

LIMCH1 specifically associates with contractile stress fibers. The N-terminus of LIMCH1 directly interacts with the head of NM-IIA. LIMCH1 depletion modulates the myosin regulatory light chain and FAK phosphorylation. Actin stress fiber and focal adhesion are decreased in LIMCH1-depleted cells, leading to increased cell migration.
Abstract
Nonmuscle myosin II (NM-II) is an important motor protein involved in cell migration. Incorporation of NM-II into actin stress fiber provides a traction force to promote actin retrograde flow and focal adhesion assembly. However, the components involved in regulation of NM-II activity are not well understood. Here we identified a novel actin stress fiber–associated protein, LIM and calponin-homology domains 1 (LIMCH1), which regulates NM-II activity. The recruitment of LIMCH1 into contractile stress fibers revealed its localization complementary to actinin-1. LIMCH1 interacted with NM-IIA, but not NM-IIB, independent of the inhibition of myosin ATPase activity with blebbistatin. Moreover, the N-terminus of LIMCH1 binds to the head region of NM-IIA. Depletion of LIMCH1 attenuated myosin regulatory light chain (MRLC) diphosphorylation in HeLa cells, which was restored by reexpression of small interfering RNA–resistant LIMCH1. In addition, LIMCH1-depleted HeLa cells exhibited a decrease in the number of actin stress fibers and focal adhesions, leading to enhanced cell migration. Collectively, our data suggest that LIMCH1 plays a positive role in regulation of NM-II activity through effects on MRLC during cell migration.
INTRODUCTION
Cell migration plays an important role in a wide variety of biological phenomena, such as embryonic development, wound healing, immune response, and cancer metastasis. Various signaling pathways involving growth factors and extracellular matrix mediate directional cell migration to regulate cytoskeletal and adhesion machinery within the cell (Ridley et al., 2003). Cell movement comprises several steps executed in distinct cellular areas: 1) formation of lamellipodia and filopodia, mediated by polymerization of actin filament, at the leading edge to extend the cell membrane; 2) assembly of adhesions at the leading edge and its release at the rear of the cell; and 3) retraction of the rear of the cell by actomyosin-mediated traction force to translocate the cell body (Lauffenburger and Horwitz, 1996; Pollard and Borisy, 2003). Optimal migration requires spatiotemporal coordination between actin filaments and nonmuscle myosin II (NM-II) to control adhesion turnover (Gupton and Waterman-Storer, 2006).
Nonmuscle myosin II, an actin-based motor protein, is a hexamer composed of two myosin heavy chains (MHCs), two essential light chains (MELCs), and two regulatory light chains (MRLCs). Each MHC bears a motor domain carrying the actin-binding site and ATPase, two IQ domains that bind the MELC and MRLC, respectively, and an α-helical coiled-coil responsible for NM-II assembly into filaments. NM-II isoforms execute various cellular functions, depending on their location (Vicente-Manzanares et al., 2007; Ronen and Ravid, 2009). For instance, NM-IIA mediates traction force to promote retrograde F-actin flow in the lamella and inhibits cell spreading (Cai et al., 2006). Localized in the rear and center of the cell, NM-IIB regulates cell polarity and tail retraction (Kolega, 2003; Vicente-Manzanares et al., 2007, 2008). Cooperative interaction between NM-IIA and NM-IIB promotes actomyosin bundling, generating stable focal adhesions (Vicente-Manzanares et al., 2011). Regulation of NM-II activity depends on phosphorylation of MRLC, which induces a conformational change in the motor domain to facilitate interaction with actin, resulting in the activation of myosin ATPase. Therefore higher phosphorylation of MRLC can enhance cell contraction. (Vicente-Manzanares et al., 2009). Phosphorylation of MRLC at Ser-19 (pMRLC-S19) activates the ATPase activity of myosin and induces NM-II assembly. In comparison to pMRLC-S19, diphosphorylation of MRLC at both Ser-19 and Thr-18 (ppMRLC-S19/T18) results in higher ATPase activity and more NM-II assembly (Ikebe and Hartshorne, 1985; Ikebe et al., 1988; Tan et al., 1992) and markedly enhances the formation of actin stress fibers (Iwasaki et al., 2001; Joo et al., 2007). Whereas diphosphorylation of MRLC is regulated by Rho kinase (ROCK), zipper-interacting protein kinase (ZIPK), and citron kinase, myosin light chain kinase (MLCK) and myotonic dystrophy kinase–related CDC42-binding kinase can phosphorylate MRLC at Ser-19 alone (Murata-Hori et al., 2001; Tan et al., 2011).
Actin stress fibers are typically composed of actomyosin filaments; these filaments are thick and stable in low-motility cells but thin, dynamic, and less developed in high-motility cells (Pellegrin and Mellor, 2007). Actin stress fibers are classified as dorsal stress fibers, ventral stress fibers, transverse arcs, and perinuclear actin cap (Heath, 1983; Small et al., 1998; Khatau et al., 2009). Unlike other fiber types, NM-II was not embedded into dorsal stress fibers, which lack contractility (Tojkander et al., 2012). Transverse arcs are formed of contractile units—the product of the endwise assembly of actinin–cross-linked actin filaments and NM-II/tropomyosin-decorated actin filaments (Hotulainen and Lappalainen, 2006; Tojkander et al., 2011). Transverse arcs may associate with dorsal stress fibers to form ventral stress fibers (Hotulainen and Lappalainen, 2006). During cell migration, the transverse arc generates retrograde flow from the leading edge toward the cell center and inhibits membrane protrusion (Vicente-Manzanares et al., 2009; Tojkander et al., 2012). Ventral stress fibers connect with focal adhesions at both ends and facilitate cell movement by the retraction of the rear of the cell (Chen, 1981; Mitchison and Cramer, 1996).
LIM and calponin-homology domains 1 (LIMCH1) was identified from a border cell migration screen based on conditional ectopic expression in Drosophila. We used the UAS-GAL4 system to evaluate a number of human genes showing altered expression patterns in hepatocellular carcinoma to determine their effects on cell migration. The border cell cluster expressing a partial limch1/kiaa1102 coding sequence exhibited a migration delay (unpublished data). Here we investigated the role of LIMCH1 in cell migration and identified its interacting protein, NM-IIA. Cellular studies have shown that LIMCH1 promotes the assembly of actin stress fiber during cell spreading, thereby accelerating cell contraction. We hypothesized that the presence of LIMCH1 on contractile stress fibers enhances the spatiotemporal regulation of NM-II activity during cell migration.
RESULTS
Characterization of LIMCH1 expression pattern
On the basis of previous experiments, we constructed a full-length clone of the predicted human LIMCH1 protein (accession number NM_014988) encompassing a single calponin homology (CH) domain at the N-terminus, LIM domain at the C-terminus, and coiled-coil domains in the center region (Figure 1A). The CH domain is believed to participate in actin binding and the LIM domain in protein interaction (Dawid et al., 1998; Gimona and Mital, 1998). Western blot analysis using anti-FLAG and anti-LIMCH1 antibodies revealed exogenous and endogenous LIMCH1 signals at ∼150 kDa (Supplemental Figure S1A). The LIMCH1 expression pattern was studied in various cancer cell lines; LIMCH1 was overexpressed in Mahlavu, J7, and HeLa cells, and MDA-MB-231 (stably overexpressing epidermal growth factor receptor) and Huh7 cells showed low-level expression (Supplemental Figure S1, B and C).
FIGURE 1:

LIMCH1 associates with actin stress fibers and binds to actin. (A) Schematic diagram of LIMCH1 exhibiting a CH domain at the N-terminus and a LIM domain at the C-terminus. The center contains the coiled-coil domains. (B) Confocal fluorescence image of HeLa cells stained with anti-LIMCH1 antibody (green) and phalloidin (magenta); bar, 20 μm. Right, magnified images. (C) HeLa cells were stained with anti-LIMCH1 antibody (green) and phalloidin (magenta) after treatment with or without 5 μM CD for 30 min. Arrowheads in the inset indicate condensed actin filaments and LIMCH1; bar, 20 μm.
