

ABSTRACT
Influenza virus assembles and buds at the plasma membrane of virus-infected cells. The viral proteins assemble at the same site on the plasma membrane for budding to occur. This involves a complex web of interactions among viral proteins. Some proteins, like hemagglutinin (HA), NA, and M2, are integral membrane proteins. M1 is peripherally membrane associated, whereas NP associates with viral RNA to form an RNP complex that associates with the cytoplasmic face of the plasma membrane. Furthermore, HA and NP have been shown to be concentrated in cholesterol-rich membrane raft domains, whereas M2, although containing a cholesterol binding motif, is not raft associated. Here we identify viral proteins in planar sheets of plasma membrane using immunogold staining. The distribution of these proteins was examined individually and pairwise by using the Ripley K function, a type of nearest-neighbor analysis. Individually, HA, NA, M1, M2, and NP were shown to self-associate in or on the plasma membrane. HA and M2 are strongly coclustered in the plasma membrane; however, in the case of NA and M2, clustering depends upon the expression system used. Despite both proteins being raft resident, HA and NA occupy distinct but adjacent membrane domains. M2 and M1 strongly cocluster, but the association of M1 with HA or NA is dependent upon the means of expression. The presence of HA and NP at the site of budding depends upon the coexpression of other viral proteins. Similarly, M2 and NP occupy separate compartments, but an association can be bridged by the coexpression of M1.
IMPORTANCE The complement of influenza virus proteins necessary for the budding of progeny virions needs to accumulate at budozones. This is complicated by HA and NA residing in lipid raft-like domains, whereas M2, although an integral membrane protein, is not raft associated. Other necessary protein components such as M1 and NP are peripherally associated with the membrane. Our data define spatial relationships between viral proteins in the plasma membrane. Some proteins, such as HA and M2, inherently cocluster within the membrane, although M2 is found mostly at the periphery of regions of HA, consistent with the proposed role of M2 in scission at the end of budding. The association between some pairs of influenza virus proteins, such as M2 and NP, appears to be brokered by additional influenza virus proteins, in this case M1. HA and NA, while raft associated, reside in distinct domains, reflecting their distributions in the viral membrane.
KEYWORDS: budozone, lateral organization of spikes, immunogold labeling, influenza virus assembly, protein organization, virus budding
INTRODUCTION
Influenza A virus is a member of the Orthomyxoviridae family of enveloped, negative-stranded, segmented RNA viruses. The viral genome is in the form of eight ribonucleoprotein (RNP) complexes, which encode 12 to 13 proteins, most of which are incorporated into progeny virions (1–4). The envelope of the virus contains the two major glycoproteins hemagglutinin (HA) and neuraminidase (NA) as well as the proton-selective ion channel protein M2 (4–7). A dense, ordered protein layer has been observed beneath the viral envelope, which is believed to be composed of the matrix protein M1 (8, 9). The eight viral RNPs are thought to be linked to the viral envelope via a bridge formed between M1 proteins and both the RNPs and the inner surface of the membrane. It is generally believed that M1 binds viral RNA (vRNA), although a direct interaction between M1 and NP has been reported (10–15).
Viral RNA is transcribed and replicated in the nucleus of virus-infected cells. The viral RNA is wound around the surface of the nucleocapsid protein (NP) subunits and associates with the RNA polymerase complex, comprised of the PA, PB1, and PB2 proteins. NP binds single-stranded RNA without sequence specificity (16, 17), but the virus packages only RNPs associated with viral RNA (18). The integrity of the viral RNP (vRNP) is maintained at least in part by NP-NP interactions, and RNP depleted of RNA maintains the characteristic morphology of vRNP (19). During virus infection, viral RNPs exit the nucleus and are transported to the plasma membrane, where the RNP becomes associated with viral components, resulting in the assembly and budding of virions at the so-called budozone. The M1 protein is thought to mediate the transport of viral RNPs out of the nucleus and their association with the plasma membrane through a direct interaction with the membrane or association with the cytoplasmic tails of glycoproteins. However, it was reported previously that NP expressed by itself is capable of membrane association, and NP has been shown to bind to the cytoplasmic face of plasma membrane raft domains (3, 20).
The multifunctional M2 protein has been shown to be a proton-selective low-pH-activated ion channel essential for the release of the M1 protein from RNPs and allowing the transport of RNPs into the nucleus (21, 22). In addition, M2 has a role in virus assembly and budding. The cytoplasmic tail of M2 is necessary for the efficient incorporation of vRNPs into progeny virions and the production of infectious particles, and a small region (residues 71 to 73) has been identified as being important for interacting with M1 (23–25). Residues 45 to 62 of the M2 cytoplasmic tail form an amphipathic helix, which has been implicated in playing a role in the budding process by inducing membrane curvature (26). Mutation of the amphipathic helix of M2 results in altered budding, as viral filament formation is impaired (25, 27, 28). A functional link between M1 and M2 was demonstrated when escape mutants of a monoclonal antibody (14C2) specific for the M2 ectodomain were found to restrict virus growth and were mapped to the M1 protein (29). The M2 protein does not associate with rafts despite possessing a CRAC domain, which has been shown in other proteins to mediate cholesterol binding (30). The CRAC domain in M2 may provide an affinity for cholesterol-rich regions of the budozone, where it would be positioned to facilitate the scission event at the end of the budding process (27).
Influenza virus buds from cholesterol-rich regions of plasma membranes that are thought to be formed by the coalescence of membrane raft domains. The glycoproteins HA and NA intrinsically concentrate within membrane rafts (31–35). When expressed by themselves, these proteins were capable of bringing together the cholesterol-rich regions into larger membrane domains (32, 34). The cytoplasmic tails of HA and NA are believed to play a role in the organization of budding. Recombinant viruses lacking the NA cytoplasmic tail or both the HA and NA cytoplasmic tails have altered morphology, and the double tail deletion mutant had reduced infectivity (36–38). Additionally, the cytoplasmic tail-minus mutations of HA and NA were found to have a reduced affinity for membrane rafts (39).
It is presumed that there is an interaction between the HA and NA cytoplasmic tails and M1 in part due to their proximal locations in the viral particle. However, efforts to show a direct interaction have yielded contradictory results. Enami and Enami (40) reported previously that the cytoplasmic tails of HA and NA stimulated the association of M1 with cellular membranes, and Ali et al. (41) showed previously that membrane-bound M1 became detergent resistant in the presence of HA and NA, suggesting an association with raft domains in which the glycoproteins are concentrated. In contrast, other reports indicated that the expression of HA and NA did not influence the membrane association of M1 (42, 43). The cytoplasmic tails of both HA and NA contain apical targeting signals, and in virus-infected polarized cells, progeny viruses bud from the apical membrane surface. However, when the cytoplasmic tail of HA was mutated to redirect the protein to the basolateral surface, influenza virus budding still occurred at the apical surface (44, 45). Additionally, redirected HA did not affect the location of NA and M2 (45).
The formation of virus-like particles (VLPs) in cells expressing influenza virus proteins has been used as a model system to identify the components sufficient to support specific budding events. Initial reports suggested that M1 was capable of producing VLPs by itself (46, 47). However, the expression systems used resulted in the overexpression of M1, which likely resulted in the nonspecific release of vesicles (exosomes) enriched in M1 protein. Zhao and coworkers (15) did not observe budding or vesicles in cells expressing M1 alone. However, when a noncytotoxic expression system was used to express amounts of each protein similar to those synthesized in influenza virus-infected cells, VLPs were produced (48). HA, when expressed alone, was sufficient to generate VLPs, although VLP release was also supported to a lesser extent by the expression of NA or M2 (48). VLP production was enhanced by the coexpression of HA, NA, and M2. However, although individual proteins may be able to mediate vesicle formation and release, this may not reflect the actual viral life cycle, where interactions between viral components as a group may modify the actions of an individual protein. For example, the deletion of the M2 cytoplasmic tail inhibits budding (23, 25), and infection with viruses containing a deletion of the cytoplasmic tail of NA, or the cytoplasmic tails of both NA and HA, resulted in drastically altered morphologies not seen in VLP populations (37, 38).
