

Abstract
Obstructive sleep apnoea (OSA) is a highly prevalent condition and recognized as a major public health burden conveying a significant risk of cardiometabolic diseases and mortality. Type 2 diabetes (T2D), insulin resistance (IR) and glucose tolerance are common in subjects with OSA and this association is at least in part independent of the effects of obesity. Continuous positive airway pressure (CPAP) is the treatment of choice for the majority of patients with OSA but the benefit of CPAP on glycaemic health is uncertain. Thus, a greater understanding of the mechanisms by which OSA leads to metabolic dysfunction might identify novel therapeutic approaches. Intermittent hypoxia (IH), a hallmark feature of OSA, likely plays a key role in the pathogenesis and experimental studies using animal and in vitro models suggest that IH leads to pancreatic β‐cell dysfunction and to insulin resistance in the insulin target organs liver, skeletal muscle and adipose tissue. In particular, IH induces a pro‐inflammatory phenotype of the visceral adipose tissue with polarization of adipose tissue macrophages towards a M1–pro‐inflammatory subtype, upregulation and secretion of numerous pro‐inflammatory adipokines and subsequent impairment of the insulin‐signalling pathway, changes which bear a striking similarity to adipose tissue dysfunction seen in obesity. In this review, the available evidence linking IH with metabolic dysfunction is explored with a special emphasis on the adipose tissue in this process.
Keywords: adipose tissue, hypoxia, sleep apnoea
Abbreviations
- BMI
body mass index
- CLS
crown‐like structure
- CPAP
continuous positive airway pressure
- ET‐1
endothelin 1
- FFA
free fatty acid
- HIF
hypoxia inducible factor
- HFD
high‐fat diet
- IH
intermittent hypoxia
- IKK
inhibitor of nuclear factor kappa B kinase
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- IR
insulin resistance
- IRS
insulin‐receptor substrate
- JNK
c‐Jun N‐terminal protein kinases
- MCP‐1
monocyte chemoattractant protein‐1
- MetS
metabolic syndrome
- NF‐κB
nuclear factor kappa B
- OSA
obstructive sleep apnoea
- PKR
protein kinase R
- ROS
reactive oxygen species
- SVF
stromal vascular fraction
- T2D
type 2 diabetes
- TNF‐α
tumour necrosis factor alpha
- WAT
white adipose tissue
Introduction
Obstructive sleep apnoea (OSA), characterized by repeated pharyngeal collapse during sleep leading to intermittent hypoxia (IH), sleep fragmentation and excessive daytime sleepiness, represents an increasing public health burden. It is a common disorder affecting about 14% of men and 5% of women (Peppard et al. 2013) and prevalence is rapidly rising due to the strong association of OSA with obesity, with at least 60% of OSA patients being obese (Peppard et al. 2000). Furthermore, OSA represents a significant risk factor for the development of numerous cardiovascular diseases, such as systemic arterial hypertension, coronary artery disease, arrhythmias and heart failure leading to increased morbidity and mortality (Ip et al. 2002; Punjabi et al. 2002; McNicholas & Bonsignore, 2007; Kent et al. 2014). There is also compelling evidence of an independent association of OSA with metabolic dysfunction, and in particular with alterations in glucose metabolism leading to type 2 diabetes (T2D), insulin resistance (IR), glucose intolerance and metabolic syndrome (MetS), and this association seems to be at least in part irrespective of the degree of obesity (Ip et al. 2002; Punjabi et al. 2002; Kent et al. 2014). Moreover, OSA and obesity may exert synergistic negative effects on glucose metabolism.
The pathophysiology underlying metabolic dysfunction in OSA is incompletely understood but a large body of data indicates that the particular form of intermittent hypoxia (IH) observed in OSA, with repetitive short cycles of desaturation followed by rapid reoxygenation, plays a pivotal role. Experimental studies indicate that IH exacerbates its detrimental effect on glucose metabolism through various mechanisms. It impairs pancreatic insulin secretion but also acts directly on insulin target organs such as liver, skeletal muscle and adipose tissue. Given the close link of OSA with obesity, the latter has attracted specific attention and indeed, studies in animals and in vitro indicate that IH leads to adipose tissue inflammation with the downstream consequence of decreased insulin sensitivity. This review summarizes the evidence of IH‐induced glucose metabolic dysfunction, focusing primarily on the role of the adipose tissue in this process.
