheng S., Wang Q., Chen R., Bioorg. Med. Chem. 2005, 13, 4760–4766. [DOI] [PubMed] [Google Scholar]
- 18. Wang X., Choe Y., Craik C. S., Ellman J. A., Bioorg. Med. Chem. Lett. 2002, 12, 2201–2204. [DOI] [PubMed] [Google Scholar]
- 19. Smith A. L., Stevenson G. I., Swain C. J., Castro J., Tetrahedron Lett. 1998, 39, 8317–8320. [Google Scholar]
- 20. Cheng X., Hii K. K. M., Tetrahedron 2001, 57, 5445–5450. [Google Scholar]
- 21. Na Y., Shen H., Byers L. D., Bioorg. Chem. 2011, 39, 111–113. [DOI] [PubMed] [Google Scholar]
- 22. Torres-García C., Díaz M., Blasi D., Farràs I., Fernández I., Ariza X., Farràs J., Lloyd-Williams P., Royo M., Nicolás E., Int. J. Pept. Res. Ther. 2012, 18, 7–19. [Google Scholar]
- 23. Atherton E., Sheppard R. C., Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, Oxford, 1989. [Google Scholar]
- 24. Albericio F., Kneib-Cordonier N., Biancalana S., Gera L., Masada R. I., Hudson D., Barany G., J. Org. Chem. 1990, 55, 3730–3743. [Google Scholar]
- 25. Ramos-Tomillero I., Mendive-Tapia L., Góngora-Benítez M., Nicolás E., Tulla-Puche J., Albericio F., Molecules 2013, 18, 5155–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parham W. E., DeLaitsch D. M., J. Am. Chem. Soc. 1954, 76, 4962–4965. [Google Scholar]
- 27. Holland G. F., Cohen L. A., J. Am. Chem. Soc. 1958, 80, 3765–3769. [Google Scholar]
- 28. Otaka A., Morimoto H., Fujii N., Koide T., Funakoshi S., Yajima H., Chem. Pharm. Bull. 1989, 37, 526–528. [Google Scholar]
- 29. Hibino H., Nishiuchi Y., Org. Lett. 2012, 14, 1926–1929. [DOI] [PubMed] [Google Scholar]
- 30. Hibino H., Miki Y., Nishiuchi Y., J. Pept. Sci. 2014, 20, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramos-Tomillero I., Rodríguez H., Albericio F., Org. Lett. 2015, 17, 1680–1683. [DOI] [PubMed] [Google Scholar]
- 32. Ramos-Tomillero I., Rodríguez H., Albericio F. in Peptides 2015. Proceedings of the 24th American Peptide Symposium (Eds.: V. Srivastava, A. Yudin, M. Lebl), American Peptide Society, San Diego, 2015, pp. 274–277. [Google Scholar]
- 33. Hiskey R. G., Tucker W. P., J. Am. Chem. Soc. 1962, 84, 4789–4794. [Google Scholar]
- 34. Miyashita M., Yoshikoshi A., Grieco P. A., J. Org. Chem. 1977, 42, 3772–3774. [Google Scholar]
- 35. Corey E., Danheiser R. L., Chandrasekaran S., Siret P., Keck G. E., Gras J. L., J. Am. Chem. Soc. 1978, 100, 8031–8034. [Google Scholar]
- 36. Liu J., Wong C.-H., Tetrahedron Lett. 2002, 43, 4037–4039. [Google Scholar]
- 37. Schwalm R., Binder H., Funhoff D., J. Appl. Polym. Sci. 2000, 78, 208–216. [Google Scholar]
- 38. Palladino P., Stetsenko D. A., Org. Lett. 2012, 14, 6346–6349. [DOI] [PubMed] [Google Scholar]
- 39. Rastetter W. H., Erickson T. J., Venuti M. C., J. Org. Chem. 1980, 45, 5011–5012. [Google Scholar]
- 40. Rastetter W. H., Erickson T. J., Venuti M. C., J. Org. Chem. 1981, 46, 3579–3590. [Google Scholar]
- 41. Yu X., Dai Y., Yang T., Gagné M. R., Gong H., Tetrahedron 2011, 67, 144–151. [Google Scholar]
- 42. Olsen R. K., Ramasamy K., Bhat K. L., Low C. L., Waring M. J., J. Am. Chem. Soc. 1986, 108, 6032–6036. [DOI] [PubMed] [Google Scholar]
- 43. Tan L., Ma D., Angew. Chem. Int. Ed. 2008, 47, 3614–3617; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3670–3673. [Google Scholar]
- 44. Mohapatra D. K., Datta A., Synlett 1996, 1129–1130. [Google Scholar]
- 45. Karmakar S., Mohapatra D. K., Synlett 2001, 1326–1328. [Google Scholar]
