

Abstract
Primary aldosteronism (PA) is a severe form of autonomous aldosteronism. Milder forms of autonomous and renin-independent aldosteronism may be common, even in normotension. We characterized aldosterone secretion in 210 normotensives who had suppressed plasma renin activity (PRA<1.0 ng/mL/h), completed an oral sodium suppression test, received an infusion of angiotensin II (AngII), and had measurements of blood pressure (BP) and renal plasma flow (RPF). Continuous associations between urinary aldosterone excretion rate (AER), renin, and potassium handling were investigated. Severe autonomous aldosterone secretion that was consistent with confirmed PA was defined based on accepted criteria of an AER >12 mcg/24h with urinary sodium excretion >200 mmol/24h. Across the population, there were strong and significant associations between higher AER and higher urinary potassium excretion, higher AngII-stimulated aldosterone, and lower PRA, suggesting a continuum of renin-independent aldosteronism and mineralocorticoid receptor activity. Autonomous aldosterone secretion that fulfilled confirmatory criteria for PA was detected in 29 participants (14%). Normotensives with evidence suggestive of confirmed PA had higher 24h urinary AER (20.2±12.2 vs. 6.2±2.9 mcg/24h, P<0.001) as expected, but also higher AngII-stimulated aldosterone (12.4±8.6 vs. 6.6±4.3 ng/dL, P<0.001) and lower 24h urinary sodium-to-potassium excretion (2.69±0.65 vs. 3.69±1.50 mmol/mmol, P=0.001); however, there were no differences in age, aldosterone-to-renin ratio, BP, or RPF between the two groups. These findings indicate a continuum of renin-independent aldosteronism and mineralocorticoid receptor activity in normotension that ranges from subtle to overtly dysregulated and autonomous. Longitudinal studies are needed to determine whether this spectrum of autonomous aldosterone secretion contributes to hypertension and cardiovascular disease.
Keywords: Primary Aldosteronism, normotensive, aldosterone, renin, hypertension, potassium
INTRODUCTION
Primary aldosteronism (PA) is the most common cause of secondary hypertension and is characterized by an autonomous secretion of aldosterone, independent of renin and sodium status, resulting in excessive activation of the mineralocorticoid receptor (MR)1. The recognition of autonomous aldosterone secretion is important since inappropriate activation of the MR is known to increase cardiovascular morbidity and mortality, and treatment can mitigate this risk1–3.
PA is classically considered to be an uncommon disease that is investigated in patients with severe hypertension and/or hypokalemia. However, emerging research suggests that the spectrum of renin-independent aldosteronism may involve a more extensive continuum4. For example, when PA screening and confirmatory testing were conducted indiscriminately in a large population of mild-to-moderate essential hypertensives, 10% were confirmed to have PA5. We recently showed that implementing unbiased confirmatory testing for PA in mild hypertensives detected biochemical confirmation of PA in nearly 20% of the population6. Moreover, among participants with a confirmed diagnosis of PA, the severity of PA could be objectively quantified based on the suppression of renin; participants with more severe PA had greater renin suppression than those with a milder PA phenotype6. The notion that autonomous aldosterone secretion may originate in normotension and progress to induce hypertension is probably best exemplified by Markou et al. who biochemically confirmed PA in 13% of their normotensive cohort, and showed that these normotensives with “subclinical” PA had a greater than 15-fold higher risk for developing incident hypertension during five years of follow-up7.
The histopathological basis for normotensive PA has recently been suggested. The discovery of aldosterone producing cell clusters (APCCs), non-neoplastic foci of autonomous aldosterone secretion in morphologically normal adrenal glands, has challenged our understanding of the pathogenesis of PA8, 9. APCCs are now regarded as a potential precursor for PA: a source of mild and autonomous aldosterone secretion that may infrequently undergo neoplastic transformation to an aldosterone-producing adenoma or hyperplasia and cause overt PA.
Given the public health importance of detecting inappropriate aldosterone secretion and MR activation early in its pathogenesis, we aimed to characterize autonomous aldosterone secretion that may be “subclinical” in normotension based on features of aldosterone autonomy, evidence for excessive MR activation, and vascular function. We hypothesized that a continuous spectrum of autonomous aldosterone secretion may exist in normotension prior to progressing to the classic and overt phenotype of severe hypertension and/or hypokalemia that we more commonly recognize. Herein, we report the results of detailed physiologic studies to characterize autonomous aldosterone secretion in healthy normotensive participants.
METHODS
Study Population and Protocol
The study was a cross-sectional analysis of normotensive participants from the Hypertensive Pathotype (HyperPATH) cohort. The HyperPATH cohort includes normotensive adults, recruited from the community, who underwent detailed profiling of the renin–angiotensin–aldosterone system (RAAS) in response to sodium intake and aldosterone secretagogues after controlling for known confounders of the RAAS including posture, circadian rhythm, anti-hypertensive medications, and dietary intakes of sodium and potassium. The details of the HyperPATH protocol have been described previously10, 11. Although studies from the HyperPATH cohort have been previously published, the current analyses are original and novel. The study protocol was approved by our institutional research and ethics board and all participants provided informed consent.