Intermittent hypoxia and glucose metabolism
Various clinical and population studies have revealed an independent association of OSA with impaired glucose haemostasis including T2D, IR, glucose intolerance and MetS and this topic has been extensively reviewed elsewhere (Levy et al. 2009; Bonsignore et al. 2013; Kent et al. 2015). Notably, in many of these studies, measures of nocturnal hypoxaemia had the most robust relationship with impaired glycaemic health, suggesting a key role of intermittent hypoxia in the pathophysiology (Punjabi et al. 2004; Sulit et al. 2006; Polotsky et al. 2009). Over the last two decades, detailed insight into the effects of IH has been provided by animal studies. In the majority of these studies, rodents are housed in specific chambers and intermittently breathe air with low oxygen concentration. Protocols of the IH treatment vary but a common model lowers the ambient oxygen content to 5% for 60 cycles h−1, mimicking severe OSA. Exposing lean C57BL/6J mice to just 9 h of such protocol of IH already resulted in decreased whole‐body insulin sensitivity as measured by hyperinsulinaemic euglycaemic clamp (Iiyori et al. 2007). Chronic IH applied for several weeks, which more realistically recapitulates OSA, leads to several metabolic alterations, including higher fasting glucose and insulin levels, insulin resistance, glucose intolerance and β‐cell dysfunction (Wang et al. 2013; Poulain et al. 2014). A small number of studies have also investigated the effects of IH in the presence of obesity in both, leptin‐deficient and diet‐induced obese mice, and consistently suggested an exacerbation of the obesity‐induced metabolic dysfunction by IH (Polotsky et al. 2003; Drager et al. 2011; Murphy et al. 2015). Moreover, in healthy volunteers, an acute protocol of IH applied for 5 h resulted in impaired insulin sensitivity without increase in compensatory insulin secretion (Louis & Punjabi, 2009).
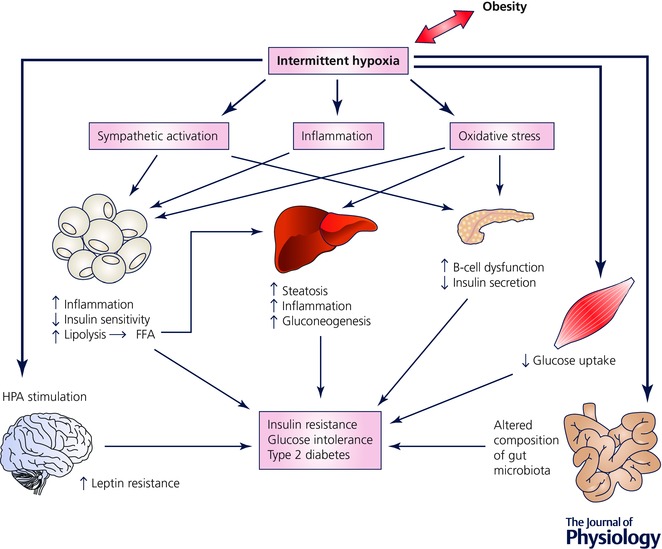
There are several potential mechanisms by which IH mediates its effect on glucose metabolic dysfunction (Fig. 1). Activation of the sympathetic nervous system in response to IH has been extensively studied in humans and rodents and has been identified to be an important pathway in the development of systemic arterial hypertension and other cardiovascular diseases in OSA (Fletcher, 2003). IH‐induced sympathetic excitation also probably impacts on glucose metabolism as catecholamines are known to decrease insulin sensitivity, to reduce insulin‐mediated glucose uptake and to stimulate hepatic gluconeogenesis (Deibert & DeFronzo, 1980; Dungan et al. 2009). Furthermore, noradrenalin (epinephrine) is a potent mediator of adipose lipolysis and through this mechanism IH may additionally impair insulin sensitivity (Carmen & Victor, 2006). However, a recent study investigated the effect of adrenal medullectomy on glucose metabolism in mice treated with IH, and although surgery led to improved insulin secretion, IH‐induced hyperglycaemia and IR remained unaffected, supporting the need for further studies on this subject (Shin et al. 2014). Furthermore, local and systemic inflammation in response to IH is central in the pathogenesis of OSA‐associated cardiometabolic processes (Ryan et al. 2009). IH is a potent pro‐inflammatory stimulus through activation of nuclear factor‐kappa B (NF‐κB), the master transcriptional regulator of inflammatory responses with the downstream consequence of the production of numerous pro‐inflammatory mediators such as tumour necrosis factor (TNF)‐α, interleukin (IL)‐6, IL‐8 or monocyte chemoattractant protein 1 (MCP1) (Ryan et al. 2005; Greenberg et al. 2006; Ryan et al. 2006). These products have detrimental effects on cardiovascular and metabolic health and documented inhibitory effects on insulin sensitivity in the liver and adipose tissue (Ouchi et al. 2011; Cooke et al. 