

Abstract
Atherosclerosis rarely develops in the region of arteries exposed to undisturbed flow (u-flow, unidirectional flow). Instead, atherogenesis occurs in the area exposed to disturbed flow (d-flow, multidirectional flow). Based on these general pathohistological observations, u-flow is considered to be athero-protective, while d-flow is atherogenic. The fact that u-flow and d-flow induce such clearly different biological responses in the wall of large arteries indicates that these two types of flow activate each distinct intracellular signaling cascade in vascular endothelial cells (ECs), which are directly exposed to blood flow. The ability of ECs to differentially respond to the two types of flow provides an opportunity to identify molecular events that lead to endothelial dysfunction and atherosclerosis. In this review, we will focus on various molecular events, which are differentially regulated by these two flow types. We will discuss how various kinases, ER stress, inflammasome, SUMOylation, and DNA methylation play roles in the differential flow response, endothelial dysfunction, and atherosclerosis. We will also discuss the interplay among the molecular events and how they coordinately regulate flow-dependent signaling and cellular responses. It is hoped that clear understanding of the way how the two flow types beget each unique phenotype in ECs will lead us to possible points of intervention against endothelial dysfunction and cardiovascular diseases.
Keywords: MAP kinase, SUMOylation, p90RSK, SENP2, DRP-1, NEMO, PKC family, AMPK, ER stress, PPARs, ERK5, DNA methylation, HATs and HDACs
Introduction
Atherosclerosis is a multi-factorial disease that is associated with risk factors, including hypercholesterolemia, hyperlipidemia, hyperglycemia, smoking, hypertension, aging, and diabetes. These risk factors are the so-called systemic factors to which the entire cardiovascular system is exposed in more or less uniformly. However, atherosclerosis is a focalized disease that develops in anatomically identifiable regions of the arterial system. It is well known that while atherogenesis rarely occurs in the region of arteries exposed to undisturbed flow (u-flow), lesions frequently arise in areas exposed to disturbed flow (d-flow). This regional specificity of atherogenesis suggests a strong correlation between the local pattern of blood flow (u-flow or d-flow) and the pathogenesis (inhibition or promotion) of atherosclerotic plaque formation. In this regard, it is important to understand how u-flow inhibits, while d-flow promotes atherogenesis, because this understanding will guide us toward therapeutic interventions against this disease.
One of the key events that cause endothelial cell (EC) dysfunction and atherosclerosis is the increased expression by ECs of cell adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin. Normally, healthy ECs do not recruit leukocytes on the luminal surface, but when ECs are placed in atherogenic environments (e.g., disturbed flow), the expression of these adhesion molecules increases, promoting leukocyte recruitment and inflammation. In contrast, u-flow inhibits these pro-atherogenic events in ECs. Recent studies have reported a number of signaling pathways that are differentially regulated by the two types of flow. In this review, we summarize flow-activated signaling pathways that are either anti-antherogenic or pro-atherogenic and discuss their regulatory mechanisms.
Mitogen-activated protein kinases (MAPKs) and extracellular signal-regulated-kinase 5 (ERK5) in endothelial cells
Mitogen-activated protein kinases (MAPKs) are highly conserved serine/threonine kinases that are activated by a wide variety of stimuli, including growth factors, G protein–coupled receptor activation, and oxidative stress. Four major groups of the MAPK family, including extracellular signal-regulated-kinase (ERK)1/2, c-Jun N-terminal kinases (JNKs), p38 kinases, and the MEK5-regulated ERK5, have been reported to regulate downstream transcription factors, which mediate cell growth and differentiation [1, 2] (Fig. 1). The activation of these MAP kinases requires dual phosphorylation of threonine and tyrosine residues within the threonine-x-tyrosine motif (T-x-Y motif), which is T-E-Y in ERK1/2, T-P-Y in JNKs, T-G-Y in p38, and T-D-Y in ERK5.
Fig. 1.

MAPK signaling. ATF2 activating transcription factor 2, BAD BCL2 associated agonist of cell death, Bim Bcl-2 interacting mediator of cell death, cdc25B cell division cycle 25B, GLK germinal center kinase-like kinase, HGK hapatocyte progenitor kinase-like/germinal center kinase-like kinase, HPK1 hematopoietic progenitor kinase1, HSF1 heat-shock transcription factor 1, LPS lipopolysaccharide, MK1/2 MAPK-activated protein kinase ½, MLK mixed lineage kinase, MNK menkes disease-associated protein, MSK1/2 mitogen-and stress-activated protein kinase1/2, NFAT nuclear factor of activated T-cells, NHE-1 Na+/H+ exchanger-1, Nur77 nuclear hormone receptor 77, PAK1/2 p21 (RAC1)-activated kinase ½, PKC protein kinase C, PPARs peroxisome proliferator-activated receptors, Sap1 serum response factor accessory protein 1, TAB 1/2 TGF-beta-activated kinase1-binding protein1/2, TAK/MAP3K7 TGF-beta-activated kinase, TRAF TNF receptor-associated factor 6
ERK1/2 activity is involved in embryogenesis, cell proliferation, differentiation, and apoptosis, whereas the overexpression or constitutive activation of ERK1/2 can result in the progression of several cancers [3]. In 1995, Bradford Berk’s group showed that flow-activated ERK1/2 [4] and that a herbimycin-sensitive tyrosine kinase and PKC may be involved in this activation [5–7]. The role of endothelial ERK1/2 in regulating atherosclerosis is not clearly defined. Chen et al. have reported that an inhibitor of mitogen-activated protein kinase kinase 1/2 (MEK1/2, the upstream kinase of ERK1/2) and a liver X receptor (LXR) ligand can synergistically reduce atherosclerotic plaque formation, but the effect of MEK1/2 inhibition on atherosclerosis was marginal [8].
JNK can phosphorylate several transcription factors, including c-Jun, ATF2, Elk-2, RXRα, NFAT4, HSF-1, and p53, which can contribute to both endothelial inflammation and apoptosis [9–13]. It has been well established that the induction of VCAM-1, ICAM-1, and E-selectin by TNF-α is regulated by JNK [14–16]. JNK can also accelerate apoptosis by the following two different ways: one mechanism is by regulating nuclear transcriptional factors, including c-Jun and p53. For example, JNK can phosphorylate p53 at Ser6, which inhibits ubiquitin-mediated p53 degradation and increases the p53 expression level, resulting in p53-mediated up-regulation of several pro-apoptotic genes, including Bax and PUMA [17, 18]. The other mechanism is by regulating mitochondrial function. It has been reported that JNK translocates to mitochondria and phosphorylates Bcl-2, Bim, and Bid, all of which are apoptosis-related molecules [9]. These molecules can induce cell apoptosis singly as well as cooperatively. It was first reported that JNK phosphorylated Bcl-2 at Ser70, which inhibited anti-apoptotic effect of Bcl-2 [19]. Then, it was discovered that JNK also phosphorylated Bim at Ser65 [20] or Thr56 [21], which up-regulated Bim’s pro-apoptotic activity via activating Bax and Bak, and subsequently inhibited the anti-apoptotic effect of Bcl-2. Finally, the contribution of Bid in JNK-mediated apoptosis was reported, although this was not by direct phosphorylation of Bid by JNK [22]. In this case, JNK played a role in cleaving Bid and generated a JNK-mediated Bid cleavage product (jBid), which then translocated to mitochondria and induced apoptosis by releasing Smac/DIABLO, not cytochrome c [22]. The role of JNK on atherosclerosis likely depends on its isoforms. Ricci et al. reported that ApoE−/− mice also lacking JNK2 (ApoE−/− JNK2−/− mice), but not ApoE−/− JNK1−/− mice, developed less atherosclerosis than ApoE−/− mice [23]. Pharmacological inhibition of JNK efficiently reduced plaque formation [23, 24], indicating that JNK is a pro-atherogenic MAPK.
Possible roles for p38α in the development and progression of atherosclerosis have been suggested. For example, the activation of p38α can up-regulate migration [25, 26], proliferation [25, 26], permeability [27], apoptosis [28], and adhesion molecule expression [29] of endothelial cells. It was reported that u-flow increased p38 activation and inhibited matrix metalloproteinase MMP-2 expression [30]. However, Kardakaris et al. reported that EC-specific p38α knockout in ApoE−/− mice had no effect on the atherosclerotic plaque formation [31]. These results suggest that the pathological role of flow-induced p38 activation in ECs during the development of atherosclerosis remains unclear.
MAPK kinase 5 (MEK5) is a dual-phosphorylation kinase and is an immediate upstream activator of ERK5. Hayashi et al. reported that the depletion of endothelial ERK5 impaired vascular integrity via enhancing EC apoptosis [32]. Although the mechanism for the anti-apoptotic property of ERK5 is unclear, it is attributed to possible phosphorylation of BAD (Bcl2 antagonist of cell death) [33].
Berk et al. have earlier found that u-flow activates ERK1/2, p38, and ERK5, while it inhibits TNF-α-induced JNK activation [34]. It appears that the inhibition of JNK signaling can be one of the mechanisms for the u-flow-induced anti-inflammatory and anti-apoptotic effects. In the following sections, we will discuss several possible mechanisms how u-flow could inhibit JNK signaling.
Role of ERK5 in s-flow-induced anti-inflammatory responses in ECs
It has been reported that u-flow increases ERK5 kinase activity [35] but not d-flow [36]. Since u-flow but not d-flow inhibits EC inflammation as we explained above, ERK5 may be involved in regulating the u-flow-induced anti-inflammatory effect. Tumor necrosis factor-alpha (TNFα) induces EC inflammation and up-regulates the expression of adhesion molecules including VCAM-1, ICAM-1, and E-selectin in ECs. JNK activation is required for TNFα-induced VCAM-1 expression, and u-flow inhibits JNK activation as well as VCAM-1 expression [37]. Li et al. showed that the MEK5 inhibitor BIX02188 completely reversed u-flow-mediated inhibitory effect without inhibiting ERK1/2, suggesting that u-flow inhibits TNFα-mediated signaling in ECs by a mechanism that depends on MEK5-ERK5 but not MEK1-ERK1/2 activation [37, 38]. The exact molecular mechanism through which u-flow inhibits TNFα-induced JNK activation via activating MEK5/ERK5 needs further investigation [14, 39].
Role of the PKC family in EC dysfunction
The protein kinase C (PKC) family consists of ubiquitously expressed homologous serine/threonine kinases, which are divided into three groups according to their second messenger requirements: classical, novel, and atypical [40–43]. Classical PKCs consist of α, βI, βII, and γ isoforms and require Ca2+, diacylglycerol (DAG), phosphatidylserine, and phorbol esters for activation. Novel PKCs are δ, ɛ, η, and θ isoforms. These isoforms only need DAG for activation and are Ca2+ insensitive. Atypical PKCs consist of ζ and ι/λ isoforms, and they lack the calcium-sensitive C2 domain and contain only one atypical C1 domain. Thus, atypical PKCs are unresponsive to DAG and calcium [40–43].
In terms of the role of classical PKCs in regulating EC functions, ox-LDL reduces PKCα activity and subsequently de-phosphorylates eNOS. De-phosphorylation of eNOS causes its dissociation from the plasma and Golgi membrane and also increases the production of eNOS-derived superoxide anion [44]. A PKCβ antagonist, CGP53353, inhibits JNK and mitochondrial gene p66Shc activation as well as ROS production in ECs [45–52]. The role of endothelial PKCβ in diabetes has been extensively studied. Baseline vascular endothelial PKCβ expression is increased in diabetes compared to non-diabetics. The reduced PKCβ expression impairs eNOS activation, which can be reversed by a PKCβ specific synthetic inhibitor, LY379196 [53]. The possible contribution of PKCβ to insulin resistance has also been suggested; insulin-induced eNOS expression is impaired by PKCβ-mediated phosphorylation of IRS2 (insulin receptor substrate 2) at S303 and S375 in vascular endothelial cells [54]. The depletion of PKCβ in ApoE−/− mice results in decelerated plaque formation with reduced expression of Egr-1 (early growth response protein-1) and inhibits the expression of endothelial cell adhesion molecules and matrix metalloproteinase-2 (MMP2) [55]. PKCβ inhibition by ruboxistaurin reduces plaque lesion size in diabetic mice [56]. However, clinical trials with ruboxistaurin have not shown improved endothelial function, including endothelium-dependent vasodilatation [57]. In terms of the novel PKCs family, treatment with a PKCδ inhibitor or a PKCɛ activator decreases eNOS phosphorylation and increases EC survival, but the role of this family of kinases in atherosclerosis remains unclear [58].