Briefly, normotensives were defined as having a mean blood pressure (BP) <140/90 mmHg measured by an automated sphygmomanometer (Dinamap, Critikon, Tampa, FL) after five minutes of quiet seated rest (mean of three consecutive measurements). In addition to a normal BP, participants could not have any first-degree relatives diagnosed with hypertension prior to the age of 60.
All normotensive participants completed a dietary intervention consisting of one week of a liberalized dietary sodium intake (LIB) (>200 mmol of sodium per day) and one week of a restricted dietary sodium intake (RES) (10 mmol of sodium per day). During each dietary phase, participants were also given 100 mmol/day of potassium and 10 mmol/day of calcium to standardize the intake of these electrolytes. Upon completing each dietary intervention, participants were admitted to the Clinical Research Center in the evening and slept there overnight while maintaining a supine posture. All participants completed a 24h urine collection which was used to measure aldosterone excretion rate, sodium, potassium, and creatinine. At 8:00AM the following morning, supine blood pressure (BP) was measured and the average of five readings were recorded. Intravenous blood sampling was obtained in the morning to measure serum aldosterone and plasma renin activity (PRA). The aldosterone-to-renin ratio (ARR) was calculated from these values. Renal plasma flow (RPF) was measured using para-aminohippurate clearance as previously described11. Participants also received a 60 minute infusion of angiotensin II (AngII) at a dose of 3 ng/kg/min and aldosterone concentrations were measured again following the infusion. The change in serum aldosterone in response to the infusion of AngII (ΔALDOAngII) was calculated as the difference between baseline and stimulated aldosterone. Upon completing the RES intervention, participants also completed a posture protocol where they maintained a standing posture for 60 minutes to maximally stimulate the RAAS and then underwent blood sampling to measure stimulated PRA and aldosterone.
For the current study, we included all normotensive HyperPATH participants who had complete data on measurements of serum aldosterone, PRA, and BP, and who completed infusions of AngII and had measurements of RPF on LIB. To focus on autonomous aldosteronism that was renin-independent, we excluded participants who had a PRA≥ 1.0 ng/mL/h while on LIB, which may have suggested a form of secondary aldosteronism. The final study population for the current analysis was 210 normotensive participants.
Severe Autonomous Aldosterone Secretion Suggestive of Primary Aldosteronism
We considered participants with severely dysregulated and autonomous aldosterone secretion to potentially have biochemically confirmed PA if they fulfilled the confirmatory criteria recommended by The Endocrine Society1. Participants with a suppressed PRA (<1.0 ng/mL/h) and evidence for severely autonomous aldosterone secretion while on the LIB intervention (24h urinary aldosterone excretion rate ≥12 mcg with a 24h urinary sodium excretion rate >200 mmol) were considered to potentially have PA1, 12. The confirmatory criteria for PA are generally recommended for hypertensives with an elevated ARR; however, we applied these confirmatory criteria to our normotensive participants irrespective of their ARR to identify the most severe and dysregulated forms of autonomous aldosterone secretion. Participants with a 24h urinary sodium excretion rate >200 mmol and a 24h urinary aldosterone excretion rate < 12mcg were considered to not have PA. However, since some experts consider an aldosterone excretion rate of 10–12 mcg/24h to be suggestive of PA, we also conducted a sensitivity analysis using a stricter definition to exclude PA: 24h urinary aldosterone excretion rate <10 mcg and a urinary sodium excretion of >200 mmol1.
Continuous Assessments of Autonomous Aldosterone Secretion, Mineralocorticoid Receptor Activity, and Vascular Dysfunction
The 24h urinary aldosterone excretion rate, the ARR, and the ΔALDOAngII on LIB conditions were considered to be representations of autonomous aldosterone secretion since all participants had a suppressed PRA. The degree of mineralocorticoid receptor (MR) activation was indirectly assessed via BP, serum potassium concentrations, 24h urinary potassium excretion, and 24h urinary sodium-to-potassium excretion, all on LIB13. Further, the inability to stimulate PRA when on RES was used as a proxy of MR activation, as we have previously demonstrated6: decreased stimulation of renin when sodium restricted is a biomarker for excessive MR activity. Evidence for vascular dysfunction was assessed via measurements of BP and RPF on LIB.
Laboratory assays
Serum aldosterone and PRA were measured in duplicate after each dietary intervention, after the posture protocol, and before and after the infusion of AngII, and averaged. The PRA assay (Diasorin, Inc., Stillwater, MN) had a dynamic range of 0.1–50 ng/mL/h; inter-assay variation is 5.6–7.6%). The serum aldosterone assay (Siemens, Los Angeles, CA) had a dynamic range of 2.5–120 ng/dL; inter-assay variation 3.8–15.7%. RPF was measured via para-aminohippurate clearance as previously described14 using the average of triplicate measures of para-aminohippurate.
Statistical analyses
Our analytic approach was two-fold. We first investigated whether there was a continuum of autonomous aldosterone secretion and MR activation across the entire study population. We assessed associations between 24h aldosterone excretion rate and AngII-stimulated aldosterone secretion (a marker of aldosterone secretion), urinary sodium-to-potassium excretion (an indirect marker of MR activity), and the inability to stimulate renin when sodium restricted (another indirect marker of MR activity). Finally, we assessed the frequency of participants with severely dysregulated aldosterone secretion that may be consistent with biochemically confirmed PA. We then investigated the defining characteristics of normotensive participants with and without evidence for PA.