2016). Some studies have also suggested that IH directly affects the hypothalamic–pituitary–adrenal axis with a downstream‐consequence of increased cortisol release, which has well‐known negative consequences on glucose metabolisms, including inhibition of insulin secretion and insulin resistance (Yokoe et al. 2008). In addition, IH has generally been associated with increased production of reactive oxygen species (ROS) promoting oxidative stress, and studies in rodents documented increased lipid peroxidation in the liver and pancreatic β‐cells (Rosa et al. 2011; Fang et al. 2014) and, hence, this pathway may also be involved in the pathogenesis of IH‐induced metabolic dysregulation (Henriksen et al. 2011).
Figure 1. Potential mechanisms of alteration in glucose metabolism by intermittent hypoxia.
A recent study also points to a potential effect of IH on the gut microbiota which has gained substantial attention over the last decade playing a pivotal role in the progression of metabolic dysfunction in obesity (Schroeder & Backhed, 2016). Using a mouse model, Moreno‐Indias et al. demonstrated substantial alteration in the microbiota composition in response to IH, but the functional implications of these changes need to be further explored (Moreno‐Indias et al. 2015).
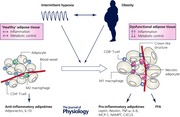
IH, obesity and adipose tissue dysfunction
As outlined above, OSA is strongly associated with obesity. According to epidemiological studies, the prevalence of OSA in obese subjects exceeds 30%, at least 60% of OSA patients are obese and the incidence of OSA increases incrementally with increasing body mass index (BMI) (Young et al. 1993; Peppard et al. 2000). Furthermore, both conditions lead to similar cardiovascular and metabolic consequences, with increasing evidence of potential synergistic effects. This intimate relationship and interaction has led to the hypothesis that OSA shares various pathophysiological mechanisms of cardiometabolic complications with obesity. The pivotal role of the white adipose tissue (WAT) in obesity‐mediated diseases is well described. Traditionally considered as an organ of pure energy storage, it has over the last two decades emerged as a key endocrine organ, participating in many physiological and pathophysiological processes mediated through the secretion of multiple hormones, cytokines, chemokines and other proteins, collectively referred to as adipokines (Ouchi et al. 2011). Obesity is characterized by an expansion of the adipose tissue which occurs via an increase in adipocyte cell size (hypertrophy), cell number (hyperplasia), or both (Jo et al. 2009). Whereas hyperplasia represents a healthy expansion of the adipose tissue, adipocyte hypertrophy leads to lipid‐laden, dysfunctional adipocytes that mediate inflammation and dysfunction. Furthermore, the distribution of excess adipose tissue has significant impact on pathophysiological consequences, wherein increased visceral fat mass to a greater extent than subcutaneous adipose tissue is associated with serious health consequences (Despres & Lemieux, 2006). In obesity, adipocyte hypertrophy and dysfunction are paralleled by quantitative and qualitative changes of the stromal vascular fraction (SVF). Immune cells are of particular relevance in this setting. Lean healthy adipose tissue is characterized by the occurrence of anti‐inflammatory regulatory T cells (Tregs) and macrophages of a M2, alternatively activated, phenotype, which promote the upregulation of anti‐inflammatory factors such as IL‐10 or adiponectin and downregulation of pro‐inflammatory cytokines, and thus are associated with tissue repair and resolution of inflammation. With increasing adipose tissue expansion, there is increased infiltration of CD8+ cytotoxic T‐cells, with a decrease in CD4+ helper and Treg cells. Macrophages polarize towards a M1, pro‐inflammatory, subtype and are arranged