LIMCH1 is an actin stress fiber association protein
We used Immunofluorescence staining to observe the subcellular localization of LIMCH1 and found that LIMCH1 exhibited a cytoskeleton pattern characterized by dotted signals (Supplemental Figure S1D). These signals were eliminated after small interfering RNA (siRNA) treatment (Supplemental Figure S1E). LIMCH1 was colocalized with actin stress fibers, as confirmed by the magnified image revealing its periodic distribution along these filaments (Figure 1B). The association of LIMCH1 with actin stress fibers was confirmed by treatment of HeLa cells with an actin filament–depolymerizing reagent, cytochalasin D (CD). Depolymerization of the actin filament was accompanied by disruption of the dotted signal that overlapped with the condensed actin foci (Figure 1C). Live imaging revealed that CD treatment altered the distribution of green fluorescent protein (GFP)–LIMCH1 in MDCK cells and directionally assembled it into compact foci (Supplemental Video S1). This is in line with myosin-II condensation during depolymerization of the actin filament (Verkhovsky et al., 1997).
LIMCH1 is colocalized with NM-II on the contractile stress fibers
In nonmuscle cells, actinin—an actin cross-linking protein—displays a periodic staining pattern along actin stress fibers (Peterson et al., 2004) and localizes adjacent to NM-II (Hotulainen and Lappalainen, 2006). We determined the relative location of LIMCH1, actinin-1, and NM-II with respect to each other in HeLa cells using specific antibodies. Of interest, LIMCH1 alternated along the actin stress fibers with actinin-1 (Figure 2A) and colocalized with MRLC (Figure 2B). Consistent with the previously defined contractile units (Tojkander et al., 2011), actinin-1 failed to overlap with MRLC (Figure 2C). Further magnification revealed that LIMCH1 displayed twin and condensed signals between the adjacent actinin-1’s. These signals partially colocalized with MRLC in a contractile unit (Figure 2, D and E). Whereas 69% of the contractile units showed a twin signal of LIMCH1, 31% of them exhibited a condensed LIMCH1 signal (Figure 2F). A similar staining pattern was observed in GFP-LIMCH1–expressing U2OS cells (Figure 2G and Supplemental Figure S1F). Furthermore, the distance between adjacent signals of actinin-1 was determined and the average length of these contractile units measured; the length of contractile units coincided with that of the actin stress fibers in Swiss 3T3 cells (Peterson et al., 2004). As indicated in Figure 2H, actinin-1 was equidistant from MRLC and LIMCH1, indicating partial colocalization of LIMCH1 with MRLC. These results suggest that LIMCH1 associated with actin stress fibers in a pattern complementary to actinin-1 and colocalized with NM-II.
FIGURE 2:

LIMCH1 colocalizes with NM-II in a contractile unit. (A, B) Confocal images of HeLa cells stained with anti-LIMCH1 (green), anti–actinin-1 (magenta in A), and anti-pMRLCS19 (magenta in B) antibodies; bar, 20 μm. Right, magnified images; bar, 2 μm. (C–E) Confocal images of HeLa cells stained with anti-LIMCH1 (green), anti–actinin-1 (magenta in C and D), and anti-pMRLCS19 (green in C, magenta in E) antibodies; bar, 0.5 μm. LIMCH1 displayed twin signals (green, dotted circles) in D and E. Right, plot profiles. Dashed lines in C and D illustrate the center-to-center space among actinin-1, MRLC, and LIMCH1 for length measurement. Arrowheads indicate a condensed LIMCH1. (F) Ratio of twin signal and condensed dot of LIMCH1 in a contractile unit measured via analysis of plot profiles of staining images. (G) Fluorescence image of U2OS cells transfected with GFP-LIMCH1 (green) and stained with antixactinin-1 antibody (magenta); bar, 0.5 μm. (H) Estimated localization of LIMCH1 between actinin-1 and assembled NM-II. The distance between the adjacent actinin-1’s was 1.29 ± 0.16 μm (n = 69 in five cells), between actinin-1 and MRLC was 0.64 ± 0.12 μm (n = 45 in three cells), and between actinin-1 and LIMCH1 was 0.52 ± 0.15 μm (n = 65 in five cells).
We examined all types of actin stress fibers with actinin-1 staining to investigate specific association between LIMCH1 and contractile stress fibers. The results clearly demonstrated the association of LIMCH1 with contractile stress fibers but not dorsal stress fibers (Supplemental Figures S2A and S1F). Furthermore, phalloidin staining revealed that during cell division, LIMCH1 showed no overlap with the contractile ring, an actomyosin structure, indicating that LIMCH1 is not involved in cytokinesis (Supplemental Figure S2B). In addition, LIMCH1 was not detected at focal adhesions and peripheral actin filaments with vinculin and actinin-4 staining, respectively (Supplemental Figure S2, C and D). These staining results confirm that LIMCH1 was specifically localized in the contractile stress fibers in the nondividing cells.
N-terminal coiled-coil domain of LIMCH1 directly interacts with the head portion of NM-IIA
Nonmuscle myosin-IIA and NM-IIB display distinct subcellular localization, as well as functions in the regulation of actin organization (Kolega, 2003; Vicente-Manzanares et al., 2007). We evaluated the role of LIMCH1 in the regulation of NM-II isoforms. HeLa cells expressing GFP-LIMCH1 were stained with anti–NM-IIA and anti–NM-IIB antibodies. NM-IIA showed even distribution in the cell (Figure 3A). In contrast, NM-IIB was primarily assembled in the cell center (Figure 3B). Costaining results revealed that LIMCH1 primarily colocalized with NM-IIA in the cell and partially overlapped with NM-IIB (Figure 3, A and B). Given the ability of MRLC to bind both NM-II and NM-18A (Tan et al., 2008), we evaluated the colocalization of LIMCH1 and NM-18A. As shown in Supplemental Figure S2E, NM-18A was mainly distributed on the lamellar and lamellipodia, which were negative for LIMCH1, indicating no role of LIMCH1 in regulation of NM-18A. We further examined the interaction of LIMCH1 with NM-IIA and NM-IIB. Immunoprecipitation assays using anti-NM-IIA antibody revealed coprecipitation of endogenous LIMCH1 with NM-IIA (Figure 3C). Endogenous LIMCH1 was less precipitated using anti-LIMCH1 antibody. Therefore we further confirmed the interaction between LIMCH1 and NM-IIA by pulling down FLAG-LIMCH1 using an anti-FLAG antibody (Figure 3D). The bidirectional interaction analysis revealed no interaction between LIMCH1 and NM-IIB (Figure 3, E and F). Taken together, the results show that LIMCH1 specifically interacts with NM-IIA.
FIGURE 3:

LIMCH1 interacts with NM-IIA. (A, B) Confocal images of GFP-LIMCH1–transfected HeLa cells (green) stained with anti–NM-IIA (magenta in A) and anti–NM-IIB (magenta in B) antibodies; bar, 20 μm. Right, plot profiles of NM-IIA, NM-IIB, and LIMCH1 in the cell, reflecting dashed lines in the staining images. Arrows indicate the direction of cell migration. (C, E) HeLa cell lysates were subjected to immunoprecipitation with anti–NM-IIA and anti–NM-IIB antibodies, respectively, followed by immunoblotting with anti-LIMCH1 antibody. (D, F) Cell extracts from HeLa cells expressing FLAG-LIMCH1 were subjected to immunoprecipitation with anti-FLAG antibody, followed by immunoblotting with anti–NM-IIA and anti–NM-IIB antibodies, respectively. A total of 5% of the input and 50% of the immunoprecipitate were loaded. Cell lysate incubated with protein G beads alone was used as lysate control (Ctrl). The NM-IIA or NM-IIB antibodies incubated with protein G beads alone were used as antibody control (antibody). LIMCH1, full-length FLAG-tagged LIMCH1. Mock, FLAG vector alone.