Among the PKC family members, PKCζ has recently emerged as an important isoform in ECs. PKCζ has three functional domains: (1) a PB1 (Phox and Bem1p) domain which constitutes a recently recognized protein–protein interaction domain found in atypical protein kinase C (aPKC) isoenzymes, PKCζ, and PKCλ/ι, certain members of MAPKs, such as MEK5, MEKK2, and MEKK3 [59, 60], and in several scaffold proteins involved in cellular signaling; (2) a zinc-finger (ZF) domain for DNA interaction; and (3) a kinase domain. The important role of PKCζ in regulating EC inflammation via nuclear factor kappa B (NF-κB) has been reported [61, 62]. Reduced PKCζ activity has been reported in ECs exposed to u-flow than those under d-flow [63]. The activity of PKCζ is regulated by proteolytic cleavage. PKCζ (72 kDa) is processed by caspase 3 to form a 50 kDa fragment (CATζ, catalytic domain of PKCζ) that consists of the catalytic domain [64]. This caspase 3-dependent processing occurs at three aspartate residues (Asp 210, 222, and 239) and frees the enzyme from the auto-inhibitory state by separating the kinase domain (aa 268–335) from the pseudosubstrate auto-inhibitory sequence (aa 116–122) [65]. Since PKCζ activity is also required for TNF-mediated activation of JNK and caspase-3 in ECs, a positive feedback loop for PKCζ-caspase 3 activation via regulating CATζ formation has been proposed [66]. The inhibitory effect of u-flow on this PKCζ-caspase 3 positive feedback loop has also been reported, but the exact regulatory mechanisms remain unclear [66] (Fig. 2).
Fig. 2.

Flow reduces catalytic domain of PKCζ (CATζ) pro-apoptotic effects. Bovine aortic endothelial cells (BAECs) overexpressing CATζ were preexposed or not to flow for 24 h before TNF/cycloheximide (CHX). a Caspase-3 activity was estimated by the detection of its cleaved form by Western blot. Anti-HA was used to detect the CATζ transfected form. b Western blots were quantified by densitometry. All figures are representative of 3 to 4 independent experiments. *P < 0.05 vs static TNF/CHX-treated cells. c Model: Flow inhibits PKCζ cleavage and prevents apoptosis. TNF/CHX, via induction of caspase-3 activation, induces PKCζ cleavage to yield CATζ. CATζ enhances TNFα–induced JNK and caspase-3 activation leading to a death pathway, where CATζ stimulates caspase-3 activation and subsequent PKCζ cleavage. In contrast, s-flow mediates prosurvival effects via inhibiting caspase-3 and JNK, thus decreasing PKCζ cleavage [66]
(Obtained permission to reproduce from Wolters Kluwer Health)
Undisturbed flow (u-flow) induces anti-inflammatory effects in ECs by inhibiting SHP2 phosphatase activity
One of the important regulators of the TNFα pathway is SHP-2, whose activity is required for the activation of JNK and NF-κB [67, 68]. SHP-2 is a ubiquitously expressed cytosolic phosphatase that contains 2 NH2-terminus tandem SH2 domains and a carboxyl-terminus catalytic domain [69]. MEKK3 is required for the TNFα-induced c-Jun and NF-κB transcriptional activity [70, 71]. Our group has found that a constitutively active form of MEKK3 (CA-MEKK3) stimulates c-Jun and NF-κB transcriptional activity and that SHP2C/S (phosphatase inactive mutant with cysteine 495 mutated to serine), but not wild-type SHP2, inhibits this transactivation [72]. We also found that Gab1 associated with MEKK3 and inhibited MEKK3-induced c-Jun and NF-κB activation [70]. Since the binding between Gab1 and MEKK3 in vitro was enhanced by the presence of SHP-2C/S, it appears that SHP-2 phosphatase activity regulates the Gab1-MEKK3 association and consequently TNFα signaling in ECs. U-flow significantly inhibited TNF-α-induced SHP-2 phosphatase activity [72]. Therefore, it is possible that u-flow inhibits SHP-2 phosphatase activity and subsequent EC inflammation via keeping the MEKK3-Gab1 interaction intact (Fig. 3).
Fig. 3.

Model of flow and TNFα-mediated signal transduction pathways to regulate adhesion molecule expression in endothelial cells.
Reprinted from Lerner-Marmarosh et al. [72], and obtained permission to reproduce from Wolters Kluwer Health
ERK5 and its transcriptional activity
Compared to other MAPKs, ERK5 has a unique feature, because it has a long COOH-terminus with transcriptional activity domains that are necessary for activating ERK5 downstream transcription factors, including peroxisome proliferator-activated receptors (PPARs) and Kruppel-like factor 2 (KLF2) (Fig. 4) [33, 34, 73]. Members of the nuclear hormone receptor family, PPARs, are known for their anti-inflammatory properties [74]. Currently, there are three known PPAR isotypes, including PPARα, PPARδ, and PPARγ, all of which are expressed in vascular ECs. Activation of PPARs via a ligand-dependent or ligand-independent mechanism has been shown to reduce inflammation by down-regulating the expression of TNF-α-induced inflammatory adhesion molecules, such as ICAM-1, VCAM-1, and E-selectin [75]. PPARs consist of two transcriptional activation domains residing in the NH2-terminus A/B domain (activation function-1) and the COOH-terminus E domain (activation function-2). MAPK family members have different effects on these receptors. While Camp et al. reported that the phosphorylation of PPARγ1 S82 by ERK1/2 decreased PPARγ1 transcriptional activity [76], our study showed that ERK5 up-regulated PPARγ transcriptional activity through binding to PPARγ and not by phosphorylating PPARγ (Fig. 5) [77]. Through intra-molecular bonds, the ERK5 NH2-terminus inhibits its own transcriptional activity located in the COOH-terminus. U-flow activates MEK5 and promotes MEK5-ERK5 binding, which disrupts the inhibitory effect of the ERK5 NH2-terminus on its COOH-terminus domain-containing PPARγ binding site and transcriptional activity. Subsequently, the ERK5 COOH-terminus can bind the hinge-helix 1 region of PPARγ, which releases SMRT co-repressor from PPARγ and increases PPARγ transcriptional activity [77]. Both releasing SMRT and increasing PPARγ transcriptional activity induced by ERK5 transcriptional activation and subsequent ERK5-PPARγ binding contribute to u-flow-induced anti-inflammatory effects and inhibit TNFα-induced adhesion molecule expression [77]. Besides its association with PPARγ, ERK5 is also reported to bind and increase PPARδ transcriptional activity in skeletal muscle. The ERK5-PPARδ association appears to be mediated by heme oxygenase 1-induced carbon monoxide, which is known to have a cytoprotective effect. Unlike PPARγ, PPARδ has no hinge-helix 1 region. However, the binding of PPARδ with the middle region of ERK5 is still required to increase PPARδ transcriptional activity. As noted, ERK5 increases transcriptional activity of both PPARγ and δ, and thus has an anti-inflammatory property [78].
Fig. 4.

Primary structure of extracellular-signal-regulated-kinase 5 (ERK5). ERK5 is the largest member of the mitogen-activated protein kinase family. This Ser/Thr protein kinase has a catalytic NH2-terminus domain, which contains the MAPK-conserved motif threonine/glutamate/tyrosine (TEY) in the activation loop that shares about 50% sequence identity with ERK1/2 and a unique long COOH-terminus tail. ERK5 activation is induced by the interaction with MEK5, which subsequently dually phosphorylates the TEY motif. In contrast, inflammatory stimuli or athero-prone flow (d-flow) leads to ERK5 deactivation via phosphorylation of Ser486 or Ser496, respectively. The K6 and K22 sites at the NH2-terminus region with small ubiquitin-like modifier (SUMO) modification inhibit ERK5 transactivation
Fig. 5.

Model of the ERK5-PPARγ1 interaction activating PPARγ1 activity. The activation of PPARγ is significantly regulated by Hinge-helix 1, which is sterically altered by ligand binding. When the PPARγ ligand binds to the receptor, Helix 12-folds back to form a part of the coactivator binding surface, and inhibits co-repressor (such as SMRT) binding to PPARγ [224]. The co-repressor interaction surface requires Helix 3–5 [225]. We found a critical role of the PPARγ Hinge-helix 1 domain on ERK5-mediated PPARγ transactivation [77]. The inactive NH2-terminus kinase domain of ERK5 inhibits its own transactivation and PPARγ binding. After ERK5 activation, the inhibitory effect of the NH2-terminus domain of ERK5 decreases, and subsequently, the middle region of ERK5 can fully interact with the Hinge-helix 1 region of PPARγ. The association of ERK5 with the Hinge-helix 1 region of PPARγ releases co-repressor of SMRT and induces full activation of PPARγ. AF-1/2: Activating Function (AF)-1/2 transactivation domain, DBD DNA-binding domain.
Reprinted and modified from Akaike et al. [77]
The KLF family is a group of zinc-finger DNA-binding transcriptional factors that have an important role in regulating vascular homeostasis and EC function [79]. The crucial role of KLF2 in regulating EC function has been well established. First, Kuo et al. have reported that KLF2 is necessary for blood vessel formation and vascular integrity, because KLF2 null mice died of embryonic hemorrhage [80]. Second, SenBanerjee et al. have demonstrated that u-flow-induced KLF2 up-regulation increases eNOS expression via binding to the eNOS promoter [81]. Therefore, KLF2 is important for regulating EC-dependent vaso-reactivity. In addition, through competition with NF-κB for transcriptional coactivator CBP/300, KLF2 inhibits the NF-κB-dependent expression of VCAM-1 and E-selectin [81]. Third, Lin et al. have reported that KLF2 inhibits inflammation-mediated vascular permeability by its effect on tight junctions [79]. Finally, statins up-regulate the eNOS and thrombomodulin expression via KLF2 [82], and KLF2 expression is induced by u-flow and inhibited by d-flow [83]. These data suggest that KLF2 mediates the u-flow- and statin-induced athero-protection.
Parmar et al. have shown that ERK5 activation is required for u-flow-induced KLF2 up-regulation using a dominant negative form of MEK5 (DN-MEK5) [84]. ERK5/Myocyte enhancing factor 2 (MEF2) is an upstream regulator of KLF2 (KLF2 promoter has a MEF2 binding site) and is activated by ERK5. Young et al. have reported that u-flow activates the AMP-activated protein kinase cascade, which then regulates KLF2 through activating MEF2 [73]. These data suggest that in addition to PPARs, the MEF2-KLF2 module is one of the important downstream targets of ERK5 and up-regulates eNOS expression and inhibits EC inflammation, especially under u-flow.
To determine the role of endothelial ERK5 in vivo, our group has generated tamoxifen (4-OHT, 4-hydroxytamoxifen)-inducible EC-specific ERK5 knock-out mice (ERK5-EKO) [85]. We found that eNOS expression in ECs was decreased, whereas the expression of VCAM-1 and E-selectin was increased in ECs isolated from 4-OHT-treated mice in comparison with vehicle-treated mice. To evaluate the functional role of endothelial ERK5, we studied leukocyte rolling and acetylcholine-induced vasodilation in mesenteric venules and arterioles of the inducible ERK5-EKO+/− mice. We found that leukocyte rolling in the venule was increased in the 4-OHT-treated group in comparison with the vehicle-treated group, and acetylcholine-induced dilation of arterioles was significantly decreased in both inducible heterozygous ERK5-EKO+/− and homozygous ERK5-EKO−/− mice in comparison with the 4-OHT-treated control mice. To examine the role of endothelial ERK5 in atherogenesis, inducible ERK5-EKO-LDLR−/− mice were fed a high-cholesterol diet for 16 weeks. We found a significant increase in both the surface area and the size of lesions in the aortic root of these induced mice in comparison with non-induced mice. These data suggest a critical role of ERK5 in EC dysfunction and atherosclerotic plaque formation. Although we have shown that ERK5 is anti-inflammatory using EC-specific ERK5 KO mice [85], Wilhelmsen et al. reported that ERK5 was pro-inflammatory when TLR2 (Toll-like receptor 2) was activated [86]. This controversy may be reconciled by different ERK5 post-translational modifications, as we will discuss in the following sections.
Flow and hippo pathway
The vital role of the hippo pathway in the regulation of organ size, cardiac development and hypertrophy, stem cell pluripotency, and tumorigenesis has been extensively studied [87–89]. However, to our knowledge, the role of this pathway in EC dysfunction as well as in atherosclerosis remains largely unknown. The core of the mammalian hippo pathway is composed of two pairs of kinases: mammalian Ste20-like kinases 1 and 2 (MST1/2) and LATS1/2, which are localized in the cytoplasm [90, 91]. MST1/2 kinases bind to their regulatory protein WW45 to form active enzymes and phosphorylate and activate LATSs [92]. In addition, MST1/2 phosphorylate MOB1 (the Mps One Binder kinase activator protein 1), which strongly interacts with LAST, thereby enhances the interaction between Mob1 and LATSs [93]. The MST1/2-LATSs Hippo pathway controls the activity of two closely related transcriptional coactivators: YAP and a transcriptional coactivator with a PDZ-binding motif (TAZ). Activated LATSs phosphorylate YAP S127, which allows 14-3-3 proteins to bind and sequester YAP to the cytoplasm [90, 91]. De (or un)-phosphorylated YAP and TAZ translocate into the nucleus and act as transcriptional coactivators by binding to the TEA domain-containing transcription factor (TEAD) and drives a transcriptional program that promotes proliferation [94] and inhibits apoptosis [90, 95, 96], which are key events for controlling organ size.
The contribution of YAP and TAZ to mechano-signaling has been suggested. Dupont et al. have reported that when a cell spreads over the extracellular matrix, the cytoskeletal adaptation to the spread-out cell shape (including formation of filamentous actin [F-actin] stress fibers) induces YAP/TAZ nuclear accumulation and activation, which promotes proliferation and inhibits apoptosis [97]. In contrast, when adhesion is restricted to a very small extracellular adhesive area, its anti-apoptotic functions are lost [88, 97]. Interestingly, the MST1/2-LATSs pathway is dispensable for the YAP/TAZ nuclear translocation and activation, and the inhibitory role of LATSs in YAP activation is cell-type dependent [87]. Since the role of LATSs and YAP/TAZ activation in EC mechano-response remains unclear, a detailed evaluation using ECs is necessary.