around necrotic adipocytes in crown‐like structures (CLS). M1 macrophages produce numerous pro‐inflammatory cytokines such as IL‐6 and TNF‐α and express inducible nitric oxide synthase (iNOS) (Ouchi et al. 2011; Cildir et al. 2013). The resulting adipose tissue inflammation leads to a release of free fatty acids (FFAs) which activate various signalling pathways, including c‐Jun N‐terminal protein kinases (JNKs), inhibitor of nuclear factor κ‐B kinase subunit β (IKKβ) and protein kinase R (PKR), collectively resulting in impairment of the insulin‐signalling pathway, with the downstream consequence of insulin resistance and metabolic dysfunction (Odegaard & Chawla, 2013).
Furthermore, there is accumulating evidence that obesity, which is characterized by excess of adipose tissue with compromised vascularity, mediates its pro‐inflammatory response of WAT through hypoxia‐derived pathways (Trayhurn et al. 2008). Hypoxia‐inducible factor (HIF)‐1 is the main transcriptional regulator of the hypoxia response. Recently Lee et al. demonstrated that genetic inhibition of HIF‐1α in adipocytes protected mice from high‐fat diet (HFD)‐induced adipose tissue inflammation and restored insulin sensitivity not only in the adipose tissue but also in liver and skeletal muscle. In accompanying in vitro mechanistic studies the authors concluded that HIF‐1α activation increased the expression of a wide array of pro‐inflammatory adipocyte genes mediated through interaction with NF‐κB (p65) gene promoters. In contrast, activation of HIF‐2α in adipocytes has opposing, protective effects with attenuation of HFD‐induced inflammation, glucose intolerance and IR (Lee et al. 2014).
Given the pivotal role of inflammation and hypoxia in obesity‐induced adipose tissue dysfunction, it has long been postulated that IH as it occurs in OSA may augment this response. Indeed, various studies using rodent or reductionist models of IH have added support to this hypothesis and, moreover, have demonstrated that IH induces inflammatory changes of the visceral adipose tissue even in the absence of obesity. Using atherosclerosis‐prone ApoE‐deficient mice, Poulain et al. demonstrated increased macrophage infiltration of adipose tissue and increased expression of monocyte chemoattractant protein (MCP)‐1, abnormalities which were associated with reduced insulin sensitivity and more advanced atherosclerotic lesions (Poulain et al. 2014). Furthermore, we and others have characterized the phenotype of adipose tissue macrophages in lean and diet‐obese C57Bl/6J wild‐type mice, confirming a shift of polarization towards a M1–pro‐inflammatory subtype in response to IH, a response partially reversible by treatment with the anti‐obesity drug resveratrol (Carreras et al. 2015; Murphy et al. 2015). Importantly, the percentage of M1 macrophages correlated directly with the level of insulin resistance and this inflammatory shift was accompanied by down‐regulation of insulin‐receptor substrate (IRS)‐1 mRNA with subsequent alteration in IRS‐1 tyrosine phosphorylation and diminished phosphorylation of Akt (Murphy AM, Thomas A, Crinion SJ, Kent BD, Tambuwala MM, Fabre A, Pepin JL, RocheHM, Arnaud C & Ryan S, unpublished data). In addition, murine adipocyte cultures in IH demonstrate greater NF‐κB activation and decreased secretion of the key anti‐inflammatory adipokine adiponectin than cells in control conditions (He et al. 2014; Carreras et al. 2015). We recently reported that primary human adipocytes treated with IH in vitro demonstrate a greater sensitivity to IH towards pro‐inflammatory pathway activation, with the downstream consequence of more enhanced expression of multiple inflammatory mediators than primary cells of non‐adipocyte lineage (Taylor et al. 2014). Collectively, these data support the critical role of adipose tissue as a potential source of pro‐inflammatory mediators in response to IH.