In an effort to shed some light on the influenza virus protein interactions that result in budding, here we describe and quantify the distribution of influenza virus protein populations in and on plasma membranes. This was done for cells transfected with plasmids expressing individual proteins or influenza virus-infected cells. Flat sheets of plasma membranes from the apical cellular surface were prepared from virus-infected or transfected cells and processed for electron microscopy by a variation of the “membrane rip” technique (49, 50). This type of analysis has been used successfully to examine the membrane distribution of a single protein, such as the IgE receptor FcεR1 and CC chemokine receptor 5 (51–53). These analyses has also been extended to compare two protein populations to each other, showing whether or not they specifically cocluster with each other, for example, prion protein, AP2, transferrin receptor (54), FcεR1, tyrosine kinase Lyn (55), lipid raft markers (56), Ras proteins (57), and a preliminary examination of influenza virus M1, M2, and HA (58). Here the influenza virus proteins HA, NA, M1, M2, and NP were identified by colloidal gold staining, and their individual distributions were analyzed by using the Ripley function (59, 60), a form of nearest-neighbor analysis. In addition, pairwise distributions of these influenza virus proteins were analyzed similarly in virus-infected and transfected cells, and distributions were compared in an effort to better understand the web of protein interactions that lead to influenza virus budding.
RESULTS
Validation of the membrane rip procedure that renders influenza virus budding visible.
CV1 cells were infected with influenza virus (A/WSN/33 [WSN]), and at 12 h postinfection (p.i.), membrane rips (planar sheets) were made and prepared for electron microscopy. Membrane microdomains have been shown to possess an inherent electron density when viewed in sheets of plasma membrane, thus appearing darker than the surrounding area (61). Additionally, the accumulated membrane-associated RNP complexes underlying the membrane surface and the concentration of viral proteins in areas of budding contribute to the electron density of budozones. Nascent influenza virus A/WSN/33 virions were observed to be associated with dark electron-dense regions in the plasma membrane from infected CV1 cells (Fig. 1, blue boxes). Budding WSN virions that were still attached to the CV1 cell plasma membrane were identified by their electron-dense RNP cores (Fig. 1, dashed red circles). WSN virions bud as regular ovoid structures, allowing easier identification of virions than the more pleomorphic A/Udorn/72 (Udorn) virions, which bud as ovoid and filamentous structures (62). The plasma membrane was orientated on the grid with the intracellular surface facing up. Despite viewing the viruses through the thickness of the plasma membrane and the density of the associated proteins, the border of viral glycoprotein spikes could frequently be identified (Fig. 1, inset, yellow arrows).
FIG 1.

Nascent influenza virus virions observed on a membrane sheet. A planar sheet of plasma membrane was prepared from CV1 cells infected with influenza virus A/WSN/33. WSN buds as spherical particles identifiable by electron-dense vRNP cores (red circles). Putative budozones with budding virions were seen as areas of high electron density (examples are in blue boxes). The cytoplasmic face of the membrane is oriented face up; frequently, the viral glycoprotein surface spikes were discernible despite viruses being viewed through the membrane sheet (inset, yellow arrows). Bar, 200 nm.
Proof of concept of immunogold labeling of HA and M2 in virus-infected MDCK cells.
To understand the viral proteins involved in organizing the budozone, planar sheets of plasma membrane were prepared from virus-infected cells or transfected cells that expressed influenza virus proteins. Planar sheets were stained with antibodies followed by immunogold reagents to show the location of proteins on either face of the plasma membrane, as detailed in Materials and Methods. Figure 2A shows a membrane sheet prepared from Udorn-infected Madin-Darby canine kidney (MDCK) cells at 12 h p.i., stained to show M2 and HA proteins (6-nm and 12-nm gold particles, respectively). The two sizes of gold particles can be seen readily in the inset in Fig. 2A, which is an enlargement of the area outlined in black. Clathrin-coated pits were frequently observed as electron-dense structures possessing the characteristic reticular pattern of the clathrin lattice (Fig. 2A, red arrows). The x and y coordinates of each gold particle were determined and used to generate a point pattern within the observation window with dimensions of 3.331 by 3.331 μm and analyzed statistically by using the Ripley K function. A schematic representation of the gold particle distribution is shown in Fig. 2B, with the inset outlining the same area in both panels. Calculation of the Ripley K function of the point pattern, as shown for individual influenza virus proteins in Fig. 3, yields a plot of the number of neighbors (y axis) that each gold particle has within a specified distance (nanometers) (x axis). A bivariate Ripley K function was used to compare the distributions of two different point patterns within the same observation window, here 6- and 12-nm gold particles, or the distribution of a single protein species, as shown in Fig. 3. A linear transformation of the Ripley K function where two populations that are distributed randomly will have a theoretical value of zero was used to present the analysis. The calculations were carried out by using the Spatstat library for R. The results were compared to those from the Ripley analysis of 100 simulated random point patterns of the same point density as the experimental data, which yielded a 99% confidence envelope that defines the limits of complete spatial randomness. This envelope is shown by dotted lines on the graphs. Data falling above the range can be said to be coclustered and not distributed randomly with respect to each other, and data within the range are randomly distributed, while data below the envelope could be said to be exclusive of each other.
FIG 2.

Example of a membrane sheet used for analysis, stained for both M2 and HA. (A) A sheet of plasma membrane from MDCK cells infected with influenza virus A/Udorn/72 at 12 h p.i. that had been stained to show M2 (6-nm gold particles) and HA (12-nm gold particles). In all figures, the proteins identified by 6-nm gold particles are listed first, while those stained with 12-nm gold particles are listed second. To allow easier visualization of gold staining, the inset shows the boxed region of the image at a higher magnification. Clathrin-coated pits with the characteristic lattice structure (red arrows) are visible. (B) The positions of all gold labeling were used to create a list of x,y coordinates for use in subsequent analyses. A diagram of the gold labeling in panel A is shown, with the same area being boxed in both panels. Bars, 200 nm.
FIG 3.

Individual distributions of influenza virus proteins HA, NA, M2, M1, and NP in membranes prepared from Udorn virus-infected cells and cells transfected with DNA expressing the viral proteins. The distributions of the individual influenza virus proteins HA, NA, M2, M1, and NP are shown for infected (left) (blue) or transfected (middle) (red) cells. The average linear transformations from the Ripley analysis for the individual protein distributions are shown for the infected (inf) and transfected (transf) samples in the right-hand column. In this and subsequent figures, “transfected” indicates cells transfected with plasmid DNA expressing the indicated viral protein(s) (n = 2 under each experimental condition). Lr-r, L(r) − r, where L stands for the linear transformation of Ripley’s K function and r is distance, in this case radius.
Individual distributions of influenza virus proteins HA, NA, M2, M1, and NP expressed via infection or transfection.
Given the different inherent properties of the influenza virus proteins, the membrane distributions of the HA, NA, M2, M1, and NP proteins were determined individually by Ripley K function analysis. HA, NA, M2, M1, and NP were expressed in MDCK cells by virus infection or in 293T cells by transfection with the expression vector pCAGGS. The transfection conditions used resulted in expression levels similar to those observed in virus-infected cell populations (48). Planar sheets of plasma membrane were prepared at 12 h p.i. or 24 h posttransfection (p.t.). The samples were stained, imaged, and analyzed as described in Materials and Methods. As expected from previous studies (32), HA was not distributed randomly in the plasma membrane and showed similar self-clustering regardless of whether it was expressed by virus infection or by transfection (Fig. 3). The average density of HA was fairly constant over the 200 nm measured. The decline in the plotted data seen for most samples over the initial 0- to 10-nm range is likely artifactual and caused by the size of the primary and secondary immune complexes. NA is known to be raft associated, and it was found to be strongly self-associated in clusters with sizes similar to those observed for HA (Fig. 3).