Flow, SREBP2, and inflammasome
The inflammasome is a protein complex that serves as a platform for the maturation of caspase-1, leading to the proteolytic maturation and secretion of IL-1β and IL-18. There are three essential components of the inflammasome, which are (1) a sensor protein, (2) the adapter protein ASC, and (3) the inflammatory protease caspase-1. The response of the inflammasome to various stresses is decided by sensor protein, such as the Nod-like receptor (NLR) family (NLRP1, NLRP3, and NLRP4) and AIM2, which function as pathogen sensors and form an inflammasome complex. NLRP3 is uniqued in that it responds to numerous diverse physical and chemical triggers, unlike other NLRP members, for example, extracellular ATP, reactive oxygen species (ROS), uric acid crystals, cholesterol crystals, and IAPP (islet amyloid polypeptide) [98, 99]. Shyy and colleagues reported that unlike u-flow, d-flow increased NLRP3 expression and subsequently cleaved caspase-1 and increased IL-1β expression [100, 101]. Not only increasing IL-1β expression but also pyroptosis (a regulated form of necrosis) can be induced by caspase-1 activation as we will explain later in this review. Shyy’s group also reported the crucial role of SREBP2 transactivation in the d-flow-induced expression of NLRP3 and Nox2 [100, 101]. In fact, they also showed that the levels of cleaved caspase-1 and expression of IL-1β and NLRP3 were higher in the aortic arch (predominantly exposed to d-flow) than in thoracic aorta (area exposed to u-flow), supporting the crucial role of d-flow–induced NLRP3 inflammasome activation in the development of atherosclerosis.
SREBPs, including SREBP-1a, -1c, and -2, are regulators of transcription of a number of genes involved in the cellular sterol and lipid homeostasis. In particular, SREBP2 is ubiquitously expressed, is a relatively selective activator of cholesterol synthesis, as opposed to fatty acid synthesis, and regulates HMG-CoA reductase and low-density lipoprotein receptor genes [102, 103]. SREBP2 regulates endothelial inflammation via increasing NLRP3 and Nox2 expression, which is not directly related to the regulation of lipid homeostasis [104]. In addition to SREBP2, d-flow can also cause sustained SREBP1 activation with nuclear localization. In contrast, u-flow induces only a transient nuclear translocation of SREBP [105]. It appears that SREBP family of proteins exhibit different responses to u-flow and d-flow, suggesting that they may be involved in determining EC responses to different flow types.
SREBPs are the bHLH-LZ family of transcription factors, and inactive newly synthesized SREBPs (pre-SREBPs) are cleaved by proteases, releasing the NH2-terminus portion (SREBP-N). This mature form of SERBPs (SREBP-N) enters the nucleus and binds the sterol regulatory element 1 (SRE-1), and activates transcription of target genes [102]. There are two possible steps at which flow can regulate SREBP activation: one is when pre-SREBPs are cleaved in the cytoplasm and the second is to regulate nuclear SREBP-N transcriptional activity. It has been suggested that d-flow can affect both steps and increase SREBPs activation. For example, d-flow may increase SREBP-N transcriptional activity via increasing Akt and mammalian target of rapamycin complex 1 (mTORC1) kinase activation [106]. In addition, Akt activation can also attenuate ubiquitination and proteosomal degradation of active SREBP-N, because Akt can inhibit GSK3 kinase activity, which directly phosphorylates SREBP-N and enhances SREBP-N degradation [106].
D-flow and regulated form of necrosis
As we discussed above, d-flow activates inflammasomes, and recently, it has been shown that activation of the caspase 1-inflammasome induces pyroptosis, a regulated form of necrosis (canonical inflammasome pathway) [107] (Fig. 1). Although pyroptosis has not been well studied in ECs, it is reasonable to discuss the possible role of the regulated form of necrosis in endothelial dysfunction. Necrosis was once recognized as an “accidental” or “mechanical” cell death induced by physio-chemical stresses [108]. This view has been greatly changed, and it is now clear that necrosis can also take place in a genetically and well controlled manner defined as the “regulated form of necrosis” [108]. Vanden Berghe et al. have defined regulated necrosis as “a genetically controlled cell death process, which leads to cellular leakage” [108]. Pyroptosis is a form of necrosis and is characterized by pore formation in the plasma membrane, cell swelling, and membrane disruption [108, 109]. Jorgensen et al. have defined pyroptosis by the following four criteria [107]: (1) programmed by an inflammatory caspase activation; (2) pore formation in the plasma membrane; (3) TUNEL positivity for DNA damage at a lower intensity than apoptosis; and (4) ADP-ribose polymerase activation following the pyroptosis-mediated DNA damage. Not only caspase 1 but also the possible involvement of caspase 4/5/11 in regulating pyroptosis was also suggested [110]. Caspase 1 can be activated by various extracellular stimuli, including ATP, cholesterol crystals, double-strand DNA, and flagellin via several different types of inflammasomes, which has recently been reviewed [107, 111]. In contrast, caspase 4/5/11 in the noncanonical inflammasome has been reported to directly sense cytosolic LPS (Fig. 6). The caspase 4/5/11 CARD binds LPS, presumably via the acidic phosphate moiety of lipid A, and causes caspase 4/5/11 CARD oligomerization and activation [107, 112, 113]. GSDMD and caspase 4/5/11 are required for cytosolic LPS-induced pyroptosis and the processing and secretion of IL-1β. Both caspase 1 and caspase 4/5/11 directly cleave the 53-kDa inactive precursor form of GSDMD (Pro-GSDMD) to generate the pro-pyroptotic NH2-terminus fragment (GSDMD p30) [109, 114].
Fig. 6.

Schemes for canonical and noncanonical inflammasome signaling. dsDNA indicates double-strand DNA, Hhcy hyperhomocysteinemia, JNK c-Jun N-terminal protein kinase, LPS lipopolysaccharide, NLRP3/ASC, NOD (nucleotide oligomerization domain)-like receptor P3/apoptosis-associated speck-like protein containing a caspase recruitment domain, pro-GSDMD 53-kDa inactive precursor form of GSDMD, and ROS reactive oxygen species. Reprinted from Abe and Morrell [226], and obtained permission to reproduce from Wolters Kluwer Health.
Modified from Kayagaki et al. [114] with permission of the publisher. Copyright ©2015, Nature Publishing Group
In the canonical pathway, caspase 1 activation induced by various types of inflammasomes triggers pyroptosis by releasing GSDMD p30, which bears intrinsic pyroptosis-inducing activity (Fig. 6) [109, 114]. In the non-canonical pathway, a direct activation of caspase 4/5/11 induced by cytosolic LPS cleaves pro-GSDMD and converts it to the mature GSDMD p30 form, which is sufficient for inducing pyroptosis [109]. However, it remains unclear how GSDMD p30 can induce pyroptosis. Pore formation in the plasma membrane is a feature of pyroptosis, and it is suggested that GSDMD p30 may directly induce the pore formation [115], but this needs further investigation. Since d-flow increases NLRP3-caspase1 activity, it will be intriguing to investigate if d-flow can induce pyroptosis.
ER stress and flow
The accumulation of misfolded proteins during protein synthesis in the endoplasmic reticulum (ER) is an endogenous cellular source of stress (ER stress). As the cellular defense mechanism against ER stress, three axes of signal transduction pathway termed the unfolded-protein response (UPR) exist in higher eukaryotic cells. In the unstressed state, immunoglobulin-heavy-chain-binding protein [BIP, also known as heat-shock protein A5 (HSPA5) and glucose-related protein 78 (GPR78)] associates with and inhibits the function of the three ER-proximal UPR transmembrane proteins (UPR’s three pronged signaling network axes), including (1) activating transcription factor 6 (ATF6), (2) inositol-requiring transmembrane kinase/endonuclease 1 (IRE1), and (3) pancreatic ER kinase (PERK, also known as EIF2AK3). Under the unstressed condition, a newly synthesized and properly folded proteins transit into the ER lumen through the SEC61 channel. When cells are stressed, unfolded, and misfolded proteins start to accumulate in the ER. This causes BIP to detach from ATF6, IRE1, and PERK, releasing its inhibitory effect on them and activates the three axes of UPR signaling.
(1) ATF6: ATF6 contains a Golgi-localization sequence (GLS), which becomes exposed after ATF6 is released from BIP, and ATF6 translocates to the Golgi apparatus, where it is cleaved by sute-1- and sute-2-proteases (S1P and S2P, respectively). The ATF6 fragment (ATF6f) transcription factor is released from the Golgi body and translocates to the nucleus, where it binds ER stress response elements (ESRE) and induces the expression of BIP, CHOP (CCAAT/enhancer-binding protein homologous protein), and X-box binding protein (XBP-1).
Civelek et al. have reported that d-flow induces the activation of adaptive UPR through ATF6α and IRE1α. In contrast, PERK, the third regulatory axe of UPR, as we will explain later, appeared not to be involved in this adaptive response [116].
(2) IRE1: Dissociation of IRE1 from BIP results in homodimerization of IRE1 and activation of its endoribonuclease activity, which excises a 26 base-pair fragment from XBP-1 mRNA and forms spliced XBP1 (sXBP1) mRNA. sXBP1 protein translocates to the nucleus and binds UPR elements (UPREs), which induces the transcription of a broad array of UPR genes, thereby increasing the synthesis and secretion of UPR proteins, including ER-associated degradation (ERAD) protein EDEM, ER-localized DNAJ4 (ERdj4), and p58IPK [117–119]. It has been suggested that d-flow-induced EC proliferation and apoptosis are mediated by sXBP1-induced VE-cadherin down-regulation. For example, in ApoE−/− mice, BrdU incorporation was significantly higher in ECs exposed to d-flow at the aortic arch than those exposed to laminar flow in the descending aorta, and sXBP-1 expression was higher in proliferating ECs than in quiescent cells. Furthermore, EC proliferation was inhibited when XBP1 and IRE1 were depleted [120]. It has been reported that d-flow decreases VE-cadherin expression via activating XBP1 splicing, leading to VE-cadherin transcriptional repression and increased degradation [120]. The reduced VE-cadherin at the adherens junction releases β-catenin which then translocates into the nucleus and promotes transcription of genes responsible for cell proliferation. In addition, the reduction of VE-cadherin can activate a multiple number of caspases and up-regulate apoptosis, which can form a positive feedback loop and further decreasing VE-cadherin expression by shedding [120–122]. In addition to its endoribonuclease activity, phosphorylated IRE1 also leads to activation of JNK, through TRAF2 (tumor necrosis factor-receptor-associated factor 2) and ASK1 (apoptosis signal-regulating kinase 1), which can lead to apoptosis via caspase 12 activation and inflammation by association of IKK with the IRE1-TRAF2 complex and activating IKK-NF-κB signaling.
(3) PERK: BIP dissociation causes dimerization and autophosphorylation of PERK and activated PERK phosphorylates Ser51 of eukaryotic translation initiation factor 2a (eIF2a), causing translation arrest of most proteins except ATF4. The phosphorylation of eIF2a up-regulates the expression of ATF4, which translocates to the nucleus and induces apoptosis via increasing CHOP expression. Phosphorylation of eIF2a induced by PERK activation induces NF-κB activation [123]. Although it has been reported that u-flow can inhibit eIF2a phosphorylation [124] and subsequent NF-κB activation, the exact mechanism remains unclear.
In summary, a low level of ER stress can be a protective mechanism for cells, because it accelerates degradation of misfolded proteins (via ERAD), impairs new protein synthesis (via PERK-mediated eIF2a phosphorylation), and induces the expression of chaperons and folders to promote protein folding (via ATF6 and IRE1-mediated sXBP-1 induction). However, excess ER stress enhances apoptosis signaling via the two axes of UPR signaling networks of PERK and ATF6/IRE1. In addition, d-flow-induced EC proliferation can be explained by sXBP-1-mediated VE-cadherin reduction.