Data are presented as means with the standard deviation of the means for continuous variables and percentages for categorical variables. Normality was assessed using the Kolmogorov–Smirnov test. In situations where a variable was not normally distributed (PRA, aldosterone, ARR), a bootstrapping procedure with 1000 iterations was performed. Non-normally distributed variables are also reported as the median and interquartile ranges. Comparisons of means and frequencies between groups were assessed using the Student’s t-test or chi-square testing. Pearson correlation and unadjusted linear regression were used to evaluate the association between urinary aldosterone excretion rate (AER) and ΔALDOAngII and the 24h urinary sodium-to-potassium ratio. We tested the interaction between AER and renin in predicting the urine sodium-to-potassium ratio using an interaction term in linear regression models that were adjusted for age, sex, race, body-mass index, and systolic blood pressure. A two-sided P-value of < 0.05 was considered statistically significant. Analyses were performed using SPSS 21 and STATA 13 statistical packages.
RESULTS
The Continuum of Autonomous Aldosterone Secretion and MR Activity
The characteristics of the study population are described in Table 1. The potential for a continuum of autonomous aldosterone secretion was investigated across all normotensive participants. A strong and significant positive association between the 24h urinary AER and the ΔALDOAngII was observed (β=0.578 ng/dL per mcg/24h; r=0.587, P<0.001), demonstrating a continuum irrespective of whether the AER was indicative of potentially confirmed “PA” (Figure 1). Similarly, a strong and significant inverse association was observed between the 24h urinary AER and the 24h urinary sodium-to-potassium excretion rate (β= −0.100 mmol/mmol per mcg/24h; r= −0.384, P<0.0001) (Figure 2). To investigate whether these findings were suggestive of a continuum of autonomous aldosteronism and excessive renal-MR activity, we assessed the interaction between 24h urinary AER and the ability to stimulate renin when sodium restricted as a predictor of urinary sodium-to-potassium excretion. We hypothesized that greater autonomous aldosterone secretion would be associated with greater MR activity, thus a greater inability to stimulate PRA when sodium restricted and a lower urinary sodium-to-potassium excretion ratio6,13. A continuous interaction was observed whereby the urinary sodium-to-potassium excretion rate was lowest when autonomous aldosterone secretion was highest and renin activity the most suppressed (Unadjusted P-interaction<0.01; Adjusted P-interaction=0.03) (Figure 3).
TABLE 1. Characteristics of the Study Population.
All values reflect measurements taken when participants were maintained on a liberalized dietary sodium intake, unless otherwise indicated.
| Characteristic | Value |
|---|---|
| Age (years) | 40.4 ± 12.4 |
| Race/Ethnicity (%Caucasian) | 72 |
| Male Sex (%) | 53 |
| Body mass index (kg/m2) | 25.7 ± 4.3 |
| Systolic BP (mmHg) | 113.8 ± 12.6 |
| ARR (ng/dL per ng/mL/h) | 15.4 ± 13.7 11.6 [6.9–25.9] |
| ARR ≥ 20 (ng/dL per ng/mL/h) (n/n; %) | 55/210; 26 |
| Supine Serum Aldosterone (ng/dL) | 3.48 ± 1.8 2.8 [2.5–4.1] |
| Supine PRA (ng/mL/h) | 0.38 ± 0.3 0.30 [0.13–0.49] |
| Supine PRA on RES (ng/mL/h) | 2.88 ± 2.1 2.27 [1.2–4.2] |
| Standing Posture PRA on RES (ng/mL/h) | 9.89 ± 6.9 8.50 [4.7–12.9] |
| Serum potassium (mmol/L) | 4.11 ± 0.3 |
| ΔALDOAngII | 7.45 ± 5.6 (+227%) |
| Renal plasma flow (mL/min/1.73m2) | 558 ± 132 |
| Urinary sodium (mmol/24h) | 248.3 ± 68.1 |
| Urinary potassium (mmol/24h) | 77.4 ± 26.7 |
| Urinary sodium-to-potassium excretion ratio (mmol/mmol per 24h) | 3.55 ± 1.45 |
| Aldosterone excretion rate (mcg/24h) | 8.13 ± 7.1 Range= 1.1–79.9 |
Data are presented as mean +/− SD for continuous variables and frequencies (%) for categorical variables. Non-normally distributed variables are also presented as median with interquartile ranges. Aldosterone excretion rate is presented as mean +/− SD and with the range of the full distribution. BP=blood pressure; ARR=aldosterone-to-renin ratio; PRA=plasma renin activity; ΔALDOAngII=the change in serum aldosterone following an infusion of angiotensin II; RES=dietary sodium restriction.
Figure 1. Association between 24h urinary aldosterone excretion and the adrenal aldosterone response to an infusion of angiotensin II.