M2 showed the strongest self-clustering of the influenza virus proteins examined, with M2 expressed as a result of infection being slightly more clustered and dense on average than M2 expressed by transfection, as shown by the magnitude of the plot (Fig. 3). M1 displayed the weakest self-clustering of the influenza virus proteins examined, showing almost the same pattern regardless of the means of expression (Fig. 3), with both plots being nearly coincident. Similarly, NP was self-clustered to the same magnitude regardless of the means of expression (Fig. 3). Presumably, the association with viral RNA in infected cells or host cell RNA in transfected cells contributed to the strong self-clustering observed for NP. The distributions of the three P proteins (PB1, PB2, and PA) and nuclear export protein (NEP) (NS2) were not examined due to a lack of suitable antibodies and the unlikelihood that these proteins have a role in budding.
Relative plasma membrane distributions of influenza virus HA, NA, and M2 proteins.
Given the complex roles that the viral surface proteins play in the viral life cycle, the relative distributions of HA and NA were compared to those of M2 and the M2 mutant mut1M2 (Ud7+UdM1/M2-Mut1), a mutant with alanine substituted at positions 71 to 73 in the M2 cytoplasmic tail. It was shown previously that this mutant greatly impairs virus replication (23). We showed previously that HA and M2 strongly coclustered in membranes prepared from virus-infected cells (23, 28). Here the same observation was made when HA and M2 were expressed transiently (Fig. 4A). M2 and HA strongly colocalized and were frequently observed as large concentrations of HA with smaller dense patches of M2 around the periphery (Fig. 2). The analysis of the coclustering of M2 and HA gave a curve indicating strong interactions over the entire distance measured. M2 is not concentrated in lipid rafts, unlike HA; thus, the distribution observed may reflect M2 interacting with a membrane domain rather than HA itself (30, 32, 39, 63, 64). While the distribution of mut1M2 compared to HA was not random in nature, it was reduced by ∼70% from that seen for wild-type (wt) M2 at 100 nm (Fig. 4A).
FIG 4.
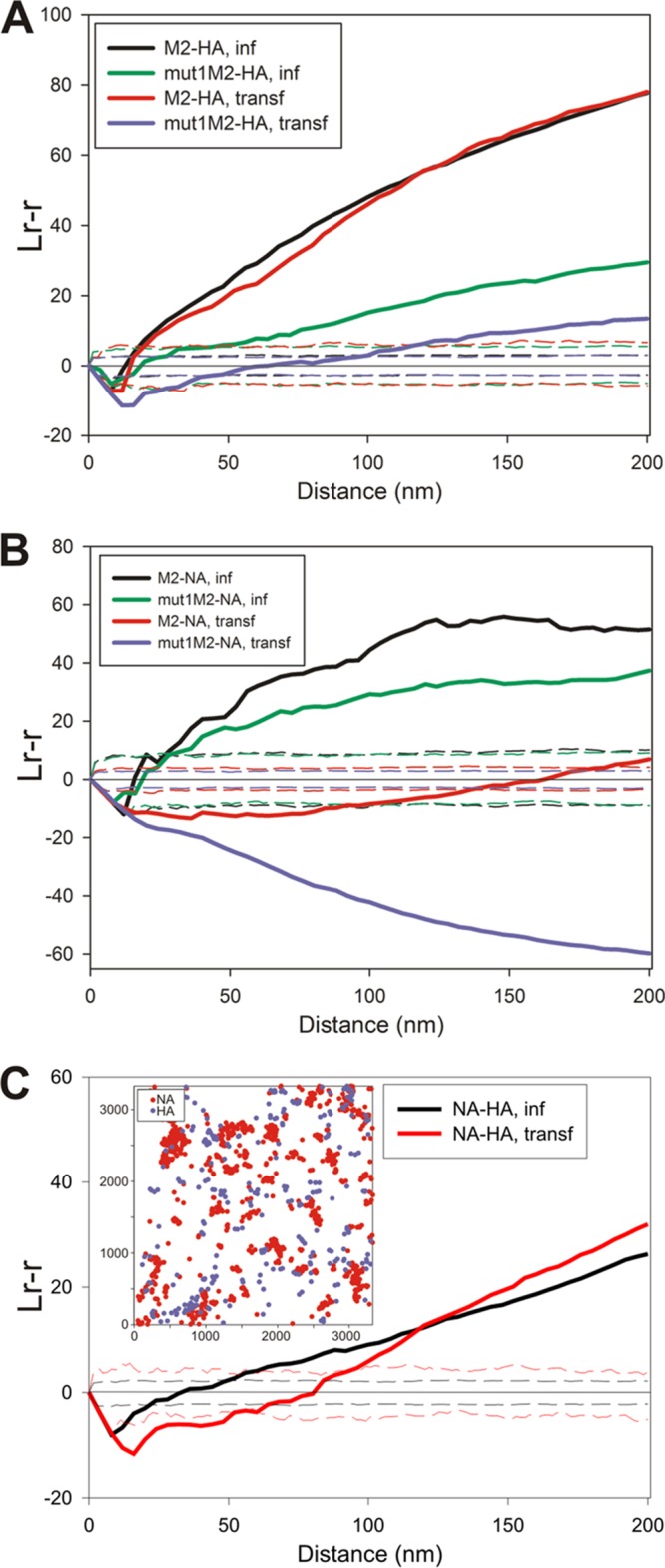
Relative distributions of influenza virus cell surface-expressed proteins. Planar sheets of plasma membrane were prepared from either WSN-infected MDCK cells at 12 h p.i. or 293T cells at 24 h posttransfection. Membranes were labeled with immune reagents as described in Materials and Methods and examined by electron microscopy. (A) Analysis of the density of HA (12-nm gold particles) surrounding each M2 protein (6-nm gold particles) for a maximum radius of 200 nm, presented as an average linear transformation of data from a bivariate Ripley analysis. The distributions of both wild-type M2 and mut1M2 relative to HA were examined in virus-infected MDCK and transfected 293T cells (n = 5 for M2-HA in infected cells, n = 4 for mut1M2-HA in infected cells, n = 4 for M2-HA in transfected cells, and n = 3 for mut1M2-HA in transfected cells). (B) Distributions of M2 or mut1M2 (6-nm gold particles) and NA (12-nm gold particles) in virus-infected and transfected cells were compared by Ripley analysis (n = 4 for M2-NA in infected cells, n = 4 for mut1M2-NA in infected cells, n = 2 for M2-NA in transfected cells, and n = 2 for mut1M2-NA in transfected cells). (C) The spatial arrangements of HA (12-nm gold particles) and NA (6-nm gold particles) were compared to each other in virus-infected and transfected cells. The inset shows a graphical representation of the gold particle distribution for a representative image showing the separate but juxtaposed domains occupied by HA and NA, as described by the bivariate Ripley analysis (n = 3 for NA-HA in infected cells, and n = 4 for NA-HA in transfected cells).
In virus-infected cells, the distribution of NA compared to M2 was similar to that observed for HA in that there was strong coclustering, and this was reduced upon infection with a virus expressing mut1M2 (Fig. 4B). However, unlike HA, when NA and M2 were coexpressed by transfection, they were distributed randomly with respect to each other. When the distributions of coexpressed NA and mut1M2 were compared, the data indicated that the two protein populations occupied distinct and different membrane regions since the data fell below the range defining a random distribution.