SUMOylation
SUMOylation is a post-translational modification in which a SUMO (small ubiquitin-like modifier) protein is covalently conjugated to a lysine residue of a target protein. SUMOylation regulates various protein functions, including sub-cellular localization, protein–protein interaction, and DNA binding amongst others. SUMOylation is also important for a wide range of cellular processes including transcriptional regulation, epigenetic regulation, signal transduction, cell proliferation, apoptosis, and cell cycle regulation [125]. There are four SUMO isoforms (SUMO 1–4) in human, and SUMO1 shares ~50% sequence identity with SUMO2, while SUMO2 and SUMO3 differ by only three NH2-terminus residues (97% identity). SUMO2/3 are considered to form a subfamily, and their function is thought to be different from that of SUMO1 based on the sequence difference. SUMO1-3 are ubiquitously expressed, whereas SUMO4 is limited to kidneys, lymph nodes, and spleen [126, 127]. The functional role of SUMO4 remains controversial, because the COOH-terminus proline residue, which is unique to SUMO4, inhibits the maturation of SUMO4 into the conjugatable form (ormodifier) [128]. SUMOylation is achieved via a three-step enzyme process that involves deconjugation and conjugation enzymes (Fig. 7) [129]. The first step is to convert precursor SUMO to mature SUMO by SUMO proteases sentrin/SUMO-specific proteases (SENPs), whose proteolytic action exposes the di-glycine COOH-terminus motif. The processed SUMO is then activated by the E1-activating enzyme (SAE1–SAE2 heterodimer) in an ATP-dependent manner [130]. In the second step, activated SUMO is transferred to the E2-conjugating enzyme (Ubc9), forming a thioester bond between Ubc9 and SUMO. In the third and final steps, SUMO E3 ligases catalyze the transfer of SUMO from the Ubc3-SUMO complex to the target substrate containing the free e-amino group of a lysine residue [131]. Reversibility of protein SUMOylation is achieved by de-SUMOylation enzymes called sentrin/SUMO-specific proteases (SENPs; SENP1-7), which are also important for SUMO processing. SENP1 and 2 contain both nuclear localization and export signal domains, and shuttling of SENPs from one compartment of the cell to another has an effect of altering SUMOylation levels in different sub-cellular regions as we will discuss in the subsequent sections in this review. SUMO modification of substrates is known to play a major role in the following molecular events and processes: (1) SUMOylation provides a platform to recruit molecules which non-covalently bind to SUMO via the SUMO-interacting motif (SIM); (2) SUMOylation can promote or block the association of molecules that interact with SUMOylated substrates; (3) SUMOylation can regulate substrate stability by competing for the lysine site with ubiquitination or degradation by recruiting the SUMO-targeted ubiquitin ligase (STUbL) family of proteins to the SUMOylated substrates; and (4) SUMOylation induces conformational changes in proteins, so that their interaction with other molecules can be regulated [132, 133].
Fig. 7.

Regulation of SUMOylation pathway. Protein SUMOylation is regulated by a recycling system that consists of conjugation and deconjugation pathways. SUMO conjugation to target substrates involves an enzymatic cascade that is comprised of three classes of enzymes (E1, E2, and E3). The sentrin/SUMO-specific proteases (SENPs) are responsible for the deconjugation pathway as well as the maturation process of newly synthesized SUMO protein. The primary sub-cellular localization of each SENP is also listed [228]. Reprinted from Heo, Berk, and Abe [36].
Modified from Woo et al. with permission of the publisher [227]
ERK5 SUMOylation
As we have summarized earlier in this review, u-flow-mediated ERK5 activation leads to the up-regulation of ERK5 downstream transcription factors KLF2 and PPARs with resulting anti-inflammatory and athero-protective effects [134]. On the contrary, hydrogen peroxide (H2O2), the most important reactive oxygen species, and advance glycation end products (AGE) promote EC inflammation via inhibiting ERK5 transcriptional activity. To elucidate the molecular mechanism of H2O2/AGE-mediated inhibition of ERK5 transcriptional activity, our group investigated the role of ERK5 SUMOylation (Fig. 8) [135]. When cultured ECs were treated with H2O2, ERK5 was SUMOylated at Lys6 and 22 (K6/22), which subsequently inhibited ERK5 transcriptional activity and the expression of KLF2 and eNOS. We also found that d-flow increased, but u-flow decreased, ERK5 SUMOylation, suggesting that ERK5 SUMOylation can be a key switch to distinguish the response of endothelial cells to these two different types of flow.
Fig. 8.
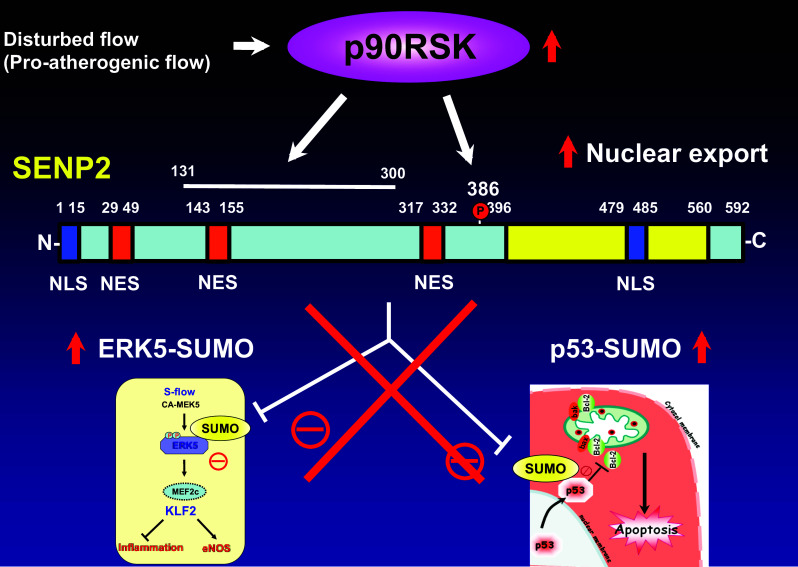
p90RSK regulates SENP2 de-SUMOylation. The increase of ERK5 transcriptional activation is vaso-protective via up-regulating KLF2 and eNOS expression. ERK5 SUMOylation inhibits ERK5/MEF2 transcriptional activity and subsequent KLF2 promoter activity and KLF2-mediated eNOS expression [134]. p53 in the nucleus can be protective against cell death by up-regulating p21 expression [139]. p53 SUMOylation mediates p53 nuclear export, and cytoplasmic p53 directly interacts with the Bcl-2 (B cell lymphoma/leukemia-2) family of proteins, and attenuates anti-apoptotic function of these proteins [137]. p90RSK regulates SENP2 de-SUMOylation function by the association at aa131–300 and phosphorylating SENP2-T368, which subsequently induces SENP2 nuclear export. The nuclear export of SENP2 increases nuclear ERK5 and p53 SUMoylation, resulting in d-flow-induced EC inflammation, apoptosis, and consequent plaque formation [144]
p53 SUMOylation
In addition to promoting EC inflammation, d-flow is known to induce EC apoptosis. The increased EC apoptosis results in increased EC turnover [136, 137]. In contrast, u-flow inhibits EC apoptosis [138]. Transcription factor p53 has been demonstrated to play a key role in promoting cell death by up-regulating the expression of pro-apoptotic factors or promoting cell arrest (for DNA repair) when DNA damage occurs to prevent the further DNA damage [139]. The role of p53 in EC proliferation under u-flow was investigated by Lin et al. who observed that the expression of p53 was increased via JNK activation with resulting increases in p21 expression and GADD45 (growth arrest and DNA damage inducible protein 45), causing EC growth arrest [140]. p21 expression is known to inhibit RB phosphorylation thus inhibiting progression of cells into S phase. GADD45 causes cell arrest in the G0–G1 and G2–M phases of the cell cycle. Of note, in addition to inhibiting cell proliferation, p21 can inhibit apoptosis via down-regulating glutathione peroxidase and superoxide dismutase anti-oxidant activity [139, 141, 142]. Taken together, p53 causes EC growth arrest during u-flow which could also inhibit EC apoptosis [140]. Importantly, it should be noted that these effects of p53 on ECs occur when p53 is localized within the nucleus.
In contrast to u-flow, when cultured ECs are exposed to d-flow, we have found that p53 is exported from the nucleus to the cytoplasm and that this nuclear export is regulated by PKCζ-mediated p53 SUMOylation (Fig. 9) [137]. First, d-flow activates PKCζ via increasing reactive oxygen species production. The activation of PKCζ enables its COOH-terminus kinase domain (aa401–587) to associate with the RING domain of PIASy (protein inhibitor of activated STATy). The PIASy RING domain contains the catalytic SUMO ligase site, and the PIASy-PKCζ association increases PIASy enzymatic activity, probably by changing its tertiary structure. As the result, the PIASy-PKCζ association causes an increase in p53 SUMOylation. Once SUMOylated, p53 is exported to the cytoplasm from the nucleus [137]. In the cytoplasm, p53 induces apoptosis through a direct interaction with Bax and Bcl-2, which blocks the Bax/Bcl-2 anti-apoptotic function and subsequently induces EC apoptosis (Fig. 8) [137, 143]. These data suggest the crucial role of p53 SUMOylation in regulating d-flow-induced EC apoptosis (Fig. 9).
Fig. 9.

Athero-prone flow (d-flow) increases p53SUMOylation via PKCζ-PIAS4 binding. a Athero-prone flow uniquely induces PKCζ activation, which increases PKCζ-PIAS4 binding and PIAS4-SUMO E3 ligase activity, and subsequently increases p53-SUMOylation. p53-SUMOylation causes p53 nuclear export and binds to Bcl-2, which inhibits anti-apoptotic function of Bcl-2 and increases apoptosis. Reprinted from Abe and Berk [229], and obtained permission to reproduce from Wolters Kluwer Health. b Athero-prone flow uniquely activates PKCζ, which increases PKCζ-PIAS4 binding at the SP-RING domain and PIAS4 small ubiquitin-like modifier (SUMO) E3 ligase activity, subsequently increasing p53 SUMOylation. PIAS protein inhibitor of activated STAT, SAP scaffold attachment factor-A/B, acinus, and PIAS domain, PINIT Pro-Ile-Asn-Ile-Thr motif, SP-RING Siz/PIAS-RING domain [227, 229].
Reprinted from Heo, Berk, and Abe [36]
Role of SENP2 in d-flow-induced ERK5 and p53 SUMOylation
SUMO-specific proteases (SENPs) are required for processing inactive pro-form SUMO proteins to mature forms for conjugation as well as controlling deconjugation from substrate proteins [129]. Of the six SENP isoforms that exist in humans (SENP 1–3 and SENP 5–7), we characterized the functional role of SENP2 in controlling d-flow-induced SUMOylation of ERK5 and p53 [135]. When SENP2 is deleted, d-flow-induced EC inflammation and apoptosis are up-regulated. In addition, the accelerated EC inflammation and apoptosis are likely due to increased SUMOylation of p53 and ERK5, respectively. This notion is supported by experiments using SUMOylation mutants of ERK5 and p53, which, when overexpressed in cultured ECs, inhibit d-flow-induced EC inflammation, EC apoptosis as well as the expression of inflammatory cell adhesion molecules. Moreover, aortas from SENP2 knock-out mice exhibit increased atherosclerotic plaque formation. As such, one might expect d-flow to decrease SENP2 expression. However, the expression of SENP2 is not regulated by d-flow. Because SENP2 has several nuclear localizations (NLS) and nuclear export signals (NES), one possibility is that SENP2 localization could affect its ability to de-SUMOylate substrates in different sub-cellular compartments. Our group has reported the significant role of p90RSK (p90 kDa ribosomal S6 kinases) in accelerating SENP2 nuclear export [144] via up-regulating SENP2 T368 phosphorylation [135, 144].
p90RSK is a unique serine/threonine kinase that contains two functional catalitic domains: the NH2-terminus and the COOH-terminus kinase domains [145]. The NH2-terminus kinase domain appears to belong to the AGC group of kinases (i.e., PKC and PKA) and phosphorylates its substrates, while the COOH-terminus kinase domain is a member of the calcium/calmodulin-dependent kinase group and regulates mainly the activation of the p90RSK NH2-terminus kinase [146]. Recently, we identified de-SUMOylation enzyme SENP2 as one of the substrates for p90RSK [144]. p90RSK phosphorylates SENP2 T368, and increases SUMOylation of p53 and ERK5 in the nucleus (Fig. 8). We assume that the nuclear export of SENP2 induced by p90RSK may also contribute to up-regulate nuclear p53 and ERK5 SUMOylation. In this study, we also established the role of the endothelial p90RSK-SENP2 module in regulating d-flow-induced atherosclerotic plaque formation in mice [144].
Other SUMOylation events, which are crucial for regulating endothelial functions
MK2 (MAPK-activated protein kinase-2)
MK2 is a pro-inflammatory kinase that enhances NF-κB activity by blocking nuclear retention of p38 and preventing excessive phosphorylation of MSK1 (mitogen- and stress-activated protein kinase 1). Since MSK1 phosphorylates p65 to facilitate the nuclear export of the latter, reduced MSK1 activity results in the nuclear retention and sustained activation of NF-κB. In fact, it has been reported that the depletion of MK2 significantly decreases TNF-α-induced inflammatory responses in ECs and plaque formation in Ldlr−/− mice with the reduced expression of VCAM-1 and MCP-1, suggesting that activation of endothelial MK2 is pro-atherogenic [147]. Our group has previously reported that MK2 K339 SUMOylation inhibits its kinase activity and subsequent actin filament remodeling and TNFα-mediated inhibition of EC migration [148]. Although the role of MK2 SUMOylation in endothelial inflammation remains unclear, these data suggest that MK2 SUMOylation can regulate endothelial functions.