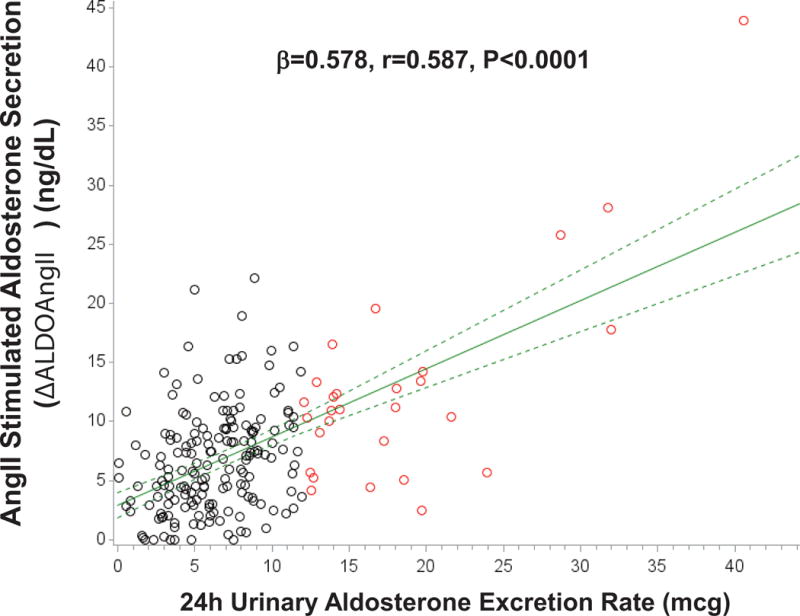
Black circles represent normotensive participants without PA (24h urinary AER<12 mcg/24), red circles represent normotensive participants who fulfill the confirmatory criteria for PA (24h urinary AER≥12 mcg/24h), solid green line represents the mean regression line, and dotted green lines represent the 95% confidence intervals for the mean regression line. (AngII=angiotensin II)
Figure 2. Association between 24h urinary aldosterone excretion and the 24h urinary sodium-to-potassium excretion ratio.
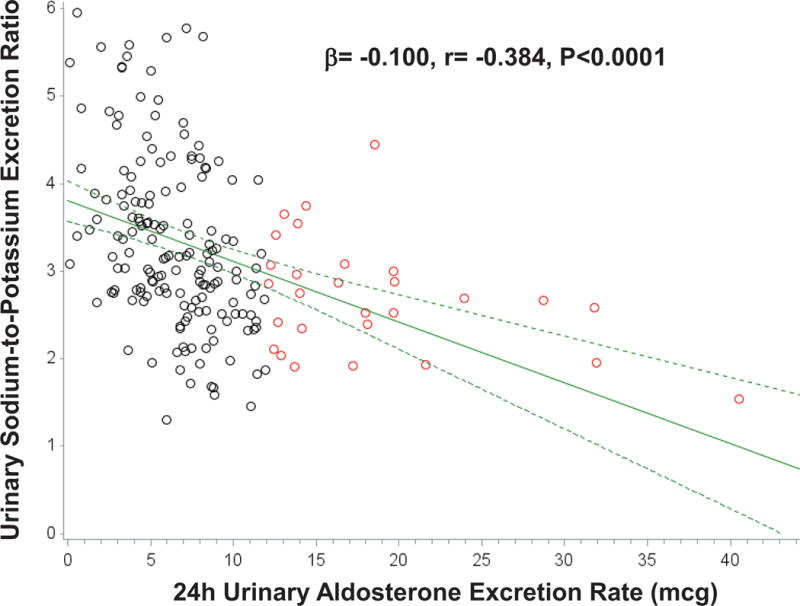
Black circles represent normotensive participants without PA (24h urinary AER<12 mcg/24), red circles represent normotensive participants who fulfill the confirmatory criteria for PA (24h urinary AER≥12 mcg/24h), solid green line represents the mean regression line, and dotted green lines represent the 95% confidence intervals for the mean regression line.
Figure 3. The Relationship Between Autonomous Aldosterone Secretion, Renin, and Urinary Sodium-to-Potassium Excretion.