When the membrane distribution of HA was compared to that of NA in virus-infected or transfected cells (Fig. 4C), there was little difference observed between the patterns. In both cases, the two proteins occupied separate membrane regions over the initial distances measured (25 to 50 nm), but at distances of more than ∼75 to 100 nm, HA and NA were then distributed in a nonrandom fashion. The shape of the bivariate Ripley K function was consistent with HA and NA occupying separate membrane domains that were adjacent and therefore could be described as being coclustered after 75 to 100 nm. The inset in Fig. 4C shows a typical point pattern, with NA in red and HA in blue. The two protein populations were not randomly distributed with respect to each other, yet the two markers clearly occupied separate but adjoining spaces.
M1 coclusters with NA and M2, but coclustering with HA occurs only in virus-infected cells.
The distribution of M1 was examined in planar sheets of plasma membrane and compared to those of HA, NA, and M2. M1 and HA coclustered in the plasma membrane of virus-infected MDCK cells but not when expressed by transfection in 293T cells (Fig. 5A), suggesting that other viral components were responsible for the close proximity of M1 to HA during infection. NA and M1 very weakly colocalized during infection, consistent with NA residing in a different membrane domain from that of HA. However, when NA and M1 were expressed together by transfection, there was strong coclustering evident (Fig. 5A), contrary to what was observed for HA. These results suggest that the presence of other viral proteins places constraints on the distribution of NA, M1, or both. M2 colocalized with M1 when expressed by infection or expressed by transfection (Fig. 5B). When M1 was expressed along with mut1M2, the two protein populations were distributed randomly with respect to each other (Fig. 5B), confirming our previous results (23). Dense concentrated areas of M1 staining were seldom observed in samples prepared from either virus-infected MDCK cells or transfected 293T cells. This was unexpected, as dense concentrations of M1 would be predicted if M1 were driving budding. It is possible that the epitopes in dense patches of M1 were inaccessible to the antibodies, although M1 staining was attempted with three different primary antibodies and was detected by using both 6-nm and 12-nm secondary gold reagents, with little difference in label densities other than the slight increase in sensitivity expected by the use of a smaller gold particle.
FIG 5.
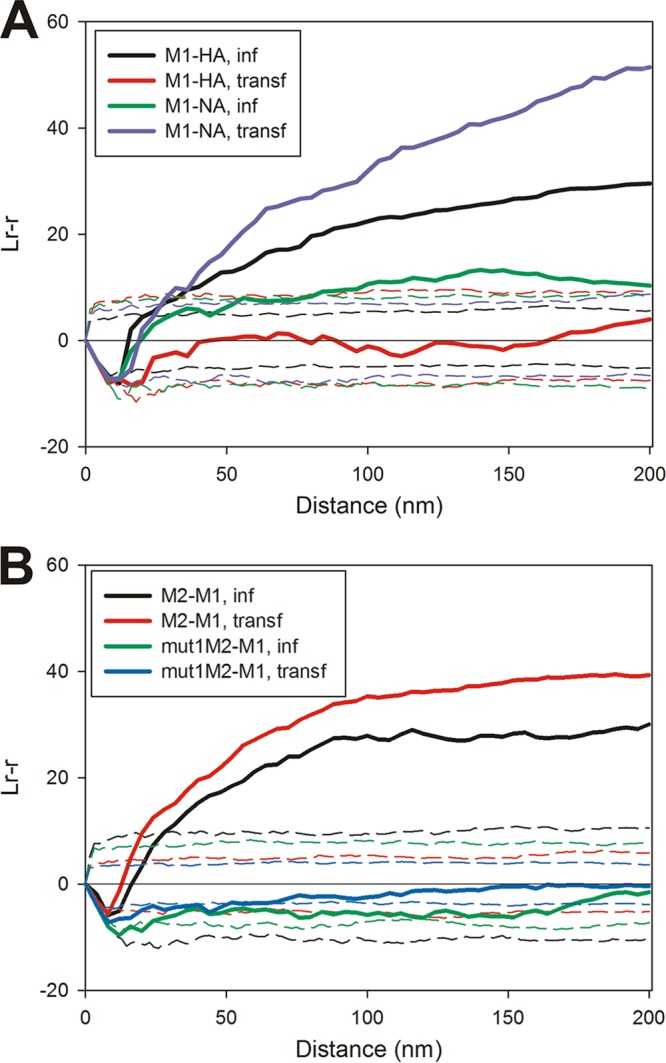
Relative membrane distribution of M1 compared to influenza virus surface proteins. (A) Codistribution of M1 (6-nm gold particles) relative to HA or NA (12-nm gold particles) determined by bivariate Ripley analysis of stained sheets of planar membrane prepared from virus-infected and transfected cells (n = 4 for M1-HA in infected cells, n = 4 for M1-HA in transfected cells, n = 3 for M1-NA in infected cells, and n = 3 for M1-NA in transfected cells). (B) Bivariate Ripley analysis comparing the distribution of membrane-associated M1 (12-nm gold particles) to that of M2 or mut1M2 (6-nm gold particles) (n = 4 for M2-M1 in infected cells, n = 2 for M2-M1 in transfected cells, n = 4 for mut1M2-M1 in infected cells, and n = 2 for mut1M2-M1 in transfected cells).
Distribution of membrane-associated NP.
When NP and HA were examined in virus-infected or transfected cells, NP and HA coclustered in virus-infected cells (Fig. 6A), but in transfected 293T cells, they were found to be distributed randomly with respect to each other: the Ripley K function fell within the envelope defining complete spatial randomness. To identify the viral proteins that are necessary to bring HA in close proximity to NP, 293T cells were transfected to express HA, NP, and M2 or HA, NP, M2, and M1, and sheets of membrane were then immunostained for NP and HA. In both cases, the populations of NP and HA were not coclustered and remained distributed randomly with respect to each other (results not shown). However, when NA was expressed along with HA, NP, M1, and M2, coclustering occurred for NP and HA although at ∼58% of the magnitude of infected samples at a radius of 100 nm. The difference in the magnitude of coclustering observed between infected and transfected samples is likely due to the contributions of other viral (and perhaps host cell) factors in organizing the budozone. We did not test any possible role, if any, of the polymerase complex or NEP (NS2) in the organization of budding due to a lack of a suitable antibodies.
FIG 6.

Spatial relationship of membrane-associated NP relative to HA, NA, M2, or M1. (A) Distribution of NP (6-nm gold particles) compared to that of HA (12-nm gold particles) in infected and transfected cells. The results of a bivariate Ripley analysis of NP and HA staining are also shown for 293T cells, which were transfected to express M1, M2, NA, NP, and HA (n = 3 for NP-HA in infected cells, n = 3 for NP-HA in transfected cells, and n = 3 for M1-M2-NA-NP-HA in transfected cells). (B and C) Relative positions of NP (6-nm gold particles) and NA (12-nm gold particles) (B) or NP (6-nm gold particles) and M1 (12-nm gold particles) (C) in membranes prepared from infected or transfected cells described by a bivariate Ripley analysis (n = 3 for NP-NA in infected cells, n = 3 for NP-NA in transfected cells, n = 4 for NP-M1 in infected cells, and n = 5 for NP-M1 in transfected cells). (D) Comparisons of the distributions of NP (12-nm gold particles) and M2 (6-nm gold particles) in infected and transfected cells. The degree of colocalization between NP and either M2 or mut1M2 cotransfected with M1 was also examined (n = 4 for M2-NP in infected cells, n = 5 for M2-NP in transfected cells, n = 2 for M1-M2-NP in transfected cells, and n = 2 for M1-mut1M2-NP in transfected cells).
When NP and NA were coexpressed by virus infection or transfection, the relative distributions of the two populations fell at the border between coclustered and random (Fig. 6B). If budding initiates within the barges of HA that define the budozone, the bivariate Ripley K function comparing NP to NA may be a result of NA occupying the periphery of the HA domains. NP and M1 were found to be coclustered when they were expressed by either infection or transfection (Fig. 6C), although the magnitude of colocalization was slightly higher when the proteins were expressed by transfection in the absence of other viral factors.