IκB and NEMO/IKKγ
In endothelial cells, NF-κB plays a crucial role in increasing EC inflammation and has a pro-atherogenic role. NF-κB is the collective name for a family of transcription factors with five members: c-Rel, relB, p65 (relA), p105/p50, and p100/p52. NF-κB dimers are kept inactive by association with inhibitory proteins, the inhibitors of NF-κB (IκBs). When inactive, NF-κB is in the cytoplasm and is bound to IκB. To activate NF-kB, IκB kinase (IKK) is first activated by pro-inflammatory stimuli, including d-flow and cytokines, and phosphorylates IκB which is then ubiquitinated and subjected to proteasomal degradation. Freed NF-κB then translocates into the nucleus and transactivates pro-inflammatory genes [149]. IKKs form a complex with two kinases, called IKK1 (or IKKα) and IKK2 (or IKKβ), and a regulatory subunit named NF-κB essential modulator (NEMO or IKKγ). NEMO has no catalytic activity but is required for both IKK activation and its subsequent phosphorylation of IκB [150]. When NEMO is deleted, IKKs cannot efficiently phosphorylate IB and activate NF-κB signaling [151]. Gareus et al. have reported that endothelial specific depletion of NEMO or dominant negative IkBa inhibited atherosclerotic plaque formation, demonstrating the crucial role of NF-κB signaling in atherosclerosis [152].
SUMOylation of IκBα and NEMO has been reported. SUMO1 modified IκBα is protected from ubiquitination and degradation, thus down-regulating NF-κB activation [153]. In contrast, modification of IκBα by SUMO2/3 confers the opposite effect and dissociates IκBα from NF-κB, leading to NF-κB activation [154]. However, the exact vascular pathophysiological role of IκBα SUMOylation remains unclear.
Miyamoto and his colleagues have reported that genotoxic stress induces SUMOylation (SUMO1) of NEMO, leading to increased IKK-NF-κB activation [155–161]. Interestingly, a de-SUMOylation enzyme, SENP2, is a downstream transcriptional target of NF-κB thereby creating a negative feedback mechanism for NF-κB activation through NEMO de-SUMOylation [162]. SENP6 inhibits Toll-like receptor-triggered inflammation via de-SUMOylation of NEMO at K277 [163]. However, the role of NEMO SUMOylation in flow-induced signaling and atherosclerotic plaque formation has not been well-characterized and awaits further investigation.
Adenosine monophosphate-activated protein kinase (AMPK) and LKB1
AMPK is a stress-activated kinase which can orchestrate the cellular response to a variety of stresses in the heart by regulating metabolism, protein synthesis, degradation, autophagy, and apoptosis [164]. AMPK is a complex of three subunits: a catalytic subunit (α) containing a serine–threonine kinase domain (KD) with a Thr172 phosphorylation site which is the target of liver kinase B1 (LKB1) and calcium-calmodulin-activated protein kinase kinase-β (CAMKKβ) and two regulatory subunits (β and γ) [164]. In unstressed cells, AMPK is mainly localized in the cytoplasm but can translocate to the nucleus after its activation [164]. It has been reported that u-flow but not d-flow activates AMPK. The s-flow-induced AMPK activation inhibits Akt-mTOR-S6K signaling and subsequently causes EC arrest in the G0/G1 phase [165, 166]. In addition, AMPK can suppress ER stress by decreasing mitochondrial ROS production and activating eNOS [166, 167]. The role of AMPK in regulating EC inflammation and subsequent atherogenesis is isoform dependent; AMPKα1 is pro-inflammatory and AMPKα2 is anti-inflammatory. Indeed, reduced atherosclerotic plaque formation was reported in AMPKα1−/−/ApoE−/− [168], while it increased in AMPKα2−/−/LDLR−/− mice [166]. One of the substrates of AMPKα2 is poly [ADP ribose] polymerase 1 (PARP1) [169]. Originally, PARP1 was recognized as the molecule that utilized NAD+ to synthesize poly(ADP)-ribose (PAR) and “PARylated” itself and other nuclear proteins [170]. In addition to PARylation, PARP1 is now known to directly associate with a specific DNA sequence TTGATATAAAT within target genes vis its zinc-finger domain [171]. PARP1 is involved in multiple cellular functions, such as repair of single- and double-strand DNA breaks, RNA interference, mitochondrial function, and cell division. In terms of inflammation, PARP1 can play as a coactivator of NF-kB and activate pro-inflammatory pathways, such as p38 MAPK and JNK pathways, and inhibit B cell lymphoma–6 (Bcl-6)-induced anti-inflammatory function [169, 172–176]. In addition, PARP-1 represses Bcl-6 transcription by sequence-specific binding to the first intron of the Bcl-6 gene, and PARP-1 knockdown induces Bcl-6 expression [171]. Shyy and his associates have reported that AMPKα2 phosphorylates PARP1, which then dissociates from the Bcl-6 intron 1. This up-regulates Bcl-6 transcription, and in ECs, the increased Bcl-6 expression inhibits the expression of VCAM-1, MCP-1, and MCP-3 and down-regulates inflammatory responses [169]. In contrast, AMPKα1 has been suggested to mediate the expression of pro-inflammatory cytokines by activating NF-κB [177, 178]. Further investigation is necessary to determine how AMPKα1 and AMPKα2 play opposite roles in regulating NF-kB activation.
U-flow increases LKB1 activity. Zhang et al. have reported that endothelium-specific LKB1 deletion decreases eNOS activity and impairs endothelial functions, causing hypertension and cardiac hypertrophy in mice [179]. They also found that the depletion of LKB1 inhibited AMPK activation and induced human antigen R (HuR) nuclear export. HuR binds to and stabilizes caveolin-1 mRNA and enhances caveolin-1 expression, and at the same time, it inhibits eNOS catalytic activity. It is unclear what the functional relationship is between LKB1 and the two AMPK isoforms in ECs exposed to d-flow.
Rubio et al. have reported that AMPKβ2 SUMOylation by SUMO2 activates AMPK and inhibits its ubiquitin-dependent degradation [180]. In contrast, AMPKα1 SUMOylation inhibits AMPK activity specifically for activating mTORC1 signaling [181]. It remains unclear how AMPKα1 and AMPKα2 SUMOylation coordinately regulate AMPK kinase activity. Yeh and his group have recently shown that LKB1 K178 SUMO1 modification promotes LKB1 association with AMPKα SUMO-interacting motif (SIM) and accelerates AMPK activation [182].
Dynamin-related protein 1 (DRP1)
It has been reported that aberrant flow inhibits the function of mitochondrial respiratory complexes I, II/III, and IV as early as 5 min from the flow onset, and this inhibition is due to increased production of ONOO− [183]. In addition, aberrant flow can induce mitochondrial fission via regulating DRP-1 sub-cellular localization. Mitochondria are dynamic organelles, and their overall morphology changes in response to cellular activity via processes called fission and fusion (i.e., elongation) [184]. These morphological changes are under the tight regulation dictated by the physiological state of the cell [185, 186]. Mitochondrial fusion is described as integrating the outer and inner membranes of one mitochondrium with those of another mitochondrium, while mitochondrial fission is achieved by pinching off of a mitochondrium. The crucial role of the GTPase called DRP1 in the process of fission has been well established [187, 188].
SUMOylation of DRP1 regulates this GTPase and thus also regulates mitochondrial fission. DRP1 SUMOylation (SUMO1) up-regulates mitochondrial fission and causes hyper-fragmentation of mitochondria [189–191]. In Cos7 and HeLa cells, mitochondrial hyper-fragmentation resulted in apoptosis [192–194]. Cytochrome c functions as the terminal trigger for apoptotic cell death and is located in the intermembrane space of mitochondria [195]. Release of cytochrome c has been shown to depend on SUMOylation (SUMO1) of DRP1 [196]. The level of DRP1 SUMOylation can be reduced by de-SUMOylation enzymes, including SENP5, SENP3, and SENP2 [197, 198]. DRP1 can be SUMOylated not only by SUMO1 but also by SUMO2 and 3. The functional consequence of DRP1 SUMO1 vs SUMO2/3 modification can be different.
Other post-translational modifications of ERK5
ERK5 S496 phosphorylation by p90RSK
Upon activation, p90RSK is able to phosphorylate transcription factors, such as CREB, NF-κB, and c-fos, and increases their transcriptional activity [85, 144, 199]. In our study, we found that H2O2 (used as ROS) activated p90RSK, leading both to p90RSK binding to the COOH-terminal transcriptional domain (aa 571–807) of ERK5 and to phosphorylation of ERK5 S496 with the subsequent inhibition of ERK5 transcriptional activity [85]. The inhibition of ERK5 transcriptional activity decreased the expression of KLF2 and eNOS but increased adhesion molecule expression, leading to EC dysfunction with accelerated atherosclerotic plaque formation. The same phenotypes were observed in EC-specific ERK5 knock-out mice. The role of p90RSK in EC dysfunction was further studied using the specific p90RSK inhibitor, FMK-MEA. In this study, we found that the inhibitor decreased adhesion molecule expression and increased eNOS expression, which are anti-atherogenic phenotypes. However, when EC-specific ERK5 knock-out mice were treated with FMK-MEA, the anti-atherogenic effects of FMK-MEA were no longer observed. Our study suggests that ERK5 is necessary for the p90RSK inhibitor to exert its anti-atherogenic effect. Taken together, we have demonstrated that d-flow-induced p90RSK activation (higher expression noted in aortic arch) inhibits ERK5 transcriptional activity, which is critical for atherosclerotic plaque formation, and the inhibition of this process by FMK-MEA prevented atherosclerotic plaque formation [85].
ERK5 S486 phosphorylation by PKCζ and p62
PKC (protein kinase C) is a family of serine–threonine kinases that phosphorylate numerous proteins, thus regulating their intercellular signaling. One isomer of this family is PKCζ, which, as previously mentioned, has been associated with d-flow-induced p53-SUMOylation with resulting EC apoptosis. In addition, we found that PKCζ can phosphorylate ERK5 S486, which then triggers degradation of eNOS protein, although the precise mechanism that ties these events will require future studies. Our group has also found that p62 is an important molecule implicated in TNFα-mediated PKCζ activation [200]. p62, a scaffold protein, contains a PB1 (Phox/Bem1p) domain on its NH2-terminus region that can interact with other PB1 domain-containing proteins [201]. PKCζ also has a PB1 domain, and we have found that TNFα induces p62 association with PKCζ, thus activating PKCζ and resulting in EC apoptosis. The p62-PKCζ interaction is required for PKCζ-induced JNK activation and caspase three cleavage, but how this association fits into the d-flow and u-flow-induced signaling needs further investigation [200]. Evidence provided by these studies suggests that PKCζ activation mediates EC inflammation and apoptosis and plays an important role in atherogenesis.
Blood flow and epigenetics
Epigenetics is defined as the modification in gene expression, usually through alterations in the DNA conformation without changing the DNA code itself [202]. The most studied epigenetic alterations are DNA methylation and histone modifications. Genomic DNA is found in two forms: heterochromatin and euchromatin. Heterochromatin is a tightly packed, highly methylated DNA associated with the events of histone deacetylation. In contrast, euchromatin consists of unmethylated DNA arranged in a more loose conformation in association with acetylated histones [203]. Fluid shear stress has been shown to regulate chromatin remodeling and histone modifications through several signaling events involving methylation, phosphorylation, SUMOylation, and acetylation [204, 205]. In this section, we will focus on the role of d-flow in regulating DNA methylation and histone acetylation.
DNA methylation
In mammals, DNA methylation occurs in a 5′ carbon of a cytosine base pair usually in the setting of CpG (cytosine-phosphate-guanine) dinucleotides [206, 207]. CpG islands are regions made of at least 200 base pairs, a CpG percentage greater than 50%, and an observed CpG/expected CpG in excess of 0.6 [208]. Around 50% of human genes contain CpG islands in their promoter regions, these islands are usually unmethylated and, therefore, transcriptionally active [209, 210]. Methylation of a cytosine residue at the promoter region results in transcriptional gene silencing [211]. Apart from CpG islands, CG sites are scant. This occurs, because genomic 5-methylcytosine residues are hotspots for spontaneous deamination into thymine, a phenomenon known as CG suppression [212]. DNA methyltransferases are the enzymes that catalyze the transfer of methyl groups to DNA. There are three main enzymes located on different chromosomes: DNMT1, DNMT3A, and DNMT3B. DNMT3A and DNMT3B are known as de novo methyltransferases. These enzymes can add methyl groups to cytosine residues of completely unmethylated DNA. DNMT1 is known as a maintenance methylase, because this enzyme can only add methyl groups to cytosine residues of partially methylated DNA [213]. DNMT1 has also been found to have de novo methyltransferase activity [214]. Jiang et al. reported a down-regulation in KLF4 expression in swine and human aortic ECs exposed to d-flow [215]. They found that d-flow increases the expression of DNMT3A, which methylates the CpG islands at the myocyte enhancer factor binding site within the KLF-4 promoter. Hyper-methylation of this site leads to decreased interaction of MEF2 with the KLF-4 promoter, abolishing KLF4 expression. In addition to KLF2, KLF4 is also one of the zinc-finger regulatory transcription factors for the gene networks that confer the athero-resistant, anti-inflammatory, and anti-thrombotic properties to the endothelium [216–218]. Therefore, the reduction of KLF4 induced by the hyper-methylation can be pro-atherogenic. The inhibitory effect of d-flow on KLF4 transcription is mitigated by RG-108 and 5-Aza (DNMT inhibitors) and DNMT3A knockout [204, 215]. In addition to DNMT3A, Dunn et al. have found that d-flow created by a partial carotid ligation in mice also up-regulates DNMT1 expression in ECs [204]. They have also reported that 5-Aza is able to inhibit d-flow-induced atherosclerosis and that d-flow induces an increase in DNA methylation in 11 gene promoters (HOXA5, KLF3, TMEM184B, ADAMTSl5, CMKRL1, PKP4, ACVRL1, DOK4, SPRY2, ZFP46, and F2RL1), which can be reversed by 5-Aza. Interestingly, the methylated region in 5 of the 11 genes (HOXA5, KLF3, CMKLR1, ACVRL1, and SPRY2) contained a cAMP response element (CRE) in their promoters, and these investigators were able to confirm that THOXA5, KLF3, CMKLR1, and ACVRL1 genes were hyper-methylated by d-flow at the CRE CG site. Zhou et al. using human umbilical vein endothelial cells (HUVECs) also found that d-flow increased DNMT1 expression and DNMT1 nuclear translocation [219]. These data suggest that DNA methylation plays a crucial role in regulating d-flow-induced endothelial inflammation and dysfunction.