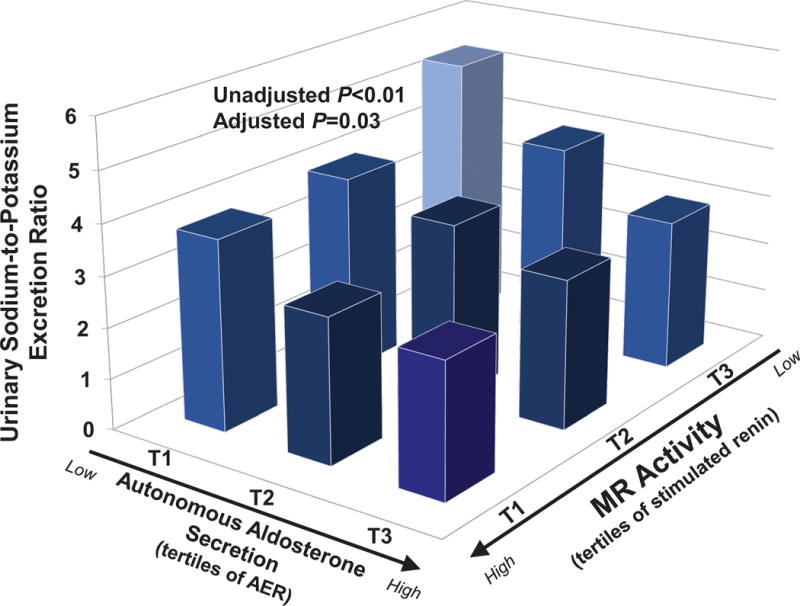
Autonomous aldosterone secretion is represented by tertiles of the 24h urinary aldosterone excretion rate measured on a liberalized dietary sodium intervention. Renin activity is represented by tertiles of renin when sodium restricted, whereby an inability to stimulate renin serves as a surrogate for excessive MR activity. The figure displays the interaction between the independent variables, aldosterone secretion and renin activity, and the dependent variable, urinary sodium-to-potassium excretion ratio. Bars represent the mean value in each tertile. The adjusted interaction model is adjusted for age, sex, race, body-mass index, and systolic blood pressure. (AER=24h aldosterone excretion rate; PRA=plasma renin activity)
Severely Dysregulated and Autonomous Aldosterone Secretion in Normotension
The frequency of participants with autonomous aldosterone secretion so severe that it fulfilled confirmatory criteria for PA was 14% (29/210) (Table 2). Among these participants, only 6/29 (21%, or 3% of the entire study population) also had an ARR≥20 (Table 2). The most notable features of these participants with dysregulated aldosterone secretion that fulfilled confirmatory criteria for PA, when compared to those who did not fulfill confirmatory criteria for PA (181/210), were an expectedly higher 24h urinary aldosterone excretion rate (20.2 ±12.2 vs. 6.2 ± 2.9 mcg/24h, P<0.001), a higher 24h urinary potassium excretion rate (86.9 ± 20.5 vs. 75.8 ± 27.2, P<0.05), a lower 24h urinary sodium-to-potassium excretion ratio (2.69 ± 0.65 vs. 3.69 ± 1.5, P=0.001), and higher aldosterone secretion in response to an infusion of AngII (ΔALDOAngII) (+352% vs +206% P<0.001) (Table 2). Further, when study participants were sodium restricted, normotensives who fulfilled the confirmatory criteria for PA had lower stimulated PRA (Table 2). There were no significant differences in age, sex, racial distribution, body-mass index, BP, RPF, serum potassium, supine serum aldosterone, and ARR between normotensives with and without potential PA (Table 2).
TABLE 2. Characteristics of Normotensive Participants with severely dysregulated autonomous aldosterone secretion fulfilling confirmatory criteria for PA.
All values reflect measurements taken when participants were maintained on a liberalized dietary sodium intake, unless otherwise indicated.
| Characteristic | Normotensives who fulfill confirmatory criteria for PA 29/210 (14%) |
Normotensives who do not fulfill confirmatory criteria for PA 181/210 (86%) |
P |
|---|---|---|---|
| Age (years) | 42.4 ± 9.9 | 39.6 ± 12.7 | 0.261 |
| Race/Ethnicity (%Caucasian) | 83 | 70 | 0.163 |
| Male Sex (%) | 38 | 55 | 0.085 |
| Body mass index (kg/m2) | 25.9 ± 4.2 | 25.6 ± 4.3 | 0.856 |
| Systolic BP (mmHg) | 110.8 ± 11.9 | 113.4 ± 12.7 | 0.333 |
| ARR (ng/dL per ng/mL/h) | 14.9 ± 15.1 8.4 [6.5–15.9] |
15.6 ± 13.5 12.4 [7.5–25] |
0.818* |
| ARR ≥ 20 (ng/dL per ng/mL/h) (n/n; %) | 6/29; 21 | 49/181; 27 | 0.382 |
| Supine Serum Aldosterone (ng/dL) | 3.98 ± 1.8 3.4 [2.5–5.1] |
3.47 ± 1.7 2.7 [2.5–3.9] |
0.170* |
| Supine PRA (ng/mL/h) | 0.42 ± 0.3 0.39 [0.20–0.63] |
0.38 ± 0.4 0.29 [0.13–0.45] |
0.368* |
| Supine PRA on RES (ng/mL/h) | 2.59 ± 2.1 2.06 [0.90–4.37] |
2.92 ± 2.1 2.27 [1.20–4.13] |
0.433* |
| Standing Posture PRA on RES (ng/mL/h) | 7.49 ± 5.5 7.60 [3.95–10.99] |
10.28 ± 7.0 8.8 [4.8–13.5] |
0.015* |
| Serum potassium (mmol/L) | 4.27 ± 0.3 | 4.34 ± 0.3 | 0.326 |
| ΔALDOAngII | 12.4 ± 8.6 (+352%) | 6.61 ± 4.3 (+206%) | <0.001 |
| Renal plasma flow (mL/min/1.73m2) | 586 ± 92 | 552 ± 143 | 0.263 |
| Urinary sodium (mmol/24h) | 225.3 ± 43.7 | 251.8 ± 71.3 | 0.058 |
| Urinary potassium (mmol/24h) | 86.9 ± 20.5 | 75.8 ± 27.2 | 0.039 |
| Urinary sodium-to-potassium excretion ratio (mmol/mmol per 24h) | 2.69 ± 0.65 | 3.69 ± 1.5 | 0.001 |
| Aldosterone excretion rate (mcg/24h) | 20.2 ± 12.2 Range: 12.1–71.9 |
6.2 ± 2.9 Range: 1.1–11.9 |
<0.001 |
Data are presented as mean +/− SD for continuous variables and frequencies (%) for categorical variables. Non-normally distributed variables are also presented as median with interquartile ranges. Aldosterone excretion rate is presented as mean +/− SD and with the range of the full distribution.