Adipose tissue inflammation and insulin resistance give rise to lipolysis with increased and persistent FFA flux into the systemic circulation which is linked to the development of hepatic and muscle insulin resistance and β‐cell dysfunction (Cooke et al. 2016). Studies using C57BL/6J wild‐type and ApoE‐deficient mice have demonstrated increased serum FFA levels with IH treatment and have proposed a pathophysiological role of this response in the development of dyslipidaemia and steatohepatitis (Savransky et al. 2007; Jun et al. 2010; Weiszenstein et al. 2016). Pharmacological inhibition of lipolysis by acipimox, a nicotinic acid analogue, ameliorated the detrimental effects on glucose metabolism induced by chronic IH exposure (Weiszenstein et al. 2016) and in a recent investigation using a combined in vivo–in vitro approach a potential mechanistic link between IH and lipolysis was proposed through upregulation of endothelin 1 (ET‐1) (Briancon‐Marjollet et al. 2016).
The detailed intermediate mechanisms by which IH contributes to adipose tissue inflammation are as‐yet unknown. However, a hypoxia‐mediated response through HIF‐1α activation is plausible. In support of this hypothesis, a study in lean and diet‐induced obese mice suggested that IH exposure results in a substantial decrease in adipose tissue oxygen tension with attenuation of the cyclical fluctuations (Reinke et al. 2011). Subsequently, adipose HIF‐1 activation in response to IH has been demonstrated, associated with accelerated atherosclerosis but the detailed role of this pathway in the IH‐induced metabolic dysfunction remains unknown (Drager et al. 2013). As outlined above, IH has been closely linked to activation of NF‐κB‐mediated inflammation, sympathetic excitation and oxidative stress and although the involvement of these pathways in IH‐induced adipose tissue inflammation and metabolic dysfunction is likely, the exact contribution and interplay of these mechanistic links in the actual process will require further detailed investigation.
Current limitations and future directions
Despite the fast growing evidence of IH‐induced adipose tissue inflammation and subsequent impairment of glucose metabolism in animals, we are as yet lacking evidence of this response in humans. The rodent models as currently used have significant limitations and thus results have to be interpreted with caution. Firstly, the epididymal fat pads are the largest and best characterized visceral adipose tissue depot in mice but there is no true equivalent in humans. In contrast, the largest visceral adipose tissue organ in humans is omental fat which is essentially absent in rodents. Omental adipose tissue shows considerably greater vascularity and also drains blood into the portal circulation unlike the murine visceral depots and hence the relevance of the pathophysiological responses seen in mice in response to obesity or IH for the human condition of OSA is as yet unclear. Given the difficulties in routine sampling of visceral adipose tissue in humans, studies are generally limited to the morbidly obese patient group undergoing bariatric surgery, which, however, only represents a limited proportion of OSA subjects. Investigating such a cohort, no relationship between the severity of IH and the total number of macrophages in omental and subcutaneous fat depots was observed but the phenotypes of adipose tissue macrophages or other metabolic indices were not investigated and thus it remains unclear if the observed adipose tissue inflammation in response to IH mimics the process in subjects with OSA (Aron‐Wisnewsky et al. 2012). Secondly, the IH rodent model has several caveats. It does not recapitulate all the effects of OSA, as besides IH there are also other potential pathophysiological triggering factors for cardiometabolic diseases including sleep fragmentation, intrathoracic pressure swings and hypercapnia. In addition, the pattern of IH in the majority of these models resembles very severe OSA and hence caution is advised in translating these findings universally to clinical OSA. Furthermore, rodents do not sleep continuously for 6–8 h as humans do. Although systems have been developed to trigger IH during sleep only, most models use IH unlinked to sleep.