Histone acetylation
Histones are the basic proteins associated with DNA, forming nucleosomes. Eight core histone proteins consisting of two H2A/H2B dimers and two H3/H4 dimers form nucleosomes. Each nucleosome is made of DNA wrapped around a spool-like structure of the eight histones. The chain of nucleosomes is then wrapped into a 30 nm spiral called a solenoid, where additional H1 histone proteins are associated with each nucleosome to maintain the chromosome structure. Post-translational modifications of the histone NN2-terminus can modulate its interaction with DNA, which is known as the “histone code”.
Certain lysine residues of histone are subjected to acetylation and deacetylation and each catalyzed by histone acetyltransferases (HATs) and deacetylases (HDACs), respectively [205, 220]. There are multiple proteins with HAT activity, which are divided into three family groups: GNAT, MYST, and CBP/p300 families. Usually, histone acetylation is associated with transcription activation. For example, the possible role of p300 HAT in s-flow-induced eNOS expression has been reported [221]. U-flow increases the ability of NF-κB subunits p50 and p65 to bind the eNOS promoter. p300 HAT activation induced by u-flow increases acetylation of both p65 and histones 3 and 4 localized in the proximity of the eNOS shear stress response element (SSRE). This acetylation of p65 and histones opens the chromatin structure at the SSRE and induces eNOS mRNA expression [221]. D-flow-induced H3 lysine 14 acetylation has been reported, [205], but the pathological meaning and regulatory mechanism remain unclear. In addition, roles of GNAT and MYST in flow-mediated signaling are not clear.
In contrast to histone acetylation, histone deacetylation is associated with gene silencing. HDACs are categorized into class I (HDAC1/2/3) and class II (HDAC5/7), and both expression and nuclear accumulation of class I and II HDACs were induced by d-flow [222]. In addition, the association of HDAC-1/2/3 with NF-E2-related factor-2 (NRF2) and HDAC-3/5/7 with myocyte enhancer factor-2 (MEF2) is induced by d-flow, and these associations down-regulate the activation of NADPH quinone oxidoreductase-1, an anti-oxidant gene, and KLF2, an anti-inflammatory gene, respectively [222]. In contrast, u-flow dissociates HDAC5 from MEF2C and promotes MEF2-dependent KLF2 gene transcription [223]. These data suggest that HDACs play crucial roles in regulating flow-induced EC responses.
Conclusion
It is clear that ECs are differently regulated by different blood flow patterns: d-flow activates pro-atherogenic signaling, while u-flow induces athero-protective responses in ECs. Each flow type activates different signaling molecules, leading to different sub-cellular localizations of the same molecule and different patterns of gene expression, both of which regulate the proliferation, differentiation, apoptosis, and inflammation of endothelial cells. Thus, the two types of flow are great experimental tools for investigating the pathogenesis of atherosclerosis and other vascular dysfunctions. In fact, as we have discussed in this review, many fruitful accomplishments have been achieved from various flow-based studies, including differential kinase activation, inflammasome activation, DNA methylation, and post-translational modifications of signaling proteins. One of the important future goals will be to determine the interplay among molecular events activated by a specific flow type. How they coordinately regulate and decide the overall cell response under u-flow and d-flow. Although this will be a very complex process, the approach by comparing the signaling events under u-flow and d-flow is still offering us a great opportunity to understand the process of atherosclerosis induced by endothelial dysfunction.
Acknowledgements
The work was supported by Grants from the National Institute of Health to Dr. Abe (HL-130193, HL-123346, HL-118462), and from American Heart Association to Dr. Le (AHA 13SDG14500033).
Compliance with ethical standards
Conflict of interest
No conflicts of interest or financial disclosures to disclose.
References
- 1.Abe J, Berk BC. Reactive oxygen species as mediators of signal transduction in cardiovascular disease. Trends Cardiovasc Med. 1998;8:59–64. doi: 10.1016/S1050-1738(97)00133-3. [DOI] [PubMed] [Google Scholar]
- 2.Abe J, Baines CP, Berk BC. Role of mitogen-activated protein kinases in ischemia and reperfusion injury: the good and the bad. Circ Res. 2000;86:607–609. doi: 10.1161/01.RES.86.6.607. [DOI] [PubMed] [Google Scholar]
- 3.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 4.Tseng H, Peterson TE, Berk BC. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ Res. 1995;77:869–878. doi: 10.1161/01.RES.77.5.869. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi M, Berk BC. Mitogen-activated protein kinase (ERK1/2) activation by shear stress and adhesion in endothelial cells. Essential role for a herbimycin-sensitive kinase. J Clin Invest. 1996;98:2623–2631. doi: 10.1172/JCI119083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishida T, Peterson T, Kovach NL, Berk BC. Integrins modulate fluid shear stress signal transduction in endothelial cells. Circulation (abstract) 1995;92:I-629. [Google Scholar]
- 7.Ishida T, Takahashi M, Corson MA, Berk BC. Fluid shear stress-mediated signal transduction: how do endothelial cells transduce mechanical force into biological responses? Ann N Y Acad Sci. 1997;811:12–23. doi: 10.1111/j.1749-6632.1997.tb51984.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Duan Y, Yang X, Sun L, Liu M, Wang Q, Ma X, Zhang W, Li X, Hu W, Miao RQ, Xiang R, Hajjar DP, Han J. Inhibition of ERK1/2 and activation of LXR synergistically reduce atherosclerotic lesions in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2015;35:948–959. doi: 10.1161/ATVBAHA.114.305116. [DOI] [PubMed] [Google Scholar]
- 9.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amini N, Boyle JJ, Moers B, Warboys CM, Malik TH, Zakkar M, Francis SE, Mason JC, Haskard DO, Evans PC. Requirement of JNK1 for endothelial cell injury in atherogenesis. Atherosclerosis. 2014;235:613–618. doi: 10.1016/j.atherosclerosis.2014.05.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, le Luong A, Mason JC, Udalova I, Gsell W, Jones H, Haskard DO, Krams R, Evans PC. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ Res. 2011;108:950–959. doi: 10.1161/CIRCRESAHA.110.233841. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhury H, Zakkar M, Boyle J, Cuhlmann S, van der Heiden K, le Luong A, Davis J, Platt A, Mason JC, Krams R, Haskard DO, Clark AR, Evans PC. c-Jun N-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arterioscler Thromb Vasc Biol. 2010;30:546–553. doi: 10.1161/ATVBAHA.109.201368. [DOI] [PubMed] [Google Scholar]
- 13.Zakkar M, Chaudhury H, Sandvik G, Enesa K, le Luong A, Cuhlmann S, Mason JC, Krams R, Clark AR, Haskard DO, Evans PC. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. 2008;103:726–732. doi: 10.1161/CIRCRESAHA.108.183913. [DOI] [PubMed] [Google Scholar]
- 14.De Cesaris P, Starace D, Starace G, Filippini A, Stefanini M, Ziparo E. Activation of Jun N-terminal kinase/stress-activated protein kinase pathway by tumor necrosis factor alpha leads to intercellular adhesion molecule-1 expression. J Biol Chem. 1999;274:28978–28982. doi: 10.1074/jbc.274.41.28978. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad M, Theofanidis P, Medford RM. Role of activating protein-1 in the regulation of the vascular cell adhesion molecule-1 gene expression by tumor necrosis factor-alpha. J Biol Chem. 1998;273:4616–4621. doi: 10.1074/jbc.273.8.4616. [DOI] [PubMed] [Google Scholar]
- 16.Min W, Pober JS. TNF initiates E-selectin transcription in human endothelial cells through parallel TRAF-NF-kappa B and TRAF-RAC/CDC42-JNK-c-Jun/ATF2 pathways. J Immunol. 1997;159:3508–3518. [PubMed] [Google Scholar]
- 17.Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26:7222–7230. doi: 10.1038/sj.onc.1210526. [DOI] [PubMed] [Google Scholar]
- 18.Wong HK, Fricker M, Wyttenbach A, Villunger A, Michalak EM, Strasser A, Tolkovsky AM. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol Cell Biol. 2005;25:8732–8747. doi: 10.1128/MCB.25.19.8732-8747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/MCB.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson Jr EM. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/S0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 21.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/S0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 23.Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, Hersberger M, Eriksson U, Eberli FR, Becher B, Boren J, Chen M, Cybulsky MI, Moore KJ, Freeman MW, Wagner EF, Matter CM, Luscher TF. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306:1558–1561. doi: 10.1126/science.1101909. [DOI] [PubMed] [Google Scholar]
- 24.Sumara G, Belwal M, Ricci R. “Jnking” atherosclerosis. Cell Mol Life Sci. 2005;62:2487–2494. doi: 10.1007/s00018-005-5253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denes L, Jednakovits A, Hargitai J, Penzes Z, Balla A, Talosi L, Krajcsi P, Csermely P. Pharmacologically activated migration of aortic endothelial cells is mediated through p38 SAPK. Br J Pharmacol. 2002;136:597–603. doi: 10.1038/sj.bjp.0704738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMullen ME, Bryant PW, Glembotski CC, Vincent PA, Pumiglia KM. Activation of p38 has opposing effects on the proliferation and migration of endothelial cells. J Biol Chem. 2005;280:20995–21003. doi: 10.1074/jbc.M407060200. [DOI] [PubMed] [Google Scholar]
- 27.Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, Verin AD. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. 2004;287:L911–L918. doi: 10.1152/ajplung.00372.2003. [DOI] [PubMed] [Google Scholar]
- 28.Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001;276:30359–30365. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- 29.Viemann D, Goebeler M, Schmid S, Klimmek K, Sorg C, Ludwig S, Roth J. Transcriptional profiling of IKK2/NF-kappa B- and p38 MAP kinase-dependent gene expression in TNF-alpha-stimulated primary human endothelial cells. Blood. 2004;103:3365–3373. doi: 10.1182/blood-2003-09-3296. [DOI] [PubMed] [Google Scholar]
- 30.Milkiewicz M, Kelland C, Colgan S, Haas TL. Nitric oxide and p38 MAP kinase mediate shear stress-dependent inhibition of MMP-2 production in microvascular endothelial cells. J Cell Physiol. 2006;208:229–237. doi: 10.1002/jcp.20658. [DOI] [PubMed] [Google Scholar]
- 31.Kardakaris R, Gareus R, Xanthoulea S, Pasparakis M. Endothelial and macrophage-specific deficiency of P38alpha MAPK does not affect the pathogenesis of atherosclerosis in ApoE−/− mice. PLoS One. 2011;6:e21055. doi: 10.1371/journal.pone.0021055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI200419890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94:362–369. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 34.Berk BC. Atheroprotective signaling mechanisms activated by steady laminar flow in endothelial cells. Circulation. 2008;117:1082–1089. doi: 10.1161/CIRCULATIONAHA.107.720730. [DOI] [PubMed] [Google Scholar]
- 35.Yan C, Takahashi M, Okuda M, Lee JD, Berk BC. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells Dependence on tyrosine kinases and intracellular calcium. J Biol Chem. 1999;274:143–150. doi: 10.1074/jbc.274.1.143. [DOI] [PubMed] [Google Scholar]
- 36.Heo KS, Berk B, Abe JI. Disturbed flow-induced endothelial pro-atherogenic signaling via regulating post-translational modifications and epigenetic events. Antioxid Redox Signal. 2015;25:435. doi: 10.1089/ars.2015.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Tatake RJ, Natarajan K, Taba Y, Garin G, Tai C, Leung E, Surapisitchat J, Yoshizumi M, Yan C, Abe J, Berk BC. Fluid shear stress inhibits TNF-mediated JNK activation via MEK5-BMK1 in endothelial cells. Biochem Biophys Res Commun. 2008;370:159–163. doi: 10.1016/j.bbrc.2008.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci U S A. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan G, Merritt SE, Kortenjann M, Shaw PE, Holzman LB. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J Biol Chem. 1996;271:24788–24793. doi: 10.1074/jbc.271.40.24788. [DOI] [PubMed] [Google Scholar]
- 40.Carter CA. Protein kinase C as a drug target: implications for drug or diet prevention and treatment of cancer. Curr Drug Targets. 2000;1:163–183. doi: 10.2174/1389450003349317. [DOI] [PubMed] [Google Scholar]
- 41.Martelli AM, Faenza I, Billi AM, Fala F, Cocco L, Manzoli L. Nuclear protein kinase C isoforms: key players in multiple cell functions? Histol Histopathol. 2003;18:1301–1312. doi: 10.14670/HH-18.1301. [DOI] [PubMed] [Google Scholar]
- 42.Shieh BH, Parker L, Popescu D. Protein kinase C (PKC) isoforms in Drosophila. J Biochem. 2002;132:523–527. doi: 10.1093/oxfordjournals.jbchem.a003252. [DOI] [PubMed] [Google Scholar]
- 43.Storz P. Targeting protein kinase C subtypes in pancreatic cancer. Expert Rev Anticancer Ther. 2015;15:433–438. doi: 10.1586/14737140.2015.1003810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleming I, Mohamed A, Galle J, Turchanowa L, Brandes RP, Fisslthaler B, Busse R. Oxidized low-density lipoprotein increases superoxide production by endothelial nitric oxide synthase by inhibiting PKCalpha. Cardiovasc Res. 2005;65:897–906. doi: 10.1016/j.cardiores.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Lei S, Su W, Liu H, Xu J, Xia ZY, Yang QJ, Qiao X, Du Y, Zhang L, Xia Z. Nitroglycerine-induced nitrate tolerance compromises propofol protection of the endothelial cells against TNF-alpha: the role of PKC-beta2 and NADPH oxidase. Oxid Med Cell Longev. 2013;2013:678484. doi: 10.1155/2013/678484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lei S, Li H, Xu J, Liu Y, Gao X, Wang J, Ng KF, Lau WB, Ma XL, Rodrigues B, Irwin MG, Xia Z. Hyperglycemia-induced protein kinase C beta2 activation induces diastolic cardiac dysfunction in diabetic rats by impairing caveolin-3 expression and Akt/eNOS signaling. Diabetes. 2013;62:2318–2328. doi: 10.2337/db12-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu M, Chen J, Jiang H, Miao C. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc Diabetol. 2013;12:13. doi: 10.1186/1475-2840-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng B, Xie S, Wang J, Xia Z, Nie R. Inhibition of protein kinase C beta(2) prevents tumor necrosis factor-alpha-induced apoptosis and oxidative stress in endothelial cells: the role of NADPH oxidase subunits. J Vasc Res. 2012;49:144–159. doi: 10.1159/000332337. [DOI] [PubMed] [Google Scholar]