Boostrapped.
BP=blood pressure; ARR=aldosterone-to-renin ratio; PRA=plasma renin activity; ΔALDOAngII=the change in serum aldosterone following an infusion of angiotensin II; RES=dietary sodium restriction.
We repeated these comparisons in a sensitivity analysis wherein normotensives without PA were defined by the stricter criteria of 24h urinary aldosterone excretion rate <10 mcg with urinary sodium excretion >200 mmol and the aforementioned findings did not materially change.
DISCUSSION
Although PA is often considered to be an infrequent and categorical disorder, compelling evidence suggests that the spectrum of autonomous aldosterone secretion extends beyond those with an overt phenotype of hypertension and/or hypokalemia. Herein, we observed a continuous spectrum of autonomous aldosterone secretion and MR activity in a normotensive population that ranged from subtle to overtly dysregulated, such that some normotensive participants even fulfilled confirmatory criteria for PA. This continuum of autonomous aldosterone secretion may have important implications for the pathogenesis of MR-mediated hypertension and perhaps even in determining the optimal method to mitigate incident cardiovascular disease.
The frequency of normotensive participants with normokalemia and no evidence of cardiovascular or renal disease that had biochemistry that was potentially consistent with PA confirmation, based on currently accepted criteria1, was 14% (29/210). The case detection for PA using an ARR that exceeds at least 20 is typically recommended for hypertensives, where this screening criterion is validated, and therefore, it cannot be concluded that any of the 29 normotensive participants in the current study had “PA” based on our current interpretation of the disease. However, it is noteworthy that these 29 participants had severely autonomous and dysregulated aldosterone secretion that could not be suppressed with sodium loading. When applying the two-step approach of an ARR screen ≥ 20 and accepted confirmatory criteria, only 21% (6/29) of these normotensives (or 3% of the entire study population of 210) would have been considered to have “PA” by conventional standards. Thus, this arbitrary threshold for the ARR may be sensitive for case detection in hypertension, but our findings suggest that it may not be calibrated or sensitive in normotensives. Notably, normotensive participants who fulfilled the confirmatory criteria for PA could not be distinguished from participants without PA using vascular parameters (such as BP and RPF) or the recommend screening ARR; both groups had apparently “normal” values for these traits. However, the subclinical nature of “normotensive PA” could be unmasked by recognizing a significantly higher urinary aldosterone excretion rate in the face of a suppressive sodium balance, a higher adrenal aldosterone secretion rate in response to angiotensin II, and a greater urinary potassium excretion rate and inability to stimulate renin as proxies for excessive MR activation. Therefore, the current findings are noteworthy because they transcend the categorical and binary nature of the “PA” diagnosis (yes vs. no), and instead suggest that there is a continuum of renin-independent aldosteronism that exists as a subclinical phenotype prior to inducing any hypertension. Whether knowledge of this underlying physiology can inform improved methods to mitigate or treat incident hypertension is an important consideration.
Although our findings are cross-sectional, they extend the insights into the temporal pathogenesis of PA. Markou et al. previously demonstrated that a substantial proportion of unsuspecting normotensives (13%) had subclinical PA and harbored a >15-fold higher risk of developing incident hypertension when compared to normotensive participants without PA7. Their observations suggested that the genesis of autonomous aldosterone secretion may begin in normotension, and with time (and aging), the sustained impact of MR-mediated plasma volume expansion and direct cardiovascular injury resulted in incident hypertension. Indeed, other larger prospective studies have shown that higher aldosterone concentrations in normotension independently predict the risk for developing incident hypertension15, and a higher risk of prevalent cardiovascular and metabolic co-morbidities11, 14. In this regard, our current findings demonstrate that normotensives exhibit a continuum of autonomous aldosterone secretion that was “subclinical” in that it did not manifest with significantly higher BP or hypokalemia. These observations may at first seem contradictory, however, normal renal-vascular and kidney function in these young and healthy normotensives may have permitted them to excrete the excessive sodium and volume loads to maintain a state of apparent normotension. Yet, despite this apparent normalcy, they clearly had evidence for autonomous aldosterone secretion (higher urinary aldosterone excretion and aldosterone stimulation to angiotensin II while on a suppressive sodium balance) and MR activity (higher urinary potassium excretion relative to sodium excretion and inability to stimulate renin). The most noteworthy aspect of our study is that irrespective of how “PA” is formally defined, we observed a continuum of autonomous aldosterone secretion in healthy normotensives that associated with a corresponding continuum of MR activation. Although not specifically investigated in the current study, we speculate that with time, and aging, these normotensives may gradually lose the compensatory ability to handle the constitutive exposure to excessive MR activity and intravascular volume expansion.