We evaluated the domain function of LIMCH1, several domain-deleted mutants, and their interactive ability with NM-IIA and subcellular localization (Figure 4A and Supplemental Figure S3A). Immunostaining results showed that the association between delLIM as well as with delCH and actin stress fibers was independent of the CH or LIM domain (Supplemental Figure S3B), and deletion of the CH or LIM domain retained the dotted signals between adjacent actinin-1’s (Supplemental Figure S3, C and D). We also examined the association between the coiled-coil domains and actin stress fibers. The 2xCoil and CHCoil associated with actin stress fibers; however, the CHCoil also displayed a strong signal in the nucleus (Supplemental Figure S3E). Of interest, NCoil was sufficient for the localization on actin stress fibers, but CoilLIM and CCoil were distributed in the cytosol (Supplemental Figure S3F). In addition, we examined the interaction between these mutants localized on the actin stress fibers and NM-IIA with immunoprecipitation. The NCoil, but not CCoil, was sufficient to interact with NM-IIA (Figure 4B). These results indicate the role of N-terminal coiled-coil domain of LIMCH1 in localization on actin stress fibers and interaction with NM-IIA.
FIGURE 4:

The N-terminus of LIMCH1 directly interacts with the head of NM-IIA. (A) Truncation mutants of LIMCH1 fused with GFP or FLAG tags on the N-terminus were tested for their subcellular localization and interaction with NM-IIA. Right, results of the association between each truncation mutant and actin stress fibers and its interaction with NM-IIA; NT, not tested. (B) Cell extracts from FLAG-truncation mutants expressed in HeLa cells were subjected to immunoprecipitation with anti-FLAG antibody and immunoblotted with anti-FLAG and anti– NM-IIA antibodies. Cell lysate incubated with protein G beads alone was used as control (Ctrl). A total of 5% of the input and 50% of the immunoprecipitate were loaded. (C) Schematic diagram of NM-IIA showing the N-terminal motor region that binds actin and the coiled-coil domain at the center with the C-terminal tail that controls myosin heavy chain assembly. Truncations of NM-IIA fused with GST on the N-terminus were tested for their ability to bind LIMCH1. Right, relative abilities of each truncation to bind with purified His-LIMCH1 or His-CoilLIM; NT, not tested. (D) Soluble His-LIMCH1 was incubated with NM-IIA truncations immobilized on a glutathione bead, and the products of in vitro pull downs were immunoblotted with anti-LIMCH1 antibody (top). Coomassie blue staining showed the input of GST-NM-IIA truncations and 10×His-LIMCH1 (bottom). (E) Soluble His-LIMCH1 or His-CoilLIM was incubated with GST-IIA-head immobilized on a glutathione bead, and the products of in vitro pull downs were immunoblotted with anti-LIMCH1 antibody (top). Coomassie blue staining showed the input of GST-IIA-head, 10× His-LIMCH1, and 20× His-CoilLIM (lower). A total of 2% of the input and 20% of pull downs were loaded for Western blot analysis. His, hexahistidine tag was fused on the N-terminus of LIMCH1 or CoilLIM.
We identified the region of NM-IIA responsible for its interaction with LIMCH1 and whether the interaction is direct. We performed an in vitro binding assay using the synthesized head and coiled-coil portions of NM-IIA (IIA-head and IIA-coil; Figure 4C). We excluded the tail section of NM-IIA because it controls the dimerization/polymerization of NM-IIA (Ikebe et al., 2001), which may render the recombinant protein insoluble and result in nonspecific binding. LIMCH1 mostly bound to IIA-head and not IIA-coil (Figure 4D). Furthermore, CoilLIM was unable to bind to IIA-head (Figure 4E). Thus LIMCH1 binds to NM-IIA head portion through its N-terminal coiled-coil domain.
LIMCH1 can interact with disassembled NM-IIA
The spatial translocation of NM-II from the cell center to the leading edge necessitates its assembly and disassembly. However, inhibition of NM-II activity results in its diffused distribution, attributed to its disassembly (Sandquist and Means, 2008; Breckenridge et al., 2009). Therefore it is important to understand the influence of NM-II activity on the association and interaction of LIMCH1 with actin stress fibers and NM-IIA, respectively. HeLa cells were treated with the ROCK inhibitor Y27632, which is known to reduce MRLC phosphorylation in the cell center, thereby affecting NM-II activity (Totsukawa et al., 2000; Vicente-Manzanares et al., 2009). The staining data revealed a periodic pattern of LIMCH1 in the cell center (Figure 5A), which it was lost when ROCK inhibitor disrupted the central stress fibers (Figure 5B). In contrast, the expression of diphosphomimetic MRLC (MRLC-DD), which mimics ppMRLC-S19/T18, prevented Y27632-mediated stress fiber loss and maintained LIMCH1 localization (Figure 5B, magnified image). Thus the localization of LIMCH1 on the stress fiber was controlled by MRLC-DD, suggesting the regulatory role of ROCK signaling in LIMCH1 localization through MRLC phosphorylation. Furthermore, we inhibited myosin ATPase with blebbistatin treatment to achieve NM-II disassembly and studied the interaction of LIMCH1 with the disassembled NM-II. The immunoprecipitation assay confirmed the hypothesis that LIMCH1 interacted with NM-IIA after blebbistatin (Figure 5C) and Y27632 treatment (unpublished data), suggesting that the association of LIMCH1 with NM-II was independent of actin stress fiber formation. Together these results indicate that LIMCH1 interacts with the assembled and disassembled NM-IIA.
FIGURE 5:
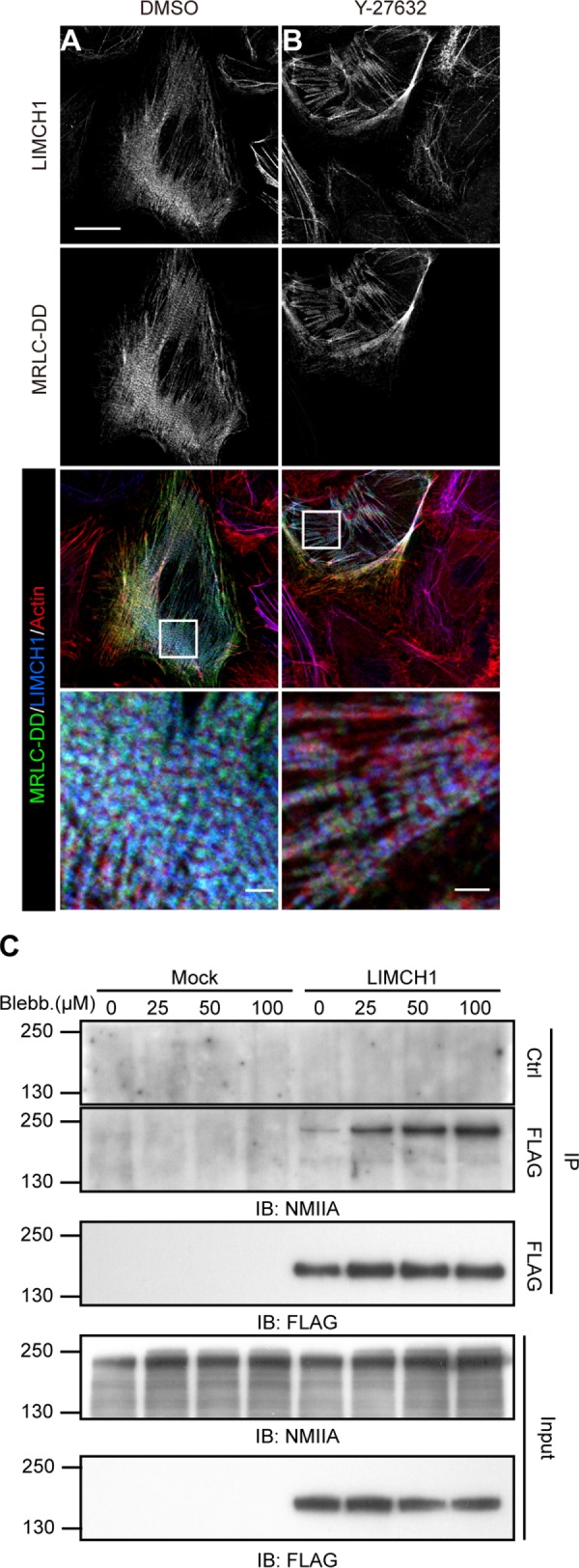
Interaction between LIMCH1 and NM-IIA is independent of myosin ATPase activity. (A, B) Confocal images of HeLa cells transfected with GFP-MRLC-DD (green) and stained with anti-LIMCH1 (blue) antibody and phalloidin (red); bar, 20 μm. These cells were treated with DMSO in A and 5 μM Y-27632 in B for 30 min. Enlarged images in insets show the colocalization of LIMCH1 and GFP-MRLC-DD; bar, 2 μm. (C) Cell extracts from FLAG-LIMCH1 expressed in HeLa cells were subjected to immunoprecipitation by anti-FLAG antibody and immunoblotted with anti-FLAG and anti–NM-IIA antibodies. These cells were treated with various concentrations of blebbistatin for 30 min. LIMCH1, FLAG-tagged LIMCH1. Mock, FLAG vector alone.