- 49.Wang F, Liu HM, Irwin MG, Xia ZY, Huang Z, Ouyang J, Xia Z. Role of protein kinase C beta2 activation in TNF-alpha-induced human vascular endothelial cell apoptosis. Can J Physiol Pharmacol. 2009;87:221–229. doi: 10.1139/Y09-004. [DOI] [PubMed] [Google Scholar]
- 50.Kouroedov A, Eto M, Joch H, Volpe M, Luscher TF, Cosentino F. Selective inhibition of protein kinase Cbeta2 prevents acute effects of high glucose on vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 2004;110:91–96. doi: 10.1161/01.CIR.0000133384.38551.A8. [DOI] [PubMed] [Google Scholar]
- 51.Ergin V, Erdogan M, Karasu C, Menevse A. 4,5-dianilinophtalimide protects neuroendocrine cells against serum deprivation-induced stress and apoptosis. Neuro Endocrinol Lett. 2013;34:359–365. [PubMed] [Google Scholar]
- 52.Zhu M, Chen J, Wen M, Sun Z, Sun X, Wang J, Miao C. Propofol protects against angiotensin II-induced mouse hippocampal HT22 cells apoptosis via inhibition of p66Shc mitochondrial translocation. Neuromol Med. 2014;16:772–781. doi: 10.1007/s12017-014-8326-6. [DOI] [PubMed] [Google Scholar]
- 53.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA, Hamburg NM. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park K, Li Q, Rask-Madsen C, Mima A, Mizutani K, Winnay J, Maeda Y, D’Aquino K, White MF, Feener EP, King GL. Serine phosphorylation sites on IRS2 activated by angiotensin II and protein kinase C to induce selective insulin resistance in endothelial cells. Mol Cell Biol. 2013;33:3227–3241. doi: 10.1128/MCB.00506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, Yan SF. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. Faseb J. 2009;23:1081–1091. doi: 10.1096/fj.08-120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong L, Shen X, Lin L, Leitges M, Rosario R, Zou YS, Yan SF. PKCbeta promotes vascular inflammation and acceleration of atherosclerosis in diabetic ApoE null mice. Arterioscler Thromb Vasc Biol. 2013;33:1779–1787. doi: 10.1161/ATVBAHA.112.301113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beckman JA, Goldfine AB, Goldin A, Prsic A, Kim S, Creager MA. Inhibition of protein kinase Cbeta does not improve endothelial function in type 2 diabetes. J Clin Endocrinol Metab. 2010;95:3783–3787. doi: 10.1210/jc.2010-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Monti M, Donnini S, Giachetti A, Mochly-Rosen D, Ziche M. deltaPKC inhibition or varepsilonPKC activation repairs endothelial vascular dysfunction by regulating eNOS post-translational modification. J Mol Cell Cardiol. 2010;48:746–756. doi: 10.1016/j.yjmcc.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 60.Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem. 2004;279:31883–31890. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- 61.Javaid K, Rahman A, Anwar KN, Frey RS, Minshall RD, Malik AB. Tumor necrosis factor-alpha induces early-onset endothelial adhesivity by protein kinase Czeta-dependent activation of intercellular adhesion molecule-1. Circ Res. 2003;92:1089–1097. doi: 10.1161/01.RES.0000072971.88704.CB. [DOI] [PubMed] [Google Scholar]
- 62.Rahman A, Anwar KN, Malik AB. Protein kinase C-zeta mediates TNF-alpha-induced ICAM-1 gene transcription in endothelial cells. Am J Physiol Cell Physiol. 2000;279:C906–C914. doi: 10.1152/ajpcell.2000.279.4.C906. [DOI] [PubMed] [Google Scholar]
- 63.Magid R, Davies PF. Endothelial protein kinase C isoform identity and differential activity of PKCzeta in an athero-susceptible region of porcine aorta. Circ Res. 2005;97:443–449. doi: 10.1161/01.RES.0000179767.37838.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith L, Chen L, Reyland ME, DeVries TA, Talanian RV, Omura S, Smith JB. Activation of atypical protein kinase C zeta by caspase processing and degradation by the ubiquitin-proteasome system. J Biol Chem. 2000;275:40620–40627. doi: 10.1074/jbc.M908517199. [DOI] [PubMed] [Google Scholar]
- 65.Smith L, Wang Z, Smith JB. Caspase processing activates atypical protein kinase C zeta by relieving autoinhibition and destabilizes the protein. Biochem J. 2003;375:663–671. doi: 10.1042/bj20030926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garin G, Abe J, Mohan A, Lu W, Yan C, Newby AC, Rhaman A, Berk BC. Flow antagonizes TNF-alpha signaling in endothelial cells by inhibiting caspase-dependent PKC zeta processing. Circ Res. 2007;101:97–105. doi: 10.1161/CIRCRESAHA.107.148270. [DOI] [PubMed] [Google Scholar]
- 67.You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. 1999;19:2416–2424. doi: 10.1128/MCB.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fukunaga K, Noguchi T, Takeda H, Matozaki T, Hayashi Y, Itoh H, Kasuga M. Requirement for protein-tyrosine phosphatase SHP-2 in insulin-induced activation of c-Jun NH(2)-terminal kinase. J Biol Chem. 2000;275:5208–5213. doi: 10.1074/jbc.275.7.5208. [DOI] [PubMed] [Google Scholar]
- 69.Neel BG. Structure and function of SH2-domain containing tyrosine phosphatases. Semin Cell Biol. 1993;4:419–432. doi: 10.1006/scel.1993.1050. [DOI] [PubMed] [Google Scholar]
- 70.Che W, Lerner-Marmarosh N, Huang Q, Osawa M, Ohta S, Yoshizumi M, Glassman M, Lee JD, Yan C, Berk BC, Abe J. Insulin-like growth factor-1 enhances inflammatory responses in endothelial cells: Role of Gab1 and MEKK3 in TNF-a-induced c-Jun and NF-kB activation and adhesion molecule expression. Circ Res. 2002;90:1222. doi: 10.1161/01.RES.0000021127.83364.7D. [DOI] [PubMed] [Google Scholar]
- 71.Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 72.Lerner-Marmarosh N, Yoshizumi M, Che W, Surapisitchat J, Kawakatsu H, Akaike M, Ding B, Huang Q, Yan C, Berk BC, Abe JI. Inhibition of tumor necrosis factor-[alpha]-induced SHP-2 phosphatase activity by shear stress: a mechanism to reduce endothelial inflammation. Arterioscler Thromb Vasc Biol. 2003;23:1775–1781. doi: 10.1161/01.ATV.0000094432.98445.36. [DOI] [PubMed] [Google Scholar]
- 73.Young A, Wu W, Sun W, Larman HB, Wang N, Li YS, Shyy JY, Chien S, Garcia-Cardena G. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. Arterioscler Thromb Vasc Biol. 2009;29:1902–1908. doi: 10.1161/ATVBAHA.109.193540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piqueras L, Sanz MJ, Perretti M, Morcillo E, Norling L, Mitchell JA, Li Y, Bishop-Bailey D. Activation of PPARbeta/delta inhibits leukocyte recruitment, cell adhesion molecule expression, and chemokine release. J Leukoc Biol. 2009;86:115–122. doi: 10.1189/jlb.0508284. [DOI] [PubMed] [Google Scholar]
- 75.Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, Stemerman MB. Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem. 2002;277:34176–34181. doi: 10.1074/jbc.M203436200. [DOI] [PubMed] [Google Scholar]
- 76.Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem. 1997;272:10811–10816. doi: 10.1074/jbc.272.16.10811. [DOI] [PubMed] [Google Scholar]
- 77.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe J. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor delta (PPARdelta) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 79.Lin Z, Natesan V, Shi H, Dong F, Kawanami D, Mahabeleshwar GH, Atkins GB, Nayak L, Cui Y, Finigan JH, Jain MK. Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol. 2010;30:1952–1959. doi: 10.1161/ATVBAHA.110.211474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuo CT, Veselits ML, Barton KP, Lu MM, Clendenin C, Leiden JM. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone Jr MA, Garcia-Cardena G, Jain MK. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- 83.Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 84.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone Jr MA, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;127:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilhelmsen K, Mesa KR, Lucero J, Xu F, Hellman J. ERK5 protein promotes, whereas MEK1 protein differentially regulates, the Toll-like receptor 2 protein-dependent activation of human endothelial cells and monocytes. J Biol Chem. 2012;287:26478–26494. doi: 10.1074/jbc.M112.359489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 89.Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras AC. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- 90.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014;588:2663–2670. doi: 10.1016/j.febslet.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 92.Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- 93.Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol. 2008;18:311–321. doi: 10.1016/j.cub.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15:1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song Q, Mao B, Cheng J, Gao Y, Jiang K, Chen J, Yuan Z, Meng S. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS One. 2015;10:e0120790. doi: 10.1371/journal.pone.0120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Campbell KN, Wong JS, Gupta R, Asanuma K, Sudol M, He JC, Mundel P. Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J Biol Chem. 2013;288:17057–17062. doi: 10.1074/jbc.C113.457390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 98.Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 100.Chen Z, Martin M, Li Z, Shyy JY. Endothelial dysfunction: the role of sterol regulatory element-binding protein-induced NOD-like receptor family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr Opin Lipidol. 2014;25:339–349. doi: 10.1097/MOL.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JH, Johnson DA, Shyy JY. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128:632–642. doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiao X, Song BL. SREBP: a novel therapeutic target. Acta Biochim Biophys Sin. 2013;45:2–10. doi: 10.1093/abbs/gms112. [DOI] [PubMed] [Google Scholar]
- 103.Daemen S, Kutmon M, Evelo CT. A pathway approach to investigate the function and regulation of SREBPs. Genes Nutr. 2013;8:289–300. doi: 10.1007/s12263-013-0342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JHF, Johnson DA, Shyy JYJ. SREBP2 Activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128:632. doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 106.Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab. 2012;16:414–419. doi: 10.1016/j.cmet.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 109.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 110.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 111.Zhao Y, Shao F. Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr Opin Microbiol. 2016;29:37–42. doi: 10.1016/j.mib.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 112.Hagar JA, Aachoui Y, Miao EA. WildCARDs: inflammatory caspases directly detect LPS. Cell Res. 2015;25:149–150. doi: 10.1038/cr.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13820. [DOI] [PubMed] [Google Scholar]
- 114.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 115.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Civelek M, Manduchi E, Riley RJ, Stoeckert Jr CJ, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 119.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 120.Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A. 2009;106:8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 122.Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol. 2008;28:223–232. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 123.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hirasawa H, Jiang C, Zhang P, Yang FC, Yokota H. Mechanical stimulation suppresses phosphorylation of eIF2alpha and PERK-mediated responses to stress to the endoplasmic reticulum. FEBS Lett. 2010;584:745–752. doi: 10.1016/j.febslet.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 125.Hilgarth RS, Murphy LA, Skaggs HS, Wilkerson DC, Xing H, Sarge KD. Regulation and function of SUMO modification. J Biol Chem. 2004;279:53899–53902. doi: 10.1074/jbc.R400021200. [DOI] [PubMed] [Google Scholar]
- 126.Guo B, Yang SH, Witty J, Sharrocks AD. Signalling pathways and the regulation of SUMO modification. Biochem Soc Trans. 2007;35:1414–1418. doi: 10.1042/BST0351414. [DOI] [PubMed] [Google Scholar]
- 127.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 128.Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun. 2005;337:517–520. doi: 10.1016/j.bbrc.2005.09.090. [DOI] [PubMed] [Google Scholar]
- 129.Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 130.Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009;284:8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16:5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Henley JM, Craig TJ, Wilkinson KA. Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol Rev. 2014;94:1249–1285. doi: 10.1152/physrev.00008.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo C, Henley JM. Wrestling with stress: roles of protein SUMOylation and deSUMOylation in cell stress response. IUBMB Life. 2014;66:71–77. doi: 10.1002/iub.1244. [DOI] [PubMed] [Google Scholar]
- 134.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 135.Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112:911–923. doi: 10.1161/CIRCRESAHA.111.300179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, Fujiwara K, Woo CH, Abe J. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15:1405–1414. doi: 10.1089/ars.2010.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garner E, Raj K. Protective mechanisms of p53-p21-pRb proteins against DNA damage-induced cell death. Cell Cycle. 2008;7:277–282. doi: 10.4161/cc.7.3.5328. [DOI] [PubMed] [Google Scholar]