M2 strongly coclustered with NP in infected cells (Fig. 6D). When M2 and NP were expressed by transfection, they did not cocluster and were observed to occupy distinct areas of the membrane. Since M1 strongly associated with both NP and M2, the ability of M1 to “bridge” an interaction between M2 and NP was tested. When M2, NP, and M1 were cotransfected into 293T cells, it was found that M2 and NP now showed a colocalization although at 24% of what was observed for infected cells at a radius of 100 nm. To test the specific nature of this change in the M2-NP distribution, mut1M2, which is defective in associating with M1, was expressed by transfection in 293T cells, along with M1 and NP. Under these conditions, the surface populations of NP and mut1M2 were distributed randomly compared to each other, even though M1 was coexpressed. The coexpression of HA and NA along with M2, NP, and M1 did not increase the magnitude of the coclustering observed for M2 and NP (results not shown). It is possible that the viral RNA polymerase or NEP could play a role in the spatial organization of M2 and NP in infected cells.
The cytoplasmic tails of the surface glycoproteins have a role in organizing the budozone.
A mutant virus lacking both the HA and NA cytoplasmic tails was previously characterized (37) and was used here to examine the effect of these deletions on the relative protein distribution in plasma membranes from virus-infected MDCK cells. The same tail deletions in HA and NA were expressed in 293T cells via transfection. In mutant virus-infected cells, tail-minus HA and tail-minus NA exhibited similarly reduced coclustering with M2 compared with wild-type HA and NA (compare Fig. 4A and 7A). This may reflect the reduced raft association of both tail-minus HA and NA (35). There was a slightly stronger colocalization between HA lacking a cytoplasmic tail and M2 when expressed by transfection, while NA lacking the cytoplasmic tail and M2 did not cocluster and can be described as being distributed exclusively of each other (Fig. 7A), similar to what was observed for wild-type NA and M2. The tail deletion mutants of HA and NA were found randomly distributed with respect to M1 in both infected and transfected cells (Fig. 7B). Wild-type NA exhibited strong coclustering with M1 when expressed by transfection (Fig. 5A); this was completely eliminated by the truncation of the cytoplasmic tail of NA. HA colocalized with M1 in infected cells only, yet this too was abrogated upon the elimination of the cytoplasmic tail of HA.
FIG 7.
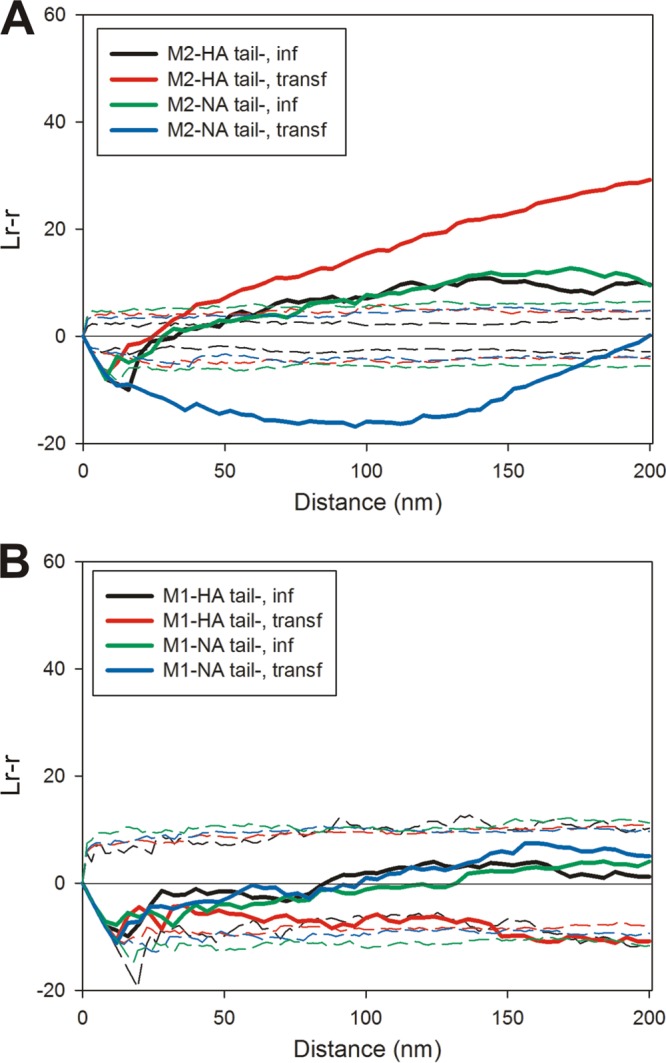
Roles of the cytoplasmic tails of HA and NA in protein colocalization. MDCK cells were infected with recombinant influenza virus encoding HA and NA lacking their cytoplasmic tails. (A) The distributions of the cytoplasmic tail-less glycoproteins (12-nm gold particles) were compared to that of M2 (6-nm gold particles). Transfected 293T cells expressing M2 and the cytoplasmic tail deletion mutants of either HA or NA were also examined (n = 2 for M2-HAtail− in infected cells, n = 2 for M2-HAtail− in transfected cells, n = 2 for M2-NAtail− in infected cells, and n = 2 for M2-NAtail− in transfected cells). (B) Spatial localization patterns of M1 (6-nm gold particles) were compared to those of the cytoplasmic tail-minus mutants of HA and NA (12-nm gold particles) in virus-infected MDCK cells and separately in transfected 293T cells by a bivariate Ripley analysis (n = 2 for M1-HAtail− in infected cells, n = 3 for M1-HAtail− in transfected cells, n = 4 for M1-NAtail− in infected cells, and n = 2 for M1-NAtail− in transfected cells).
DISCUSSION
Influenza virus buds from regions of the plasma membrane that are sphingomyosin rich, cholesterol rich, and detergent resistant, called lipid rafts. HA and NA are known to associate with lipid rafts (31, 34, 35, 65–67). As the area of HA or NA concentration greatly exceeds previously reported size estimates for lipid rafts (commonly <100 nm but ranging from <5 nm to ∼700 nm) (68, 69), it is believed that these proteins possess the inherent ability to coalesce these specialized domains into so-called “barges of rafts” (63), likely through protein-protein interactions. The size of the HA clusters was variable and spanned a large range of sizes, unlike the traditional definition of lipid rafts; this is likely driven by strong protein-lipid and protein-protein interactions and is consistent with data from previous reports (31, 32). The M2 protein, whose cytoplasmic tail amphipathic helix has a major role in creating curvature of the membrane and in the scission of budding particles (26, 70–73), is not raft associated but can bind cholesterol and yet does not cluster with raft markers (26, 30, 32, 39, 70). Many roles for the M1 protein in budding have been proposed, especially in binding to lipid bilayers, binding to the RNPs, and interacting with the cytoplasmic tails of HA, NA, and M2. However, evidence for these interactions, although indicated by genetic experiments, have been difficult to demonstrate at the biochemical level. A role for host cell proteins in budding has been proposed, e.g., RAB11A, (74), tetraspanin CD81 (75), the UBR4 ubiquitin ligase (which associates with the M2 cytoplasmic tail and promotes apical transport) (76), and a serpin necessary to cleave HA (64).