It is important to consider how and why autonomous aldosterone secretion may exist in normotension. First, screening for PA is not recommended in normotension1; the variability of the ARR and responses to confirmatory testing in normotension are not as well studied as they are in hypertension. Second, the molecular basis for our observation of a continuum of aldosterone secretion may be explained by the recent discovery of APCCs. APCCs represent large foci of abnormal CYP11B2 staining that invade the zona fasciculata in morphologically normal (non-neoplastic) adrenal glands, and harbor somatic mutations known to result in constitutive aldosterone secretion8, 9. The remarkable observation that >50% of normal adrenal glands (without an adenoma or hyperplasia) are found to have APCCs suggests that mild autonomous aldosterone secretion may be far more common than currently perceived8, 9. Since APCCs represent a non-neoplastic abnormality, it has been proposed that they may be common contributors to mild autonomous aldosterone secretion; the acquisition of subsequent proliferative/neoplastic abnormalities may result in the less common transformation to an aldosterone-producing adenoma or hyperplasia and clinically evident PA16.
Our study must be interpreted in the context of its strengths and limitations. A major strength includes the multiple physiologic and controlled interventions we employed to phenotype aldosterone autonomy, MR activation, and vascular function. Further, the study participants were not patients in a tertiary care medical center where a referral bias is often implied, but rather members of the local community recruited specifically for this study. A number of major limitations must be acknowledged. The objective of our study was to characterize the phenotypic continuum of autonomous aldosterone excess, not the prevalence of PA in normotension based on existing definitions; therefore our results are not suitable for incorporation directly into clinical care. Although we attempted to identify participants that met the accepted criteria for PA, we acknowledge that current PA definitions are calibrated for hypertensives and not normotensives. Therefore our data on the frequency of PA in our population should not be extrapolated to represent the epidemiology of PA in normotension, but rather used to contextualize the severity spectrum of renin-independent aldosteronism that appears to exist in healthy normotensives and how it may influence incident cardiovascular disease. The study was cross-sectional and therefore longitudinal studies are warranted to investigate if and how the spectrum of (dysregulated) physiology we observed relates to clinical outcomes. The measurements of serum aldosterone and PRA were obtained from a supine posture rather than the conventional seated posture, and therefore may appear lower than usual. However, our method of controlling posture for a prolonged duration added to the reliability of the results. Further, the ARR ensured a relative metric between aldosterone and PRA that was independent of posture, and we also assessed 24h urinary aldosterone excretion and angiotensin II stimulated aldosterone secretion which were not dependent on posture. Our study conditions included controlled intakes of sodium and potassium to standardize these confounders and permit inter-individual comparisons of renin and aldosterone physiology; however, since clinical patients are usually studied on ad libitum diets, we recognize that our findings may not be generalizable to the clinical setting. We did not have imaging or adrenal venous sampling results on to determine the anatomic subtype or localization of autonomous aldosterone secretion. Our main metric for autonomous aldosterone secretion was the 24h urinary aldosterone excretion rate on a sodium loaded balance, rather than circulating serum aldosterone levels; however, this methodology is well-recognized, frequently used, and a recommend confirmatory testing approach by The Endocrine Society1. Lastly, we utilized only one of the four recommended confirmatory tests to assess for PA and therefore recognize that the use of other tests (such as captopril, fludrocortisone, or saline suppression) may have yielded slightly different results. However, the major conclusion of our study should not be the focus on the categorical definition of PA, rather the continuum of autonomous aldosterone secretion we observed, which was independent of any biochemical definition of PA.
PERSPECTIVES
We identified a continuum of renin-independent aldosteronism in normotension that ranged from subtle to overtly dysregulated and autonomous aldosterone secretion. These findings suggest that aldosterone and MR mediated “essential” hypertension and cardiovascular disease may be more common than currently perceived, and provide an impetus for future longitudinal and intervention studies to investigate the clinical relevance of this physiology and potential efficacy of phenotype-driven MR antagonist treatment. Finally, these observations may support the recent histopathological discovery of APCCs which also implicate a common syndrome of autonomous aldosterone secretion.
NOVELTY AND SIGNIFICANCE.
What is new? There is a continuum of renin-independent aldosteronism in normotension that ranges from subtle to overtly dysregulated and autonomous.
What is Relevant? A substantial proportion of normotensives may have excessive autonomous aldosteronism that may contribute to the development of mineralocorticoid receptor mediated hypertension and cardiovascular disease.
Summary: Our findings demonstrate a spectrum of subclinical and autonomous aldosteronism that originates in normotension.
Acknowledgments
We thank our funding sources and research volunteers and staff.
SOURCES OF FUNDINGS
This study was supported by FONDECYT 1150327(RB), 1150437 (RB, CF), 1160836 (RB, CF), 1160695 (RB, CF) and CORFO 13CTI-21 526 -P1 (RB, CF). GH was supported by the National Institutes of Diabetes and Digestive and Kidney Disease of the National Institutes of Health under Award Number 5T32DK007527-31. AV was supported by the National Institutes of Diabetes and Digestive and Kidney Disease of the National Institutes of Health under Award Number R01DK107407, by grant 2015085 from the Doris Duke Charitable Foundation, and by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number K23HL111771.