In conclusion, despite fast‐emerging evidence of metabolic dysfunction in response to IH arising predominantly from animal and reductionist models, careful translation of these findings into the clinical setting is essential. Thus, future clinical, basic and translational studies defining the detailed mechanisms and consequences of IH‐induced adipose tissue inflammation are warranted and will potentially identify novel therapeutic targets.
Biography
Silke Ryan is a Consultant in Respiratory and Sleep Medicine and Research Fellow at University College Dublin. She graduated in medicine in 1998 and was awarded her PhD degree in 2006. Her primary research interests include the broad subject of cardiovascular disease mechanisms of obstructive sleep apnoea and in particular, inflammatory responses to intermittent hypoxia. Her research has been published widely with over 40 original manuscripts and her achievements to date have been recognized by numerous international prizes including the ATS James Skatrud New Investigator Award in 2010 and the ERS Cournand Lecture Award in 2015.

This review was presented at the symposium “Physiological gases in health and disease”, which took place at Physiology 2016, Dublin, Ireland, 29–31 July 2016.
This is an Editor's Choice article from the 15 April 2017 issue.
References
- Aron‐Wisnewsky J, Minville C, Tordjman J, Levy P, Bouillot JL, Basdevant A, Bedossa P, Clement K & Pepin JL (2012). Chronic intermittent hypoxia is a major trigger for non‐alcoholic fatty liver disease in morbid obese. J Hepatol 56, 225–233. [DOI] [PubMed] [Google Scholar]
- Bonsignore MR, Borel AL, Machan E & Grunstein R (2013). Sleep apnoea and metabolic dysfunction. Eur Respir Rev 22, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briancon‐Marjollet A, Monneret D, Henri M, Hazane‐Puch F, Pepin JL, Faure P & Godin‐Ribuot D (2016). Endothelin regulates intermittent hypoxia‐induced lipolytic remodelling of adipose tissue and phosphorylation of hormone‐sensitive lipase. J Physiol 594, 1727–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen GY & Victor SM (2006). Signalling mechanisms regulating lipolysis. Cell Signal 18, 401–408. [DOI] [PubMed] [Google Scholar]
- Carreras A, Zhang SX, Almendros I, Wang Y, Peris E, Qiao Z & Gozal D (2015). Resveratrol attenuates intermittent hypoxia‐induced macrophage migration to visceral white adipose tissue and insulin resistance in male mice. Endocrinology 156, 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cildir G, Akincilar SC & Tergaonkar V (2013). Chronic adipose tissue inflammation: all immune cells on the stage. Trends Mol Med 19, 487–500. [DOI] [PubMed] [Google Scholar]
- Cooke AA, Connaughton RM, Lyons CL, McMorrow AM & Roche HM (2016). Fatty acids and chronic low grade inflammation associated with obesity and the metabolic syndrome. Eur J Pharmacol 785, 207–214. [DOI] [PubMed] [Google Scholar]
- Deibert DC & DeFronzo RA (1980). Epinephrine‐induced insulin resistance in man. J Clin Invest 65, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres JP & Lemieux I (2006). Abdominal obesity and metabolic syndrome. Nature 444, 881–887. [DOI] [PubMed] [Google Scholar]
- Drager LF, Li J, Reinke C, Bevans‐Fonti S, Jun JC & Polotsky VY (2011). Intermittent hypoxia exacerbates metabolic effects of diet‐induced obesity. Obesity 19, 2167–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drager LF, Yao Q, Hernandez KL, Shin MK, Bevans‐Fonti S, Gay J, Sussan TE, Jun JC, Myers AC, Olivecrona G, Schwartz AR, Halberg N, Scherer PE, Semenza GL, Powell DR & Polotsky VY (2013). Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin‐like 4. Am J Respir Crit Care Med 188, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungan KM, Braithwaite SS & Preiser JC (2009). Stress hyperglycaemia. Lancet 373, 1798–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Zhang Q, Tan J, Li L, An X & Lei P (2014). Intermittent hypoxia‐induced rat pancreatic beta‐cell apoptosis and protective effects of antioxidant intervention. Nutr Diabetes 4, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC (2003). Sympathetic over activity in the etiology of hypertension of obstructive sleep apnea. Sleep 26, 15–19. [DOI] [PubMed] [Google Scholar]
- Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T & Liu SF (2006). Chronic intermittent hypoxia activates nuclear factor‐kappaB in cardiovascular tissues in vivo . Biochem Biophys Res Commun 343, 591–596. [DOI] [PubMed] [Google Scholar]
- He Q, Yang QC, Zhou Q, Zhu H, Niu WY, Feng J, Wang Y, Cao J & Chen BY (2014). Effects of varying degrees of intermittent hypoxia on proinflammatory cytokines and adipokines in rats and 3T3–L1 adipocytes. PLoS One 9, e86326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen EJ, Diamond‐Stanic MK & Marchionne EM (2011). Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med 51, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iiyori N, Alonso LC, Li J, Sanders MH, Garcia‐Ocana A, O'Doherty RM, Polotsky VY & O'Donnell CP (2007). Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med 175, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip MS, Lam B, Ng MM, Lam WK, Tsang KW & Lam KS (2002). Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 165, 670–676. [DOI] [PubMed] [Google Scholar]
- Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, Cushman SW & Periwal V (2009). Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol 5, e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J, Reinke C, Bedja D, Berkowitz D, Bevans‐Fonti S, Li J, Barouch LA, Gabrielson K & Polotsky VY (2010). Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E‐deficient mice. Atherosclerosis 209, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent BD, Grote L, Ryan S, Pepin JL, Bonsignore MR, Tkacova R, Saaresranta T, Verbraecken J, Levy P, Hedner J, McNicholas WT & collaborators (2014). Diabetes mellitus prevalence and control in sleep‐disordered breathing: the European Sleep Apnea Cohort (ESADA) study. Chest 146, 982–990. [DOI] [PubMed] [Google Scholar]
- Kent BD, McNicholas WT & Ryan S (2015). Insulin resistance, glucose intolerance and diabetes mellitus in obstructive sleep apnoea. J Thorac Dis 7, 1343–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh da Y, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS & Olefsky JM (2014). Increased adipocyte O2 consumption triggers HIF‐1alpha, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy P, Bonsignore MR & Eckel J (2009). Sleep, sleep‐disordered breathing and metabolic consequences. Eur Respir J 34, 243–260. [DOI] [PubMed] [Google Scholar]
- Louis M & Punjabi NM (2009). Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol (1985) 106, 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas WT & Bonsignore MR (2007). Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 29, 156–178. [DOI] [PubMed] [Google Scholar]
- Moreno‐Indias I, Torres M, Montserrat JM, Sanchez‐Alcoholado L, Cardona F, Tinahones FJ, Gozal D, Poroyko VA, Navajas D, Queipo‐Ortuno MI & Farre R (2015). Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur Respir J 45, 1055–1065. [DOI] [PubMed] [Google Scholar]
- Murphy AM, Thomas A, Tambuwala MM, McNicholas WT, Roche HM, Taylor CT, Pepin JL, Arnaud C & Ryan S (2015). Chronic intermittent hypoxia contributes to pro‐inflammatory macrophage alteraltion in visceral adipose tissue of lean and obese mice. Am J Respir Crit Care Med 191, A2691. [Google Scholar]
- Odegaard JI & Chawla A (2013). Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Parker JL, Lugus JJ & Walsh K (2011). Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW & Hla KM (2013). Increased prevalence of sleep‐disordered breathing in adults. Am J Epidemiol 177, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Palta M, Dempsey J & Skatrud J (2000). Longitudinal study of moderate weight change and sleep‐disordered breathing. JAMA 284, 3015–3021. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR & O'Donnell CP (2003). Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol 552, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, Steele KE, Schweizter MA, Clark JM, Torbenson MS & Schwartz AR (2009). Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med 179, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain L, Thomas A, Rieusset J, Casteilla L, Levy P, Arnaud C & Dematteis M (2014). Visceral white fat remodelling contributes to intermittent hypoxia‐induced atherogenesis. Eur Respir J 43, 513–522. [DOI] [PubMed] [Google Scholar]
- Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE & Sleep Heart Health Study I (2004). Sleep‐disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 160, 521–530. [DOI] [PubMed] [Google Scholar]
- Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR & Smith PL (2002). Sleep‐disordered breathing and insulin resistance in middle‐aged and overweight men. Am J Respir Crit Care Med 165, 677–682. [DOI] [PubMed] [Google Scholar]
- Reinke C, Bevans‐Fonti S, Drager LF, Shin MK & Polotsky VY (2011). Effects of different acute hypoxic regimens on tissue oxygen profiles and metabolic outcomes. J Appl Physiol (1985) 111, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa DP, Martinez D, Picada JN, Semedo JG & Marroni NP (2011). Hepatic oxidative stress in an animal model of sleep apnoea: effects of different duration of exposure. Comp Hepatol 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan S, Taylor CT & McNicholas WT (2005). Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 112, 2660–2667. [DOI] [PubMed] [Google Scholar]
- Ryan S, Taylor CT & McNicholas WT (2006). Predictors of elevated nuclear factor‐kappaB‐dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 174, 824–830. [DOI] [PubMed] [Google Scholar]
- Ryan S, Taylor CT & McNicholas WT (2009). Systemic inflammation: a key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Thorax 64, 631–636. [DOI] [PubMed] [Google Scholar]
- Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, Torbenson MS & Polotsky VY (2007). Chronic intermittent hypoxia predisposes to liver injury. Hepatology 45, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Schroeder BO & Backhed F (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 22, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Shin MK, Han W, Bevans‐Fonti S, Jun JC, Punjabi NM & Polotsky VY (2014). The effect of adrenal medullectomy on metabolic responses to chronic intermittent hypoxia. Respir Physiol Neurobiol 203, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulit L, Storfer‐Isser A, Kirchner HL & Redline S (2006). Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 29, 777–783. [DOI] [PubMed] [Google Scholar]
- Taylor CT, Kent BD, Crinion SJ, McNicholas WT & Ryan S (2014). Human adipocytes are highly sensitive to intermittent hypoxia induced NF‐kappaB activity and subsequent inflammatory gene expression. Biochem Biophys Res Commun 447, 660–665. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Wang B & Wood IS (2008). Hypoxia and the endocrine and signalling role of white adipose tissue. Arch Physiol Biochem 114, 267–276. [DOI] [PubMed] [Google Scholar]
- Wang N, Khan SA, Prabhakar NR & Nanduri J (2013). Impairment of pancreatic beta‐cell function by chronic intermittent hypoxia. Exp Physiol 98, 1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiszenstein M, Shimoda LA, Koc M, Seda O & Polak J (2016). Inhibition of lipolysis ameliorates diabetic phenotype in a mouse model of obstructive sleep apnea. Am J Respir Cell Mol Biol 55, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoe T, Alonso LC, Romano LC, Rosa TC, O'Doherty RM, Garcia‐Ocana A, Minoguchi K & O'Donnell CP (2008). Intermittent hypoxia reverses the diurnal glucose rhythm and causes pancreatic β‐cell replication in mice. J Physiol 586, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S & Badr S (1993). The occurrence of sleep‐disordered breathing among middle‐aged adults. N Engl J Med 328, 1230–1235. [DOI] [PubMed] [Google Scholar]