LIMCH1 depletion attenuates the formation of actin stress fibers and regulates MRLC phosphorylation
We further investigated whether LIMCH1 was involved in the formation of actin stress fibers. LIMCH1 depletion showed no significant disassembly of established actin stress fibers in quiescent cells (unpublished data). Therefore we examined the initial actin stress fibers during the cell spreading state. LIMCH1-depleted HeLa cells showed reduction in the number of actin stress fibers during cell spreading (Figure 6, A and B), particularly in the cell center (Figure 6A, asterisk). We further defined the cell outline and central area by staining with phalloidin and NM-IIB, respectively (Supplemental Figure S4). In comparison with the control and rescued cells, LIMCH1-depleted HeLa cells exhibited a 20% reduction in central stress fibers (Figure 6, C and D). Studies reported that increase in the NM-II activity promotes the formation of actin stress fibers (Joo et al., 2007; Vallenius et al., 2011). Therefore we analyzed the levels of pMRLC-S19 and ppMRLC-S19/T18 to examine the activity of NM-II. LIMCH1-depleted HeLa cells showed a decrease in ppMRLC-S19/T18 level, but pMRLC-S19 level was unaltered (Figure 6, E and F). In addition, reexpression of siRNA-resistant LIMCH1 in LIMCH1-depleted HeLa cells restored the diphosphorylation of MRLC (Figure 6, G and H). These results suggest that LIMCH1 promoted the initial assembly of actin stress fibers during cell spreading and regulated MRLC phosphorylation.
FIGURE 6:

Central stress fibers and MRLC phosphorylation are reduced in LIMCH1-depleted cells. (A) Confocal images of siRNA-treated HeLa cells stained with phalloidin and anti-vinculin antibody after cell spreading on a fibronectin-coated coverslip for 60 min; bar, 20 μm. Asterisks indicate the reduced central stress fibers and focal adhesions. (B) Quantification of intensity of total stress fibers. (C) Binary images of actin stress fibers stained with phalloidin in siRNA-treated HeLa cells were processed using ImageJ software. Cell outlines (red) and central areas (green) were defined by staining with phalloidin and NM-IIB, respectively. (D) Quantification of intensity of central stress fibers. In B and D, results were analyzed using ImageJ software (n = 80–100 cells in three independent experiments, mean ± SD, *p < 0.05, one-way ANOVA, Tukey’s multiple comparison test). (E) Cell extracts from siRNA-treated HeLa cells were probed with anti-pMRLCS19, ppMRLCS19/T18, and MRLC antibodies. (F) Relative levels of ppMRLCS19/T18 shown in E (n = 6, mean ± SD, normalized to control siRNA, *p < 0.05, two-tailed t test). (G) Cell extracts from LIMCH1-depleted and siRNA-resistant HeLa cells were probed with pMRLCS19/T18 and MRLC antibodies. (H) Relative levels of pMRLCS19/T18 shown in G (n = 3, mean ± SD, normalized to siRNA rescue, *p < 0.05, two-tailed t test).
LIMCH1 regulates the number of focal adhesions in HeLa cells
Nonmuscle myosin-II controls cell migration not only through regulation of actin retrograde flow but also through stability of focal adhesions (Cai et al., 2006; Choi et al., 2008; Aguilar-Cuenca et al., 2014). Given that MRLC diphosphorylation was regulated by LIMCH1, we hypothesized that LIMCH1 might also modulate the stability of focal adhesion. The intensity of focal adhesions in LIMCH1-depleted HeLa cells was decreased in the quiescence state (Supplemental Figure S5, A and B), and the number of central focal adhesions was reduced in the spreading state compared with the control (Figure 7A). However, the difference in the total number of focal adhesions between control and LIMCH1-depleted HeLa cells was not statistically significant (Figure 7B). LIMCH1 depletion led to an alteration of focal adhesion distribution, which located more in the peripheral areas than in the central areas of the cell (Figure 7C). The role of LIMCH1 in the maturation of focal adhesions was confirmed by the evaluation of the phosphorylation of FAK on Tyr-397, a marker for detection of focal adhesion turnover (Zaidel-Bar et al., 2007). Western blot analysis revealed that LIMCH1 depletion increased FAK phosphorylation (Figure 7, D and E), indicating a high-turnover state of focal adhesions. These data suggest that LIMCH1 could be involved in the maturation of focal adhesions in the cell center and in the formation of new peripheral focal adhesions.
FIGURE 7:

LIMCH1 depletion affects the formation of focal adhesions in the cell center and phosphorylation of FAK. (A) Binary images of focal adhesions stained with anti-paxillin antibody in siRNA-treated HeLa cells were processed using ImageJ software. Cell outlines (red) and central areas (green) were defined by staining with phalloidin and NM-IIB, respectively. (B) Quantification of the number of total focal adhesions. (C) The ratio of focal adhesions was measured by dividing the number of central or peripheral adhesions by the number of total focal adhesion. In B and C, results were analyzed using ImageJ software (n = 118–142 cells in three independent experiments, mean ± SD, *p < 0.001, one-way ANOVA, Tukey’s multiple comparison test). (D) Cell extracts from siRNA-treated cells were probed with anti-pFAKY397 and FAK antibodies. (E) Relative levels of pFAKY397 shown in D (n = 4, mean ± SD, normalized to siRNA control, **p < 0.001, two-tailed t test).
LIMCH1 depletion in HeLa cells increases cell motility
We showed that depletion of LIMCH1 attenuates actin stress fibers. Actin stress fibers are typically not prevalent and quite dynamic in high-motility cells (Pellegrin and Mellor, 2007). In relation to the regulatory effect of LIMCH1 on NM-II activity, we examined cell functions such as cell contractility and cell migration ability. The three-dimensional collagen-matrix contraction assay showed that LIMCH1 depletion reduced HeLa cell contraction in the collagen matrix (Figure 8, A–C). The serum-stimulated Transwell assay revealed that LIMCH1-depleted HeLa cells displayed a remarkable increase in the cell number compared with control cells. However, siRNA-resistant LIMCH1 nearly restored the effect on cell migration (Figure 8, D and E). In addition, we measured the cell migratory speed by following these cell tracks over time. In line with the Transwell assay results, LIMCH1-depleted cells showed a higher velocity of 15.9 ± 6.4 μm/h, with the control and rescued cells at 9.2 ± 3.6 and 10.8 ± 5.3 μm/h, respectively (Figure 8F and Supplemental Videos 2–4). Thus LIMCH1 expression enhanced cell contractility and decreased cell migration.