Footnotes
DISCLOSURES
Nothing to disclose
References
- 1.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF., Jr The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. The Journal of Clinical Endocrinology and Metabolism. 2016;101:1889–1916. doi: 10.1210/jc.2015-4061. [DOI] [PubMed] [Google Scholar]
- 2.Xanthakis V, Vasan RS. Aldosterone and the risk of hypertension. Current Hypertension reports. 2013;15:102–107. doi: 10.1007/s11906-013-0330-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu VC, Wang SM, Chang CH, Hu YH, Lin LY, Lin YH, Chueh SC, Chen L, Wu KD. Long term outcome of aldosteronism after target treatments. Scientific Reports. 2016;6:32103. doi: 10.1038/srep32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piaditis G, Markou A, Papanastasiou L, Androulakis II, Kaltsas G. Progress in aldosteronism: A review of the prevalence of primary aldosteronism in pre-hypertension and hypertension. European Journal of Endocrinology. 2015;172:R191–203. doi: 10.1530/EJE-14-0537. [DOI] [PubMed] [Google Scholar]
- 5.Mosso L, Carvajal C, Gonzalez A, Barraza A, Avila F, Montero J, Huete A, Gederlini A, Fardella CE. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42:161–165. doi: 10.1161/01.HYP.0000079505.25750.11. [DOI] [PubMed] [Google Scholar]
- 6.Baudrand R, Guarda FJ, Torrey J, Williams G, Vaidya A. Dietary sodium restriction increases the risk of misinterpreting mild cases of primary aldosteronism. The Journal of Clinical Endocrinology and Metabolism. 2016;101:3989–3996. doi: 10.1210/jc.2016-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markou A, Pappa T, Kaltsas G, Gouli A, Mitsakis K, Tsounas P, Prevoli A, Tsiavos V, Papanastasiou L, Zografos G, Chrousos GP, Piaditis GP. Evidence of primary aldosteronism in a predominantly female cohort of normotensive individuals: A very high odds ratio for progression into arterial hypertension. The Journal of Clinical Endocrinology and Metabolism. 2013;98:1409–1416. doi: 10.1210/jc.2012-3353. [DOI] [PubMed] [Google Scholar]
- 8.Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, Shibata H, Itoh H, Mitani F, Yamazaki T, Ogishima T, Suematsu M, Mukai K. Adrenocortical zonation in humans under normal and pathological conditions. The Journal of Clinical Endocrinology and Metabolism. 2010;95:2296–2305. doi: 10.1210/jc.2009-2010. [DOI] [PubMed] [Google Scholar]
- 9.Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, Liu CJ, Sanjanwala AR, Edwards MA, Gomez-Sanchez CE, Nanba K, Rainey WE. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E4591–4599. doi: 10.1073/pnas.1505529112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baudrand R, Pojoga LH, Vaidya A, Garza AE, Vohringer PA, Jeunemaitre X, Hopkins PN, Yao TM, Williams J, Adler GK, Williams GH. Statin use and adrenal aldosterone production in hypertensive and diabetic subjects. Circulation. 2015;132:1825–1833. doi: 10.1161/CIRCULATIONAHA.115.016759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaidya A, Underwood PC, Hopkins PN, Jeunemaitre X, Ferri C, Williams GH, Adler GK. Abnormal aldosterone physiology and cardiometabolic risk factors. Hypertension. 2013;61:886–893. doi: 10.1161/HYPERTENSIONAHA.111.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishizaka MK, Pratt-Ubunama M, Zaman MA, Cofield S, Calhoun DA. Validity of plasma aldosterone-to-renin activity ratio in african american and white subjects with resistant hypertension. American Journal of Hypertension. 2005;18:805–812. doi: 10.1016/j.amjhyper.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Eudy RJ, Sahasrabudhe V, Sweeney K, Tugnait M, King-Ahmad A, Near K, Loria P, Banker ME, Piotrowski DW, Boustany-Kari CM. The use of plasma aldosterone and urinary sodium to potassium ratio as translatable quantitative biomarkers of mineralocorticoid receptor antagonism. Journal of Translational Medicine. 2011;9:180. doi: 10.1186/1479-5876-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown JM, Underwood PC, Ferri C, Hopkins PN, Williams GH, Adler GK, Vaidya A. Aldosterone dysregulation with aging predicts renal vascular function and cardiovascular risk. Hypertension (Dallas, Tex.: 1979) 2014;63:1205–1211. doi: 10.1161/HYPERTENSIONAHA.114.03231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, Benjamin EJ, Levy D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. The New England Journal of Medicine. 2004;351:33–41. doi: 10.1056/NEJMoa033263. [DOI] [PubMed] [Google Scholar]
- 16.Lalli E, Barhanin J, Zennaro MC, Warth R. Local control of aldosterone production and primary aldosteronism. Trends in endocrinology and metabolism: TEM. 2016;27:123–131. doi: 10.1016/j.tem.2016.01.003. [DOI] [PubMed] [Google Scholar]