To understand the assembly of the budozone, we have characterized the distributions of the major influenza proteins HA, NA, M1, M2, and NP, singly and pairwise, using planar sheets of plasma membrane prepared from virus-infected or transfected cells expressing one or more of the viral proteins. Spatial relationships between influenza virus proteins were quantified via immunogold staining, and distribution analysis of immunogold particles was done by using the Ripley K function (59, 60). Our findings are summarized in Fig. 8. HA and M2 were observed to strongly cocluster. It is not clear whether this coclustering was due solely to protein-protein interactions or whether protein-lipid interactions also played a role. M2, while not raft associated like HA, contains a cholesterol binding motif. Despite being raft associated, NA was present in membrane regions that were distinct and separate from those containing HA yet juxtaposed to the HA microdomains. The separate nature of the HA and NA distributions mirrors the distribution of NA in nascent virions, where it is frequently found in a discrete, homogeneous patch. As expected, NA colocalizes with M2 during infection; however, when the two proteins were coexpressed by transfection, they did not cocluster. During infection, M1 coclustered with HA but only weakly with NA, yet when these proteins were expressed by transfection, almost the opposite was observed: M1 strongly colocalized with NA but not at all with HA. A possible explanation for this is that M1 is drawn away from NA by other viral proteins expressed during infection, such as M2, with which M1 strongly colocalizes during infection or transfection. The coclustering of M1 and M2 could also explain the colocalization between M1 and HA during infection if M2 served as a bridge between the two proteins. NP is distributed randomly with respect to HA when cotransfected; however, the coexpression of other viral proteins can at least partially restore the coclustering observed during infection. NA has an extremely weak interaction with NP whether during infection or upon transfection, perhaps reflecting the unusual distribution of NA with respect to the distribution of HA. M1 strongly colocalizes with NP regardless of the means of expression. M2 strongly coclusters with NP during infection, but these proteins occupy distinct membrane regions when expressed alone. The colocalization of M2 and NP observed during infection might be facilitated by the presence of M1. The nature of the described protein-protein coclustering may be the result of interactions between proteins; however, lipid-lipid and lipid-protein interactions may also play a role. We were unable to assess any contributions of the viral RNA polymerase complex of the NEP protein to the organization of the influenza virus budozone since we lacked suitable immunological reagents for these proteins. Steps were taken to minimize the chances of antibody-induced clustering; however, the possibility remains that it contributed to the distributions observed. The protein expression levels and distributions of the individual proteins were very similar in both virus-infected and transfected cells (48).
FIG 8.
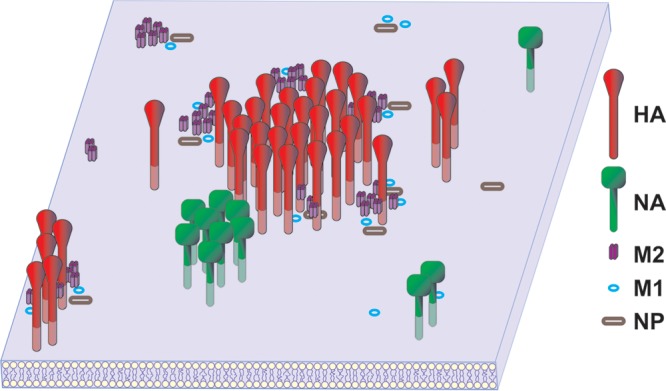
Model of influenza virus protein distribution in an infected cell membrane. Shown is a graphic representation of the distributions of the HA, NA, M1, M2, and NP proteins in the membrane of an influenza virus-infected cell. While some pairs of proteins, such as HA-M2, NA-NA, M2-M1, and NP-M1, cocluster, other protein pairs, such as HA-M1, NA-M2, NP-HA, and M2-NP, may be brought together at the site of budding by indirect protein or lipid interactions. For instance, the colocalization of M2 and NP may be facilitated by both proteins coclustering with M1. While HA, NA, and M2 are integral membrane proteins, both M1 and NP are represented by open symbols to denote that they are associated with the cytoplasmic face of the plasma membrane.
We and others showed previously that HA and M2 exhibit an intrinsic ability to strongly cocluster in the presence or absence of other viral proteins (23, 77) By using fluorescence lifetime imaging-fluorescence resonance energy transfer (77), it was found that while M2 and HA cocluster, M2 does not segregate with markers for the lipid rafts in which HA is concentrated. M2 is found in small, dense clusters generally on the periphery of areas of concentrated HA (Fig. 2). This may be due to M2 binding cholesterol at the edge of raft domains due to a CRAC domain in the M2 cytoplasmic tail amphipathic helix (26, 30, 70, 78), although a disruption of the CRAC domain in M2 of influenza virus A/WSN/33 does not affect virus replication (79).
The strong colocalization of HA with M2 that we observed is not reflected in the final protein composition of the viral membrane, where HA is estimated to comprise ∼80% of the viral surface protein, NA is estimated to comprise ∼17%, and M2 is estimated to comprise only 4 to 15 tetramers per virion (reviewed in reference 7). However, the pattern of coclustering of HA and M2 may reflect the role that M2 is believed to play in scission; by occupying the periphery of HA patches, M2 is positioned to concentrate at the base of nascent budding virions (70). The amphipathic helix in the cytoplasmic tail of M2 has been shown to modify membrane curvature and is necessary for efficient membrane scission and virus release (28, 70–72). When the distribution of HA or NA is compared to that of a mutant M2 protein with cytoplasmic tail residues 71 to 73 replaced with alanine, a mutation known to impair virus growth, likely by interrupting the interaction of M2 with M1 (23), the magnitude of coclustering is dramatically reduced, suggesting a direct or indirect impact on HA or NA interactions (Fig. 4A and B).
Mutant HA and NA proteins lacking their cytoplasmic tails showed reduced coclustering with M2 during virus infection, and this could be the result of a reduced raft association of the tail-minus forms of these proteins (39). Surprisingly, when the distribution of HA was compared to that of NA, the glycoproteins were found to occupy distinct but adjacent membrane domains, even though both proteins are found in rafts (Fig. 4C). This was shown by HA and NA exhibiting coclustering only at a radius that exceeded 75 to 100 nm, thus suggesting that the 2 proteins are not intermixed over a short distance, i.e., in the same membrane domain, but rather reside in adjacent domains. It is not known if HA and NA are separated because they occupy lipid domains of different compositions or if the separation is due to protein-protein interactions.
We reported previously that HA and NA resided in the same plasma membrane domains (80). This discrepancy is likely due to limitations of the previous method of quantitation in analyzing areas of dense gold labeling where cell profiles in thin sections were used. The density of the label when seen in profile necessitated the use of cells at 6 h p.i. or sooner. The observed distribution of NA in separate discrete domains observed here seems to reflect the nature of the viral membrane, where NA has been characterized as residing in discrete homogeneous patches distinct from HA (8, 81).
The expression of HA alone (together with sialidase activity supplied by NA or added exogenously) was sufficient to generate VLPs. However, the expression of NA and M2 together with HA enhanced the release of VLPs, and although M1 was not necessary for the production of VLPs, its presence enhanced the formation of infectious VLPs (48). M1 is reported to form a dense layer immediately beneath the lipid membrane of the virus and by virtue of its location in a position to link the glycoproteins with the vRNP. Supporting the idea of a structural role is the observation of ordered assemblies of what is believed to be M1 in fractions prepared from detergent-solubilized virus (13, 82). Biochemical experiments examining the possible interaction of M1 with the cytoplasmic tails of HA and NA have yielded contradictory results (39–42). In planar sheets of membrane from virus-infected or transfected cells, we did not observe areas of dense M1 staining that would be expected if M1 drives the organization and budding of nascent virions. Furthermore, M1 strongly colocalizes with NA but is distributed randomly with respect to HA when the proteins are expressed by transfection. The cytoplasmic tail of NA plays a role in its association with M1; when the NA cytoplasmic tail is deleted, the two proteins are no longer colocalized (Fig. 7). We confirmed data from our previous studies, which showed that the strong coclustering observed between M1 and M2 was eliminated when mut1 M2 is expressed, which was expected, since the mutation spans the sequence reported to be responsible for interacting with M1 (23, 39). Furthermore, the omission of M2 from a VLP system resulted in an ∼50% decrease in M1 incorporation, and M1 could be coimmunoprecipitated with M2 (23).