FIGURE 8:

LIMCH1 depletion decreases cell contraction and increases cell migration. (A) Cell extracts from siRNA-treated HeLa cells were immunoblotted with anti-LIMCH1 antibody. (B) Contraction images of HeLa cells grown in collagen matrix were captured by microscopy after 24 h of incubation. (C) Gel areas were measured at 6, 24, and 48 h (n = 3, mean ± SD, *p < 0.05, **p < 0.001, two-tailed t test). (D) Fluorescence images of the migrating HeLa cells (on the filters) stained with DAPI. (E) Migratory cells in D were quantified with Image-Pro software (n = 3, mean ± SD, ***p < 0.0001, two-tailed t test). (F) Distribution of migration speeds of HeLa cells on the fibronectin-coated coverslip. Migration speed was measured by tracking the path during an 8-h period. The average velocity is presented as scatter plots; the middle line shows the median value (n = 101–127 cells in three independent experiments, *p < 0.05, one-way ANOVA, Tukey’s multiple comparison test). (G) Binary images of HeLa cells were processed by ImageJ software. Cells were incubated on fibronectin-coated coverslips with CellTracker for 30 min; bar, 40 μm. (H) Cell areas in G were measured at 20 and 30 min (n = 126–140 cells in three independent experiments, mean ± SD, *p < 0.05, one-way ANOVA, Tukey’s multiple comparison test). (I) Cell attachment on fibronectin-coated coverslips was assessed at indicated time points with crystal violet staining.
To determine specific cell migration processes affected by LIMCH1, we examined cell adhesion ability and cell spreading activity by plating HeLa cells onto fibronectin-coated plates. In comparison to the control, LIMCH1 depletion enhanced HeLa cell spreading after 20- and 30-min incubation. In contrast, the spreading areas of cells expressing the siRNA-resistant construct were similar to those of control cells (Figure 8, G and H). However, no significant difference was recorded between the cell adhesion ability of the control and LIMCH1-depleted HeLa cells (Figure 8I). We conclude that LIMCH1 plays a negative role in cell migration, as depletion of LIMCH1 promoted cell migration and cell spreading.
DISCUSSION
Actin stress fibers regulate cell morphology, polarity, and migration in numerous cell culture systems. Recent studies have improved our knowledge about the molecular mechanisms of NM-II assembly into actin stress fibers. However, the regulation of NM-II activity is not well understood. We showed that a new protein, LIMCH1, is recruited into contractile stress fibers through its interaction with NM-IIA. Loss of LIMCH1 expression reduces MRLC phosphorylation and assembly. Furthermore, LIMCH1 depletion affects the formation of actin stress fibers as well as the stability of focal adhesions, leading to increase in cell spreading and migration. Our data highlight the positive regulatory role of LIMCH1 in NM-II activity and actin stress fiber formation.
Contractile stress fibers are formed by NM-II/tropomyosin-containing actin filaments and exhibit endwise assembly with actinin–cross-linked actin filaments (Hotulainen and Lappalainen, 2006; Tojkander et al., 2011). LIMCH1 displayed a complementary pattern to actinin-1 and colocalized with MRLC, suggesting its association with the NM-II/tropomyosin-containing actin filament. The absence of LIMCH1 at dorsal stress fibers indicates specific association between LIMCH1 and contractile stress fibers. However, not all types of actomyosin-constructed contractile fibers show association with LIMCH1 (e.g., the contractile ring). Inhibition of myosin ATPase activity disrupted the contractile units but failed to alter the interaction between LIMCH1 and NM-IIA. Thus the assembly of N-IIA into contractile stress fibers is accompanied by LIMC1. Furthermore, the N-terminal coiled-coil domain is involved in the localization of LIMCH1 on actin stress fibers, as well as its interaction with NM-IIA. The CH, LIM, and N-terminal coiled-coil domains of LIMCH1 are highly conserved and display homologies of 64, 60, and 59%, respectively, with LIM only 7 (LMO7; Friedberg, 2009). LIMCH1 and LMO7 are proposed to exhibit similar roles in actin cytoskeleton organization and signal transduction. Previous studies reported the association of LMO7 with actinin at the cell–cell junction for regulation of cell signaling (Ooshio et al., 2004; Yamada et al., 2004). In addition, LMO7 regulates gene expression during muscle regeneration, which is inhibited by interactions with adhesion and nuclear proteins (Dedeic et al., 2011; Wozniak et al., 2013). LOM7 is also involved in Emery–Dreifuss muscular dystrophy and lung cancer progression (Nakamura et al., 2011; Mull et al., 2014). We showed that LIMCH1 is involved in NM-II regulation, whereas LMO7 was previously implicated in α-actinin regulation.
Nonmuscle myosin-II controls contractility of actin stress fibers through myosin ATPase activity, which is regulated by MRLC phosphorylation (Vicente-Manzanares et al., 2009). Our results revealed the association between LIMCH1 and actin stress fibers in the internal region of the cell, where diphosphorylated MRLC is predominantly localized. However, LIMCH1 was absent at the distal lamella, where monophosphorylated MRLC is localized (Komatsu and Ikebe, 2004). Moreover, LIMCH1 regulates the diphosphorylation of MRLC, as indicated by the decrease in the level of ppMRLC-S19/T18, but not pMRLC-S19, upon LIMCHC1 depletion. In comparison to its monophosphorylation, diphosphorylation of MRLC results in stronger ATPase activity of myosin (Ikebe and Hartshorne, 1985; Ikebe et al., 1988). Thus LIMCH1 may enhance cell contraction through regulation of MRLC diphosphorylation and myosin assembly.
Several kinases, such as ROCK, citron kinase, and ZIPK, increase MRLC diphosphorylation (Amano et al., 1996; Murata-Hori et al., 2001; Yamashiro et al., 2003). However, their interactions with LIMCH1 have not been detected using immunoprecipitation assay followed by mass spectrometry or yeast two-hybrid assay (unpublished data). Citron kinase, responsible for the diphosphorylation of MRLC during cytokinesis, is probably not involved in the regulation of LIMCH1-mediated diphosphorylation (Madaule et al., 2000) because of the absence of LIMCH1 expression in the contractile ring. ROCK directly phosphorylates MRLC or indirectly regulates MRLC phosphorylation through myosin phosphatase inhibition in the cell center (Totsukawa et al., 2000; Riento and Ridley, 2003). Increase in myosin phosphatase activity reduces MRLC phosphorylation not only on Ser-19/Thr-18 but also on Ser-19 (Koga and Ikebe, 2005; Vallenius et al., 2011). However, LIMCH1 showed no effect on MRLC monophosphorylation, suggesting that the regulatory role of LIMCH1 in actin stress fiber formation does not involve myosin phosphatase. Therefore future studies should focus on the candidates involved in LIMCH1-mediated regulation of MRLC phosphorylation.
The initiation of cell movement at the leading edge of the cell forms broad lamellipodia, driven by the polymerization of actin filaments and stabilized by adhesion proteins connected to the extracellular matrix (Small et al., 2002). In contrast, NM-II–generated actin retrograde flow in the lamella counters membrane extension through retraction of the leading edge (Giannone et al., 2007). The present study found that LIMCH1 primarily interacts with NM-IIA and not NM-IIB. Moreover, NM-IIA is a major isoform that generates the contractile force that drives the retrograde flow in the lamella and regulates membrane retraction (Cai et al., 2006; Vicente-Manzanares et al., 2007; Betapudi, 2010). We showed that LIMCH1-depleted cells extended over a greater area during the cell-spreading period, indicating that NM-IIA–mediated retrograde flow is interfered with by LIMCH1 depletion. In addition, NM-IIA controls the initiation of focal adhesion formation, and NM-IIB enhances NM-IIA–mediated actomyosin bundling, which stabilizes focal adhesions (Betapudi, 2010; Vicente-Manzanares et al., 2011). LIMCH1 depletion resulted in reduction in focal adhesions in the cell center; thus LIMCH1 enhances the activity of NM-IIA to promote maturation of focal adhesions. Given its role as an adaptor protein, the possibility that LIMCH1 regulates the NM-II activity through recruitment of other regulatory proteins cannot be excluded.