- 140.Lin K, Hsu PP, Chen BP, Yuan S, Usami S, Shyy JY, Li YS, Chien S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci U S A. 2000;97:9385–9389. doi: 10.1073/pnas.170282597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Garner E, Martinon F, Tschopp J, Beard P, Raj K. Cells with defective p53-p21-pRb pathway are susceptible to apoptosis induced by p84N5 via caspase-6. Cancer Res. 2007;67:7631–7637. doi: 10.1158/0008-5472.CAN-07-0334. [DOI] [PubMed] [Google Scholar]
- 142.Chen A, Huang X, Xue Z, Cao D, Huang K, Chen J, Pan Y, Gao Y. The Role of p21 in apoptosis proliferation, cell cycle arrest, and antioxidant activity in UVB-irradiated human HaCaT keratinocytes. Med Sci Monit Basic Res. 2015;21:86–95. doi: 10.12659/MSMBR.893608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/S1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 144.Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K, Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. 2015;125:1299–1310. doi: 10.1172/JCI76453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/S0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 146.Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci U S A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jagavelu K, Tietge UJ, Gaestel M, Drexler H, Schieffer B, Bavendiek U. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circ Res. 2007;101:1104–1112. doi: 10.1161/CIRCRESAHA.107.156075. [DOI] [PubMed] [Google Scholar]
- 148.Chang E, Heo KS, Woo CH, Lee H, Le NT, Thomas TN, Fujiwara K, Abe J. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood. 2011;117:2527–2537. doi: 10.1182/blood-2010-08-302281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2014;306:H317–H325. doi: 10.1152/ajpheart.00182.2013. [DOI] [PubMed] [Google Scholar]
- 150.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:981–992. doi: 10.1016/S1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 152.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, Winther de MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 153.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/S1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 154.Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol. 2010;30:4901–4921. doi: 10.1128/MCB.00409-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. 2011;43:180–191. doi: 10.1016/j.molcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31:2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 158.Wuerzberger-Davis SM, Nakamura Y, Seufzer BJ, Miyamoto S. NF-kappaB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene. 2007;26:641–651. doi: 10.1038/sj.onc.1209815. [DOI] [PubMed] [Google Scholar]
- 159.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 160.Wu ZH, Mabb A, Miyamoto S. PIDD: a switch hitter. Cell. 2005;123:980–982. doi: 10.1016/j.cell.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 161.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/S0092-8674(03)00895-X. [DOI] [PubMed] [Google Scholar]
- 162.Miyamoto S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–130. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu X, Chen W, Wang Q, Li L, Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9:e1003480. doi: 10.1371/journal.ppat.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res. 2007;100:564–571. doi: 10.1161/01.RES.0000259561.23876.c5. [DOI] [PubMed] [Google Scholar]
- 166.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, Kassab GS, Garland Jr T, Shyy JY. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006;26:1281–1287. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- 168.Wang Q, Zhang M, Liang B, Shirwany N, Zhu Y, Zou MH. Activation of AMP-activated protein kinase is required for berberine-induced reduction of atherosclerosis in mice: the role of uncoupling protein 2. PLoS One. 2011;6:e25436. doi: 10.1371/journal.pone.0025436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gongol B, Marin T, Peng IC, Woo B, Martin M, King S, Sun W, Johnson DA, Chien S, Shyy JY. AMPKalpha2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc Natl Acad Sci U S A. 2013;110:3161–3166. doi: 10.1073/pnas.1222051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ambrose HE, Papadopoulou V, Beswick RW, Wagner SD. Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene. 2007;26:6244–6252. doi: 10.1038/sj.onc.1210434. [DOI] [PubMed] [Google Scholar]
- 172.Onyango IG, Tuttle JB, Bennett Jr JP. Activation of p38 and N-acetylcysteine-sensitive c-Jun NH2-terminal kinase signaling cascades is required for induction of apoptosis in Parkinson’s disease cybrids. Mol Cell Neurosci. 2005;28:452–461. doi: 10.1016/j.mcn.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 173.Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta-Leal A, Quiles-Perez R, Rodriguez-Vargas JM, Ruiz de Almodovar M, Conde C, Ruiz-Extremera A, Oliver FJ. Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem. 2007;14:1179–1187. doi: 10.2174/092986707780597998. [DOI] [PubMed] [Google Scholar]
- 174.Altmeyer M, Hottiger MO. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging (Albany NY) 2009;1:458–469. doi: 10.18632/aging.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 176.Mangerich A, Burkle A. Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation. Oxid Med Cell Longev. 2012;2012:321653. doi: 10.1155/2012/321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tang CH, Chiu YC, Tan TW, Yang RS, Fu WM. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J Immunol. 2007;179:5483–5492. doi: 10.4049/jimmunol.179.8.5483. [DOI] [PubMed] [Google Scholar]
- 178.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem. 2010;285:15346–15355. doi: 10.1074/jbc.M110.102491. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 179.Zhang W, Wang Q, Wu Y, Moriasi C, Liu Z, Dai X, Wang Q, Liu W, Yuan ZY, Zou MH. Endothelial cell–specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation. 2014;129:1428–1439. doi: 10.1161/CIRCULATIONAHA.113.004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rubio T, Vernia S, Sanz P. SUMOylation of AMPKbeta2 subunit enhances AMP-activated protein kinase activity. Mol Biol Cell. 2013;24(1801–11):S1–S4. doi: 10.1091/mbc.E12-11-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yan Y, Ollila S, Wong IP, Vallenius T, Palvimo JJ, Vaahtomeri K, Makela TP. SUMOylation of AMPK alpha 1 by PIAS4 specifically regulates mTORC1 signalling. Nat Commun. 2015;6:8979. doi: 10.1038/ncomms9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ritho J, Arold ST, Yeh ET. A Critical SUMO1 Modification of LKB1 Regulates AMPK Activity during Energy Stress. Cell Rep. 2015;12:734–742. doi: 10.1016/j.celrep.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 183.Han Z, Chen YR, Jones CI, Meenakshisundaram G, Zweier JL, Alevriadou BR. Shear-induced reactive nitrogen species inhibit mitochondrial respiratory complex activities in cultured vascular endothelial cells. Am J Physiol Cell Physiol. 2007;292:C1103–C1112. doi: 10.1152/ajpcell.00389.2006. [DOI] [PubMed] [Google Scholar]
- 184.Jayashankar V, Mueller IA, Rafelski SM. Shaping the multi-scale architecture of mitochondria. Curr Opin Cell Biol. 2016;38:45–51. doi: 10.1016/j.ceb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 185.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ong SB, Kalkhoran SB, Cabrera-Fuentes HA, Hausenloy DJ. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol. 2015;763:104–114. doi: 10.1016/j.ejphar.2015.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci. 2007;120:1178–1188. doi: 10.1242/jcs.03418. [DOI] [PubMed] [Google Scholar]
- 191.Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zungu M, Schisler J, Willis MS. All the Little Pieces-Regulation of Mitochondrial Fusion and Fission by Ubiquitin and Small Ubiquitin-Like Modifer and their Potential Relevance in the Heart. Circ J. 2011;75:2513–2521. doi: 10.1253/circj.CJ-11-0967. [DOI] [PubMed] [Google Scholar]
- 193.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 195.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 196.Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell. 2015;59:941–955. doi: 10.1016/j.molcel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 197.Mendler L, Braun T, Muller S. The Ubiquitin-Like SUMO System and Heart Function: From Development to Disease. Circ Res. 2016;118:132–144. doi: 10.1161/CIRCRESAHA.115.307730. [DOI] [PubMed] [Google Scholar]
- 198.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 199.Heo KS, Berk BC, Abe JI (2016) Disturbed Flow-Induced Endothelial Proatherogenic Signaling Via Regulating Post-Translational Modifications and Epigenetic Events. Antioxid Redox Signal [DOI] [PMC free article] [PubMed]
- 200.Kim GY, Nigro P, Fujiwara K, Abe J, Berk BC. p62 binding to protein kinase C zeta regulates tumor necrosis factor alpha-induced apoptotic pathway in endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:2974–2980. doi: 10.1161/ATVBAHA.112.300054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Moscat J, Diaz-Meco MT, Wooten MW. Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex. Cell Death Differ. 2009;16:1426–1437. doi: 10.1038/cdd.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 203.Hamm CA, Costa FF. Epigenomes as therapeutic targets. Pharmacol Ther. 2015;151:72–86. doi: 10.1016/j.pharmthera.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 204.Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, Fan Y, Jordan IK, Jo H. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. 2014;124:3187–3199. doi: 10.1172/JCI74792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Illi B, Nanni S, Scopece A, Farsetti A, Biglioli P, Capogrossi MC, Gaetano C. Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ Res. 2003;93:155–161. doi: 10.1161/01.RES.0000080933.82105.29. [DOI] [PubMed] [Google Scholar]
- 206.Guza R, Kotandeniya D, Murphy K, Dissanayake T, Lin C, Giambasu GM, Lad RR, Wojciechowski F, Amin S, Sturla SJ, Hudson RH, York DM, Jankowiak R, Jones R, Tretyakova NY. Influence of C-5 substituted cytosine and related nucleoside analogs on the formation of benzo[a]pyrene diol epoxide-dG adducts at CG base pairs of DNA. Nucleic Acids Res. 2011;39:3988–4006. doi: 10.1093/nar/gkq1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 208.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 209.Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, Jones PA. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33:e176. doi: 10.1093/nar/gni180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 212.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 213.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 214.Hsieh CL. The de novo methylation activity of Dnmt3a is distinctly different than that of Dnmt1. BMC Biochem. 2005;6:6. doi: 10.1186/1471-2091-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jiang YZ, Jimenez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res. 2014;115:32–43. doi: 10.1161/CIRCRESAHA.115.303883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, Feaver RE, Gao H, Desai A, Schmaier A, Gerson SL, Wang Y, Atkins GB, Blackman BR, Simon DI, Jain MK. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J Clin Invest. 2012;122:4727–4731. doi: 10.1172/JCI66056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 218.Villarreal Jr G, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, Garcia-Cardena G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem Biophys Res Commun. 2010;391:984–989. doi: 10.1016/j.bbrc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhou J, Li YS, Wang KC, Chien S. Epigenetic mechanism in regulation of endothelial function by disturbed flow: induction of DNA hypermethylation by DNMT1. Cell Mol Bioeng. 2014;7:218–224. doi: 10.1007/s12195-014-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Iordache F, Buzila C, Constantinescu A, Andrei E, Maniu H. Histone deacetylase (HDAC) inhibitors down-regulate endothelial lineage commitment of umbilical cord blood derived endothelial progenitor cells. Int J Mol Sci. 2012;13:15074–15085. doi: 10.3390/ijms131115074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chen W, Bacanamwo M, Harrison DG. Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J Biol Chem. 2008;283:16293–16298. doi: 10.1074/jbc.M801803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee DY, Lee CI, Lin TE, Lim SH, Zhou J, Tseng YC, Chien S, Chiu JJ. Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc Natl Acad Sci U S A. 2012;109:1967–1972. doi: 10.1073/pnas.1121214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 2010;115:2971–2979. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Schulman IG, Juguilon H, Evans RM. Activation and repression by nuclear hormone receptors: hormone modulates an equilibrium between active and repressive states. Mol Cell Biol. 1996;16:3807–3813. doi: 10.1128/MCB.16.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 226.Abe J, Morrell C. Pyroptosis as a Regulated Form of Necrosis: PI+/Annexin V−/High Caspase 1/Low Caspase 9 Activity in Cells = Pyroptosis? Circ Res. 2016;118:1457–1460. doi: 10.1161/CIRCRESAHA.116.308699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Woo CH, Abe J. SUMO–a post-translational modification with therapeutic potential? Curr Opin Pharmacol. 2010;10:146–155. doi: 10.1016/j.coph.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hickey CM, Wilson NR, Hochstrasser M. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol. 2012;13:755–766. doi: 10.1038/nrm3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol. 2014;34:2378–2386. doi: 10.1161/ATVBAHA.114.303428. [DOI] [PMC free article] [PubMed] [Google Scholar]