M1 binds to vRNP complexes and associates with the cytoplasmic tail of NA, consistent with the idea that M1 serves as a linker between the viral genome and other viral membrane components (10, 12, 83, 84). Although HA and NP strongly colocalize in infected cells, when expressed by itself, HA is distributed randomly with respect to NP. The coexpression of NP and HA along with M1, or M2 and M1, did not alter their distributions (data not shown). Even though M1 was shown to strongly cocluster with both NP and M2, and M2 strongly colocalizes with HA, M1 was unable to bridge HA to NP in the presence of M2. The expression of NP-HA-M1-M2-NA together resulted in the coclustering of NP and HA, indicating that the budozone is the product of a complex, interwoven network of interactions. The weak coclustering observed for NA and NP in both virus-infected and transfected cells may reflect the different membrane distribution of NA compared to that of HA and the lower copy number of NA than of HA in nascent virions. This could also be reflective of a reduced role for NA in the budding process compared to HA. In a VLP system, the release of NA was greatly enhanced by the expression of HA (48). In addition, the data presented here suggest that NP and M2 do not inherently directly associate. Deletions of portions of the M2 cytoplasmic tail reduce RNP incorporation into virions, likely by eliminating the interaction between M1 and M2 (25, 85). We show that the coexpression of M1 brokers an interaction between M2 and NP although not at the levels observed in virus-infected cells, suggesting that other viral or cellular factors could play a role. This bridge could be disrupted by the expression of a mutant M2 protein containing substitutions at the site in the M2 cytoplasmic tail believed to be responsible for interacting with M1.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
MDCK cells, human embryonic kidney 293T cells, and monkey kidney CV1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Cells were maintained in a humidified incubator at 37°C with 5% CO2. Influenza virus A/Udorn/72 was propagated in MDCK cells by standard methods (86). Recombinant influenza virus A/Udorn/72 encoding alanine at positions 71 to 73 of the M2 cytoplasmic tail (mut1M2) was propagated as described previously (23). Recombinant A/Udorn/72 expressing both HA and NA lacking cytoplasmic tails was previously described and characterized (37). CV1 cells were infected with influenza virus A/WSN/33 at a multiplicity of infection (MOI) of 3 and MDCK cells were infected with influenza virus A/Udorn/72 at an MOI of 5 by using standard protocols (86). After the virus was allowed to attach for 1 h at 4°C with gentle rocking, the inoculum was replaced with prewarmed medium, and the cultures were returned to 37°C and used at the indicated times postinfection. Viral proteins were expressed transiently in 293T cells by using the eukaryotic expression vector pCAGGS (87). Transfection was carried out by using Fugene 6 (Roche Applied Science, Indianapolis, IN) according to the manufacturer's directions. The amount of each plasmid as well as the total amount of DNA transfected were optimized to yield protein expression levels comparable to those found in virus-infected cells (48). Protein expression levels in transfected cell populations were monitored by Western blotting.
Antibodies.
HA was detected by using goat anti-influenza virus [A/Equine/Miami/1/63 (H3 HA); American Type Culture Collection (ATCC)] or by using mouse monoclonal antibody C45/3 (ATCC). NA was labeled by using goat anti-influenza virus NA N2 (A/Singapore/1/1957; ATCC). M2 was stained by using mouse monoclonal antibody 14C2 (6). M1 was detected by using goat anti-matrix protein antibody or mouse monoclonal antibody JZc8 specific for M1 (88). NP was stained by using mouse monoclonal antibody 20343 (Abcam, Cambridge, MA) or rabbit polyclonal serum produced by using purified WSN NP and custom prepared (Harlan Laboratories, Indianapolis, IN). Optimal working dilutions for each antibody were determined based upon the observed signal-to-background ratio. Host-specific secondary antibodies conjugated to either 6-nm or 12-nm colloidal gold were purchased from Jackson ImmunoResearch (West Grove, PA) and used at a dilution of 1:30. Gold conjugates were checked for aggregates and size distribution by negative staining and transmission electron microscopy.
Preparation and analysis of membrane sheets.
MDCK cells at 12 h p.i. or 293T cells 24 h p.t. were used for the preparation of planar sheets of plasma membrane. The cells were grown on acid-cleaned, sterile glass coverslips that had been treated with poly-l-lysine. Membrane sheets were prepared by the so-called rip technique first described by Sanan and Anderson (49). Briefly, cells were blocked at 4°C by incubation in radioimmunoprecipitation (RIP) buffer (100 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4-H2O [pH 7.3]) containing 0.2% ovalbumin and 0.1% normal donkey serum. Cell surface antigens were labeled with specific immunoglobulins followed by host-specific gold-conjugated secondary antibodies. Antibodies were diluted in blocking buffer with the addition of 0.1% partially linearized, acylated bovine serum albumin (BSA) (Electron Microscopy Sciences, Hatfield, PA). Following washing in blocking buffer, the cells were washed 4 times and incubated for 15 min in HKM buffer [25 mM HEPES (pH 7.1), 25 mM KCl, 25 mM Mg(CH3COO)2] (89). Formvar- and carbon-coated 300-mesh nickel grids were glow discharged immediately before use and poly-l-lysine treated. A piece of nitrocellulose membrane was placed upon chilled plate glass, and several electron microscope grids were placed, face up, on the filter. The nitrocellulose provided a smooth surface that absorbed excess buffer and helped minimize any sideways motion when pressure was applied. A coverslip with adherent cells was drained of excess buffer and placed cell side down against the grids. A small piece of filter paper was placed over the coverslip, followed by a rubber stopper, and a 100-ml Erlenmeyer flask containing sufficient water to bring its weight to 100 g was centered on the stopper and left there for 10 to 15 s. The coverslip with grids adherent to the cell monolayer was placed cell side up on a piece of filter paper, HKM buffer was pipetted carefully around the edges of the grids, and the grids were floated off the coverslip. If it was necessary, the bevel of a fine-gauge needle was inserted under the edge of the grid to help release it: care was taken to avoid any sideways shearing motion. The grids now had the apical cell membrane adherent to the support film, thus exposing the cytoplasmic face of the membrane. Samples were mildly fixed by floating the grids face down on drops of 2% formaldehyde in 0.1 M phosphate buffer (pH 7.3) for 15 min. For samples where antigens on the inner face of the plasma membrane were to be stained, grids were washed with phosphate buffer, blocked, and reacted with the specified primary and secondary antibodies. Finally, grids were fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3), washed, treated with freshly prepared 1% aqueous tannic acid for 10 min, washed twice for 5 min with distilled water, and air dried. In the cases where only proteins exposed on the external membrane surface were stained, grids were fixed directly with glutaraldehyde following isolation of the membrane sheets and processed from there. Procedures reported previously by others included postfixation with osmium tetroxide and staining with uranyl acetate (49, 50). In the cell lines that we used, the latter treatments increased the electron density of the isolated membranes, making the identification of gold particles more difficult; therefore, these steps were not used. Omitting these treatments did not affect the morphology or stability of the membranes when exposed to the electron beam. When membranes were stained with secondary gold-conjugated antibodies alone, the average backgrounds from 10 images were 2.28 ± 1.59 particles per μm2 for 6-nm gold and 0.15 ± 0.11 particles per μm2 for 12-nm gold. The level of background staining varied from a low level of 1.72 ± 1.4 particles per μm2 to a high level of 11.54 ± 4.51 particles per μm2 depending upon the primary antibody used and the size of the gold particle used for detection. For the proteins examined in the cell types used, it was necessary for the membrane rip protocol to be carried out as a continuous procedure. The introduction of a break, even when isolated membranes were fixed with formaldehyde and floated on buffer at 4°C overnight, resulted in the partial loss and rearrangement of the label. In addition, the analysis of pairwise distributions was not affected by the size of the gold particles. For instance, pairs of proteins were stained with 6-nm and 12-nm immunogold reagents and also the reverse, with no alteration in the outcome of the distribution analysis. The exception was for M2 in the membranes of virus-infected MDCK cells, where bound M2-specific antibodies could not be efficiently stained by 12-nm gold particles. This was likely due to the density of HA and NA in the membrane surrounding the short ectodomain of M2, combined with the relatively reduced sensitivity of the larger 12-nm gold conjugate.