A spatiotemporal balance between actomyosin activity and focal adhesion assembly promotes optimal cell migration. An imbalance in the activity of NM-II can alter focal adhesion dynamics, thereby decreasing cell migration (Gupton and Waterman-Storer, 2006). In contrast, relaxation of NM-II contractility can increase cell migration (Dulyaninova et al., 2007; Tripathi et al., 2015). LIMCH1 depletion provides an optimal threshold of traction for increasing cell migration through regulation of focal adhesion turnover, as determined by the phosphorylation of FAK on Tyr-397. These findings suggest that LIMCH1 increases actomyosin activity, resulting in the assembly of actin stress fibers and stabilization of focal adhesions, thereby impeding cell migration. Taken together, our results demonstrate that LIMCH1 plays a positive role in the spatiotemporal regulation of NM-II activity to modulate cell membrane protrusion and focal adhesion dynamics during cell migration. Future studies should examine whether LIMCH1 plays a role in cancer progression and normal tissue development.
MATERIALS AND METHODS
Cell culture, transfection, and reagents
HeLa and U2OS cells were cultured using DMEM supplemented with 10% fetal bovine serum (FBS; Biological Industries, Kibbutz Beit-Haemek, Israel) in a humidified incubator with 5% CO2. HeLa cells were transfected with siRNA or plasmid DNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions. U2OS cells were transfected with plasmid DNA using the calcium phosphate transfection method. For transient transfection, cells were incubated with plasmid DNA for 48 h. Stable transfection was achieved by treating transfected cells with 0.5 mg/ml Geneticin (Life Technologies, Grand Island, NY) for 1 wk, followed by flow cytometry analysis for sorting stably transfected cells. For knockdown experiments, cells were plated at low confluence (40%) and transfected with siRNA for 48 h. Pharmacological inhibitors used were 50 μM blebbistatin, 10 μM Y-27632 ROCK inhibitor, and 2 μM CD (Sigma-Aldrich, St. Louis, MO).
Plasmids
Full-length LIMCH1 gene encompassing amino acid residues 1–1083 was PCR amplified from kiaa1102/limch1 cDNA (Kazusa DNA Research Institute, Chiba, Japan) and subcloned into pEGFP (Clontech, Mountain View, CA). Deletion mutants of LIMCH1 derived by PCR amplification or by enzyme digestion from the GFP-LIMCH1 construct were subcloned into either p3×FLAG-Myc-CMV-26 (Sigma-Aldrich) or pET-32 (Novagen). GFP-NM-IIA (Addgene plasmid #11347) obtained through Addgene (Cambridge, MA) was a kind gift from Robert Adelstein (National Heart, Lung, and Blood Institute, National Institutes of Health). Deletion mutants of NM-IIA were derived by PCR amplification of the domain from the GFP-NM-IIA construct and its subcloning into pGEX-4T (GE Healthcare, Little Chalfont, United Kingdom). Point mutations for siRNA-resistant GFP-LIMCH1 and diphosphomimetic GFP-MRLC-S19D/T18D were introduced using site-directed mutagenesis. Cloning for the construction of truncated LIMCH1 and NM-IIA is described in detail in the Supplemental Materials.
Antibodies and siRNAs
The anti-LIMCH1 polyclonal antibody was obtained from rabbits immunized with a truncated LIMCH1 protein corresponding to the amino acid residues 441–940 from the human LIMCH1 sequence. The following antibodies were purchased: rabbit anti-MRLC (3672), mouse anti-pMRLCS19 (3675), and rabbit anti-pMRLCS19/T18 (3674) from Cell Signaling (Danvers, MA); mouse anti–actinin-1 (A7811), mouse anti-vinculin (V9264), mouse anti-FLAG (F1804), rabbit anti–NM-IIA (M8064), and rabbit anti-NMIIB (M7939) from Sigma-Aldrich; goat anti–actinin-4 (sc-49333), rabbit anti-paxillin (sc-5574), and rabbit anti-FAK (sc-558) from Santa Cruz Biotechnology (Santa Cruz, CA); and rabbit anti-pFAKY397 (700255) from Invitrogen (Carlsbad, CA); rabbit anti-NM18A was a kind gift from Jau-Song Yu (Chang Gung University, Taipei, Taiwan). These antibodies were used for immunofluorescence staining, Western blot analysis, and immunoprecipitation assay. LIMCH1 siRNAs (LIMCH1.1 and LIMCH1.2) targeted the coding regions with the following sequences: LIMCH1.1 as 5′-GAGTGGGACGGATGTTAGGATTCGA-3′; and LIMCH1.2 as 5′-CAGCTGAGGGAAGAGGACGACAAAT-3′.
Cell adhesion, spreading, and migration assay
The cell adhesion assay was performed as follows: cells were harvested with trypsin, treated with trypsin inhibitor (for trypsin neutralization; Life Technologies), and replated on a 20 μg/ml fibronectin-coated 96-well plate in 10% FBS-DMEM. Nonadherent cells were discarded by washing after incubation for 5 and 10 min. The adherent cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) and stained with crystal violet. The stained cells were dissolved in 33% acetic acid and analyzed by measuring absorbance at 550 nm.
Cell spreading assay was performed by incubating the harvested cells on 20 μg/ml fibronectin-coated coverslips in 10% FBS-DMEM in the presence of 10 μM CellTracker (Invitrogen). Cells were fixed with 4% PFA in PBS after incubation for 20 and 30 min. Fluorescence images were captured from four randomly selected fields using an Axio Imager Z1 fluorescence microscope (Zeiss, Jena, Germany) equipped with a Plan-Neofluar 20×/0.50 objective (Zeiss) and an AxioCamMR3 digital camera and processed into binary images. The cell area was measured using ImageJ software.
The Transwell migration assay was carried out using a chemotaxis chamber combined with a Transwell filter with 8-μm pores (Neuro Probe, Gaithersburg, MD). The harvested cells (1 × 104 cells) were plated into the upper well in serum-free DMEM, and the bottom chambers contained 10% FBS-DMEM. After a 6-h incubation, cells on the filter were fixed with 100% ice-cold ethanol. The upper-side cells were scraped with a cotton swab, and the under-side cells were stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) in PBS. Fluorescence images were captured from five randomly selected fields per well using an Axio Imager Z1 microscope and a Plan-Neofluar 10×/0.30 objective. The number of cells on the filter was determined using Image-Pro Plus image analysis software.
The individual cell migration assay was performed on a 42-mm-diameter coverslip (placed in a 6-cm2 plate) treated with 2 μg/ml fibronectin in 5 ml of PBS at 37°C. After overnight incubation, the coverslip was blocked with 0.2% (wt/vol) heat-inactivated bovine serum albumin (BSA) in PBS (heated at 80°C for 10 min) for 2 h at 37°C, followed by rinsing twice with PBS. The harvested cells were seeded on the fibronectin-coated glass coverslips for 60 min. After incubation, the coverslip was placed in a POC-R chamber (Zeiss), and time-lapse images were captured using an Axiovert 200 M inverted microscope equipped with an environmental chamber (5% CO2, 37°C). Phase-contrast images were acquired every 10 min for 8 h using an Achroplan 10×/0.45 objective. Migration paths were tracked by monitoring the position of the cell nuclei manually using ImageJ software. Cell velocities were derived for each cell by dividing the path length with the elapsed time. Directional persistence was calculated as the ratio of the direct distance and the total length of the migration path.
Immunofluorescence microscopy
LIMCH1 staining was carried out by fixing cells adhered to fibronectin-coated coverslip with 4% PFA in cytoskeleton buffer (CB; 10 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 1 mM ethylene glycol tetraacetic acid [EGTA]) containing 0.1% Triton X-100 for 15 min. For GFP-fused protein staining, cells were fixed with 4% PFA in CB for 10 min, followed by permeabilization using 0.1% Triton X-100 in CB. The fixed cells were incubated with the blocking solution (2% BSA in PBS) for 1 h. These coverslips were treated with various primary antibodies diluted in the blocking solution. After a 2-h incubation, the coverslips were treated with fluorescence-conjugated secondary antibodies and phalloidin (Invitrogen) diluted in the blocking solution for 1 h. After incubation, the cells were mounted in Mowiol mounting medium (Polysciences, Warrington, PA).