- 46. Nudelman A., Marcovici-Mizrahi D., Nudelman A., Flint D., Wittenbach V., Tetrahedron 2004, 60, 1731–1748. [Google Scholar]
- 47. Krishnamoorthy R., Vazquez-Serrano L. D., Turk J. A., Kowalski J. A., Benson A. G., Breaux N. T., Lipton M. A., J. Am. Chem. Soc. 2006, 128, 15392–15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iijima Y., Kimata O., Decharin S., Masui H., Hirose Y., Takahashi T., Eur. J. Org. Chem. 2014, 4725–4732. [Google Scholar]
- 49. Thompson L. A., Ellman J. A., Tetrahedron Lett. 1994, 35, 9333–9336. [Google Scholar]
- 50. Jones R. G., Mann M. J., J. Am. Chem. Soc. 1953, 75, 4048–4052. [Google Scholar]
- 51. Ott A. C., Murray M. F., Pederson R. L., J. Am. Chem. Soc. 1952, 74, 1239–1241. [Google Scholar]
- 52. Alper H., Dinkes L., Synthesis 1972, 81–81. [Google Scholar]
- 53. Torii S., Inokuchi T., Kondo K., Ito H., Bull. Chem. Soc. Jpn. 1985, 58, 1347–1348. [Google Scholar]
- 54. Bongini A., Cardillo G., Orena M., Sandri S., Synthesis 1979, 618–620. [Google Scholar]
- 55. Olah G. A., Husain A., Singh B. P., Synthesis 1983, 892–895. [Google Scholar]
- 56. Hoyer S., Laszlo P., Orlović M., Polla E., Synthesis 1986, 655–657. [Google Scholar]
- 57. Kumar P., Dinesh C. U., Reddy R. S., Pandey B., Synthesis 1993, 1069–1070. [Google Scholar]
- 58. Campelo J. M., Garcia A., Lafont F., Luna D., Marinas J. M., Synth. Commun. 1994, 24, 1345–1350. [Google Scholar]
- 59. Miura T., Masaki Y., Synth. Commun. 1995, 25, 1981–1987. [Google Scholar]
- 60. Iselin B., Schwyzer R., Helv. Chim. Acta 1956, 39, 57–64. [Google Scholar]
- 61. López-Macià À., Jiménez J. C., Royo M., Giralt E., Albericio F., J. Am. Chem. Soc. 2001, 123, 11398–11401. [DOI] [PubMed] [Google Scholar]
- 62. Pelay-Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9022–9054. [Google Scholar]
- 63.A. Sharma, I. Ramos-Tomillero, A. El-Faham, H. Rodríguez, B. G. de la Torre, F. Albericio, ChemistryOpen 2017, DOI: 10.1002/open.201600157. [DOI] [PMC free article] [PubMed]
- 64. Graham K. A., Wang Q., Eisenhut M., Haberkorn U., Mier W., Tetrahedron Lett. 2002, 43, 5021–5024. [Google Scholar]
- 65. Villorbina G., Canals D., Carde L., Grijalvo S., Pascual R., Rabal O., Teixidó J., Fabriàs G., Llebaria A., Casas J., Bioorg. Med. Chem. 2007, 15, 50–62. [DOI] [PubMed] [Google Scholar]
- 66. Olah G. A., Husain A., Singh B. P., Synthesis 1985, 703–704. [Google Scholar]
- 67. Hirschmann R., Schwam H., Strachan R., Schoenewaldt E., Barkemeyer H., Miller S., Conn J. B., Garsky V., Veber D. F., Denkewalter R. G., J. Am. Chem. Soc. 1971, 93, 2746–2754. [DOI] [PubMed] [Google Scholar]
- 68. Bombardelli E., Bonati A., Gabetta B., Martinelli E. M., Mustich G., Danieli B., J. Chem. Soc. Perkin Trans. 1 1976, 1432–1438. [PubMed] [Google Scholar]
- 69. Kalivretenos A. G., Nakanishi K., J. Org. Chem. 1993, 58, 6596–6608. [Google Scholar]
- 70. Ramaseshan M., Robitaille M., Ellingboe J. W., Dory Y. L., Deslongchamps P., Tetrahedron Lett. 2000, 41, 4737–4742. [Google Scholar]
- 71. Pappo R., Collins P. W., Tetrahedron Lett. 1972, 13, 2627–2630. [Google Scholar]
- 72. Bernady K. F., Floyd M. B., Poletto J. F., Weiss M. J., J. Org. Chem. 1979, 44, 1438–1447. [Google Scholar]
- 73. Wipf P., Kim H., J. Org. Chem. 1993, 58, 5592–5594. [Google Scholar]
- 74. Holden K. G., Mattson M. N., Cha K. H., Rapoport H., J. Org. Chem. 2002, 67, 5913–5918. [DOI] [PubMed] [Google Scholar]
- 75. Mattson M. N., Rapoport H., J. Org. Chem. 1996, 61, 6071–6074. [Google Scholar]