Image analysis of actin stress fibers and focal adhesions
Immunofluorescence images of actin stress fibers and focal adhesions were processed by ImageJ software for quantification of intensity as well as number, as previously described (Pasapera et al., 2010). Harvested cells were seeded on 20 μg/ml fibronectin-coated coverslips in 10% FBS-DMEM and incubated for 60 min. After incubation, the cells were fixed with 4% PFA in PBS and stained with anti-paxillin, anti–NM-IIB, and anti-vinculin antibodies, as well as phalloidin. Fluorescence images of paxillin, NM-IIB, and phalloidin were processed using a median filter with 13-pixel-square kernel as the background image. The background images were used to create binary images of NM-IIB and phalloidin (dividing the original image with the background image) and to define the cell outline and central region. The intensity of actin stress fibers was evaluated by subtracting the phalloidin-background image from the original phalloidin image. Focal adhesions were counted by processing the paxillin-stained image using a median filter with 1-pixel-square kernel. The background image was subtracted from the paxillin-processed image to create a binary image of focal adhesions. Automatic particle analysis was applied to count these adhesions. Central focal adhesions were quantified with normalization of the value of central area to the value of total area. Processed images are shown in Supplemental Figure S4. The intensity of focal adhesions was quantified by the selection of a 0.3- to 19-μm2 area of focal adhesions.
Western blot, immunoprecipitation, and glutathione S-transferase pull-down assay
Cells were lysed using immunoprecipitation (IP) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1.5 mM MgCl2, 1% Triton X-100, 50 mM NaF, 20 mM Na4P2O7, and 1 mM NaVO4) supplemented with a 1× protease inhibitor cocktail (Roche, Basel, Switzerland). Analysis of protein phosphorylation was performed by treatment of cells with cold 10% trichloroacetic acid on ice for 10 min, followed by three washes with PBS. The cells were lysed with IP buffer containing 0.1% SDS and sonicated. The cell lysate was centrifuged and the supernatant subjected to SDS–PAGE. The gel was transferred onto a polyvinylidene difluoride membrane and the membrane blocked with 7% nonfat dry milk in a TBS-T buffer (20 mM Tris, 137 mM NaCl, and 0.1% Tween-20), followed by incubation with desired primary antibody. Horseradish peroxidase–conjugated secondary antibodies were detected with an enhanced chemiluminescence reagent (Millipore, Billerica, MA).
Immunoprecipitation was carried out as follows: the cells were lysed with IP buffer and clarified by centrifugation. The cell lysate was precleared with protein G beads (Millipore) for 30 min. The supernatant containing 500 μg of total protein (total volume 1 ml) was incubated with 1 μg anti-FLAG, anti–NM-IIA, or anti–NM-IIB antibodies at 4°C for 2 h. After incubation, the reaction mixture was mixed with 20 μl of protein G beads at 4°C for 1 h. The beads were washed six times with IP buffer and resuspended in 30 μl of 2× sample buffer. Samples were subjected to Western blot analysis.
For glutathione S-transferase (GST) pull-down assay, recombinant protein expression and purification were carried out as per the manufacturer’s handbook (GE Healthcare). Histidine (His)-tagged domain truncations of LIMCH1 were purified from BL-21 (DE3) cells using Ni-conjugated agarose beads (GE Healthcare). The GST-tagged NM-IIA fragment was expressed in BL-21 (DE3) cells and purified using glutathione (GSH)-conjugated agarose beads (GE Healthcare). The in vitro binding between LIMCH1 and NM-IIA was evaluated by mixing GST protein or GST-tagged NM-IIA fragments immobilized on 5 μl of GSH-conjugated agarose with binding buffer (30 mM Tris, pH 7.4, 50 mM KCl, 2 mM EDTA, 2 mM EGTA, 1 mM MgCl2, 1 mM CaCl2, 1 mM dithiothreitol, 0.5% Triton X-100, 0.2% Nonidet P-40, 0.05% Tween-20, 0.2% BSA, 0.2% gelatin, 1× protease inhibitor) containing 100 ng of His-tagged LIMCH1 or His-tagged CoilLIM (total volume 0.2 ml). After incubation at 4°C for 2 h, beads were washed five times using binding buffer without protease inhibitor. Pull-down products were dissolved in 2× sample buffer and subjected to Western blot analysis.
Contraction assay
Cell contraction was assessed using a cell contraction assay kit (Cell Biolabs, San Diego, CA). A total of 1.5 × 105 siRNA-transfected cells resuspended in 80 μl of 10% FBS-DMEM was mixed with 320 μl of collagen solution. The cell–collagen mixture was carefully loaded into the wells of a 24-well culture plate and incubated at 37°C for 1 h for matrix polymerization. The polymerized matrix was detached from the edge of the wells using a needle and with 1 ml of 10% FBS-DMEM. The diameter of the collagen matrix was imaged and measured at 0, 6, 24, and 48 h.
Statistical analysis
The results were compared using two-tailed t test and one-way analysis of variance (ANOVA), Tukey’s multiple comparison. p < 0.05 was considered statistically significant. All experiments were conducted independently at least in triplicate, unless indicated otherwise.
Supplementary Material
Acknowledgments
We thank J. T. Horng, J. S. Yu, A. J. Cheng, and K. H. Lin for cell lines and reagents, M. J. Tang, Y. T. Yan, H. V. Wang, J. S. Yu, C. M. Lin, and T. L. Shen for comments on the manuscripts, and the members of the Pai lab for the valuable discussions. This work was supported by grants to L-M.P. from the National Science Council of Taiwan (MOST 103-2311-B-182-004-MY3), the Chang Gung Memorial Hospital (CMRPD1E0272), and the Ministry of Education, Taiwan (EMRPD1E1421).
Abbreviations used:
- LIMCH1
LIM and calponin-homology domains 1
- NM-II
nonmuscle myosin II
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-04-0218) on February 22, 2017.
REFERENCES
- Aguilar-Cuenca R, Juanes-Garcia A, Vicente-Manzanares M. Myosin II in mechanotransduction: master and commander of cell migration, morphogenesis, and cancer. Cell Mol Life Sci. 2014;71:479–492. doi: 10.1007/s00018-013-1439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Betapudi V. Myosin II motor proteins with different functions determine the fate of lamellipodia extension during cell spreading. PLoS One. 2010;5:e8560. doi: 10.1371/journal.pone.0008560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge MT, Dulyaninova NG, Egelhoff TT. Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol Biol Cell. 2009;20:338–347. doi: 10.1091/mbc.E08-04-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman J, Wiggins C, Silberzan P, Buguin A, Ladoux B, Sheetz M. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. Mechanism of retraction of the trailing edge during fibroblast movement. J Cell Biol. 1981;90:187–200. doi: 10.1083/jcb.90.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Vicente-Manzanares M, Zareno J, Whitmore L, Mogilner A, Horwitz A. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawid I, Breen J, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998;14:156–162. doi: 10.1016/s0168-9525(98)01424-3. [DOI] [PubMed] [Google Scholar]
- Dedeic Z, Cetera M, Cohen T, Holaska J. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J Cell Sci. 2011;124:1691–1702. doi: 10.1242/jcs.080259. [DOI] [PubMed] [Google Scholar]
- Dulyaninova N, House R, Betapudi V, Bresnick A. Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol Biol Cell. 2007;18:3144–3155. doi: 10.1091/mbc.E06-11-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg F. Alternative splicing for members of human mosaic domain superfamilies. I. The CH and LIM domains containing group of proteins. Mol Biol Rep. 2009;36:1059–1081. doi: 10.1007/s11033-008-9281-9. [DOI] [PubMed] [Google Scholar]
- Giannone G, Dubin-Thaler B, Rossier O, Cai Y, Chaga O, Jiang G, Beaver W, Dobereiner H, Freund Y, Borisy G, Sheetz M. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M, Mital R. The single CH domain of calponin is neither sufficient nor necessary for F-actin binding. J Cell Sci. 1998;111:1813–1821. doi: 10.1242/jcs.111.13.1813. [DOI] [PubMed] [Google Scholar]
- Gupton S, Waterman-Storer C. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125:1361–1374. doi: 10.1016/j.cell.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Heath J. Behaviour and structure of the leading lamella in moving fibroblasts. I. Occurrence and centripetal movement of arc-shaped microfilament bundles beneath the dorsal cell surface. J Cell Sci. 1983;60:331–354. doi: 10.1242/jcs.60.1.331. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol. 2006;173:383–394. doi: 10.1083/jcb.200511093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikebe M, Hartshorne D. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–10031. [PubMed] [Google Scholar]
- Ikebe M, Komatsu S, Woodhead J, Mabuchi K, Ikebe R, Saito J, Craig R, Higashihara M. The tip of the coiled-coil rod determines the filament formation of smooth muscle and nonmuscle myosin. J Biol Chem. 2001;276:30293–30300. doi: 10.1074/jbc.M101969200. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Koretz J, Hartshorne D. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J Biol Chem. 1988;263:6432–6437. [PubMed] [Google Scholar]
- Iwasaki T, Murata-Hori M, Ishitobi S, Hosoya H. Diphosphorylated MRLC is required for organization of stress fibers in interphase cells and the contractile ring in dividing cells. Cell Struct Funct. 2001;26:677–683. doi: 10.1247/csf.26.677. [DOI] [PubMed] [Google Scholar]
- Joo E, Surka M, Trimble W. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev Cell. 2007;13:677–690. doi: 10.1016/j.devcel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Khatau S, Hale C, Stewart-Hutchinson P, Patel M, Stewart C, Searson P, Hodzic D, Wirtz D. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci USA. 2009;106:19017–19022. doi: 10.1073/pnas.0908686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga Y, Ikebe M. p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J Biol Chem. 2005;280:4983–4991. doi: 10.1074/jbc.M410909200. [DOI] [PubMed] [Google Scholar]
- Kolega J. Asymmetric distribution of myosin IIB in migrating endothelial cells is regulated by a rho-dependent kinase and contributes to tail retraction. Mol Biol Cell. 2003;14:4745–4757. doi: 10.1091/mbc.E03-04-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu S, Ikebe M. ZIP kinase is responsible for the phosphorylation of myosin II and necessary for cell motility in mammalian fibroblasts. J Cell Biol. 2004;165:243–254. doi: 10.1083/jcb.200309056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger D, Horwitz A. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Madaule P, Furuyashiki T, Eda M, Bito H, Ishizaki T, Narumiya S. Citron, a Rho target that affects contractility during cytokinesis. Microsc Res Tech. 2000;49:123–126. doi: 10.1002/(SICI)1097-0029(20000415)49:2<123::AID-JEMT3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Cramer L. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mull A, Kim G, Holaska J. Lmo7-null mice exhibit phenotypes consistent with Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2014;51:222–228. doi: 10.1002/mus.24286. [DOI] [PubMed] [Google Scholar]
- Murata-Hori M, Fukuta Y, Ueda K, Iwasaki T, Hosoya H. HeLa ZIP kinase induces diphosphorylation of myosin II regulatory light chain and reorganization of actin filaments in nonmuscle cells. Oncogene. 2001;20:8175–8183. doi: 10.1038/sj.onc.1205055. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Hori K, Tanaka-Okamoto M, Higashiyama M, Itoh Y, Inoue M, Morinaka S, Miyoshi J. Decreased expression of LMO7 and its clinicopathological significance in human lung adenocarcinoma. Exp Ther Med. 2011;2:1053–1057. doi: 10.3892/etm.2011.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooshio T, Irie K, Morimoto K, Fukuhara A, Imai T, Takai Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J Biol Chem. 2004;279:31365–31373. doi: 10.1074/jbc.M401957200. [DOI] [PubMed] [Google Scholar]
- Pasapera A, Schneider I, Rericha E, Schlaepfer D, Waterman C. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J Cell Biol. 2010;188:877–890. doi: 10.1083/jcb.200906012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120:3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- Peterson L, Rajfur Z, Maddox A, Freel C, Chen Y, Edlund M, Otey C, Burridge K. Simultaneous stretching and contraction of stress fibers in vivo. Mol Biol Cell. 2004;15:3497–3508. doi: 10.1091/mbc.E03-09-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T, Borisy G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Ridley A, Schwartz M, Burridge K, Firtel R, Ginsberg M, Borisy G, Parsons J, Horwitz A. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Riento K, Ridley A. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Ronen D, Ravid S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J Biol Chem. 2009;284:24948–24957. doi: 10.1074/jbc.M109.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandquist JC, Means AR. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol Biol Cell. 2008;19:5156–5167. doi: 10.1091/mbc.E08-05-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small J, Rottner K, Kaverina I, Anderson K. Assembling an actin cytoskeleton for cell attachment and movement. Biochim Biophys Acta. 1998;1404:271–281. doi: 10.1016/s0167-4889(98)00080-9. [DOI] [PubMed] [Google Scholar]
- Small J, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- Tan I, Lai J, Yong J, Li S, Leung T. Chelerythrine perturbs lamellar actomyosin filaments by selective inhibition of myotonic dystrophy kinase-related Cdc42-binding kinase. FEBS Lett. 2011;585:1260–1268. doi: 10.1016/j.febslet.2011.03.054. [DOI] [PubMed] [Google Scholar]
- Tan I, Yong J, Dong J, Lim L, Leung T. A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell. 2008;135:123–136. doi: 10.1016/j.cell.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Tan J, Ravid S, Spudich J. Control of nonmuscle myosins by phosphorylation. Annu Rev Biochem. 1992;61:721–759. doi: 10.1146/annurev.bi.61.070192.003445. [DOI] [PubMed] [Google Scholar]
- Tojkander S, Gateva G, Lappalainen P. Actin stress fibers–assembly, dynamics and biological roles. J Cell Sci. 2012;125:1855–1864. doi: 10.1242/jcs.098087. [DOI] [PubMed] [Google Scholar]
- Tojkander S, Gateva G, Schevzov G, Hotulainen P, Naumanen P, Martin C, Gunning P, Lappalainen P. A molecular pathway for myosin II recruitment to stress fibers. Curr Biol. 2011;21:539–550. doi: 10.1016/j.cub.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne D, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi B, Lowy D, Zelenka P. The Cdk5 activator P39 specifically links muskelin to myosin II and regulates stress fiber formation and actin organization in lens. Exp Cell Res. 2015;330:186–198. doi: 10.1016/j.yexcr.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenius T, Vaahtomeri K, Kovac B, Osiceanu A, Viljanen M, Makela T. An association between NUAK2 and MRIP reveals a novel mechanism for regulation of actin stress fibers. J Cell Sci. 2011;124:384–393. doi: 10.1242/jcs.072660. [DOI] [PubMed] [Google Scholar]
- Verkhovsky A, Svitkina T, Borisy G. Polarity sorting of actin filaments in cytochalasin-treated fibroblasts. J Cell Sci. 1997;110:1693–1704. doi: 10.1242/jcs.110.15.1693. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Koach M, Whitmore L, Lamers M, Horwitz A. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol. 2008;183:543–554. doi: 10.1083/jcb.200806030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein R, Horwitz A. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Newell-Litwa K, Bachir A, Whitmore L, Horwitz A. Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J Cell Biol. 2011;193:381–396. doi: 10.1083/jcb.201012159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi C, Horwitz A. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M, Baker B, Chen C, Wilson K. The emerin-binding transcription factor Lmo7 is regulated by association with p130Cas at focal adhesions. Peer J. 2013;1:e134. doi: 10.7717/peerj.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Irie K, Fukuhara A, Ooshio T, Takai Y. Requirement of the actin cytoskeleton for the association of nectins with other cell adhesion molecules at adherens and tight junctions in MDCK cells. Genes Cells. 2004;9:843–855. doi: 10.1111/j.1365-2443.2004.00768.x. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Totsukawa G, Yamakita Y, Sasaki Y, Madaule P, Ishizaki T, Narumiya S, Matsumura F. Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol Biol Cell. 2003;14:1745–1756. doi: 10.1091/mbc.E02-07-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007;120:137–148. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.