Grids were examined in a JEOL 1230 electron microscope operating at 80 kV, and images were taken with either a Hamamatsu ORCA or a Gatan 831 charge-coupled-device (CCD) camera. Each image encompassed an area of plasma membrane of ∼11.1 μm2, and each image was taken for a separate membrane sheet, thus most likely originating from a different individual cell. Since previous work showed that some influenza virus membrane proteins are organized into large patches on the cell surface (31–34), the area of the membrane analyzed was relatively large, with a radius of 200 nm surrounding each gold particle. The analysis of the influenza virus protein distribution had to consider the possibility of relationships between membrane domains and not only protein-protein interactions, which would be expected to occur over a smaller distance. Under each experimental condition, 9 to 12 representational images were selected from individual experiments, given by n in the figure legends, and analyzed, and the data are presented as average L(r) − r values (where L stands for the linear transformation of Ripley’s K function and r is distance, in this case radius). To determine the limits of complete spatial randomness, 100 Monte Carlo simulations were run at the same point density as the experimental images.
Data analysis.
The MDCK cell line used had regions of plasma membrane and some cellular structures such as clathrin-coated pits that exhibited a high degree of electron density. This, coupled with the relatively low magnification of the images, made it necessary for the position of gold particles to be marked manually by using Photoshop (Adobe, San Jose, CA). The resulting images were segmented, and an x,y coordinate list of gold particle positions in each image was generated by using Image J (90). Ripley (59, 60) described a means of analysis of spatial point patterns that goes beyond the expected average density per unit area. This analysis tests spatial point patterns in a defined sampling window for randomness based upon nearest-neighbor distances. This analysis was first devised to describe the nonrandom distribution of a stand of naturally occurring redwoods (59). The Spatstat package (91) in R (http://www.R-project.org/) was used for the statistical analysis of the distributions of gold particles. The particle coordinates were analyzed by using the Ripley K function, where the expected number of neighbors for each point is given within a specified radius. The Ripley K function describes the distribution of a single population over a specified distance, while the bivariate Ripley analysis compares the relative distributions of two populations by mapping distances from each large gold particle (here 12 nm) to each small gold particle (6 nm) and vice versa. If a gold particle has more neighbors than would be expected for a given label density, the two markers are not distributed randomly with respect to each other, and the two populations can be described as coclustering or being colocalized. Complete spatial randomness is described by performing Monte Carlo simulations using the same number of points as those that are contained in the experimental data set. The data are presented as a linear transformation, where complete spatial randomness has a theoretical value of zero. Envelopes surrounding the theoretical value of complete spatial randomness (a horizontal line extending from zero on the y axis) are derived from 100 Monte Carlo simulations; if data are within the envelope, they are considered to be distributed randomly. If the experimental data fall outside the envelope, there is 99% confidence that the points are not distributed randomly. Data with values above the envelope are said to cocluster, while if data fall below the envelope, they can be said to occupy distinct membrane domains exclusive of each other.
ACKNOWLEDGMENTS
We thank Bridget S. Wilson, University of New Mexico School of Medicine, for extensive help with the statistical analysis.
This work was supported in part by NIH NIAID research grant R01 AI-20101. G.P.L. is a Howard Hughes Medical Institute (HHMI) specialist, and R.A.L. is an HHMI investigator.
REFERENCES
- 1.Shaw ML, Palese P. 2013. Orthomyxoviridae, p 1151–1185. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrasco M, Amorim MJ, Digard P. 2004. Lipid raft-dependent targeting of the influenza A virus nucleoprotein to the apical plasma membrane. Traffic 5:979–992. doi: 10.1111/j.1600-0854.2004.00237.x. [DOI] [PubMed] [Google Scholar]
- 4.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamb RA, Zebedee SL, Richardson CD. 1985. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 40:627–633. doi: 10.1016/0092-8674(85)90211-9. [DOI] [PubMed] [Google Scholar]
- 6.Zebedee SL, Lamb RA. 1988. Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J Virol 62:2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nayak DP, Balogun RA, Yamada H, Zhou ZH. 2009. Influenza virus morphogenesis and budding. Virus Res 143:147–161. doi: 10.1016/j.virusres.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calder LJ, Wasilewski S, Berriman JA, Rosenthal PB. 2010. Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107:10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruigrok RW, Schoehn G, Dessen A, Forest E, Volchkov V, Dolnik O, Klenk HD, Weissenhorn W. 2000. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J Mol Biol 300:103–112. doi: 10.1006/jmbi.2000.3822. [DOI] [PubMed] [Google Scholar]
- 10.Elster C, Larsen K, Gagnon J, Ruigrok RWH, Baudin F. 1997. Influenza virus M1 protein binds to RNA through its nuclear localization signal. J Gen Virol 78:1589–1596. doi: 10.1099/0022-1317-78-7-1589. [DOI] [PubMed] [Google Scholar]
- 11.Huang X, Liu T, Muller J, Levandowski RA, Ye Z. 2001. Effect of influenza virus matrix protein and viral RNA on ribonucleoprotein formation and nuclear export. Virology 287:405–416. doi: 10.1006/viro.2001.1067. [DOI] [PubMed] [Google Scholar]
- 12.Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. 2007. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 88:2280–2290. doi: 10.1099/vir.0.82809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruigrok RW, Barge A, Durrer P, Brunner J, Ma K, Whittaker GR. 2000. Membrane interaction of influenza virus M1 protein. Virology 267:289–298. doi: 10.1006/viro.1999.0134. [DOI] [PubMed] [Google Scholar]
- 14.Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA. 1999. Association of influenza virus matrix protein with ribonucleoproteins. J Virol 73:7467–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Ekstrom M, Garoff H. 1998. The M1 and NP proteins of influenza A virus form homo- but not heterooligomeric complexes when coexpressed in BHK-21 cells. J Gen Virol 79(Part 10):2435–2446. [DOI] [PubMed] [Google Scholar]
- 16.Baudin F, Bach C, Cusack S, Ruigrok RWH. 1994. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J 13:3158–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamanaka K, Ogasawara N, Yoshikawa H, Ishihama A, Nagata K. 1991. In vivo analysis of the promoter structure of the influenza virus RNA genome using a transfection system with an engineered RNA. Proc Natl Acad Sci U S A 88:5369–5373. doi: 10.1073/pnas.88.12.5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.York A, Hengrung N, Vreede FT, Huiskonen JT, Fodor E. 2013. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc Natl Acad Sci U S A 110:E4238–E4245. doi: 10.1073/pnas.1315068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruigrok RWH, Baudin F. 1995. Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J Gen Virol 76:1009–1014. [DOI] [PubMed] [Google Scholar]
- 20.Avalos RT, Yu Z, Nayak DP. 1997. Association of influenza virus NP and M1 proteins with cellular cytoskeletal elements in influenza virus-infected cells. J Virol 71:2947–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinto LH, Holsinger LJ, Lamb RA. 1992. Influenza virus M2 protein has ion channel activity. Cell 69:517–528. doi: 10.1016/0092-8674(92)90452-I. [DOI] [PubMed] [Google Scholar]
- 22.Martin K, Helenius A. 1991. Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67:117–130. doi: 10.1016/0092-8674(91)90576-K. [DOI] [PubMed] [Google Scholar]
- 23.Chen BJ, Leser GP, Jackson D, Lamb RA. 2008. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCown MF, Pekosz A. 2005. The influenza A virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J Virol 79:3595–3605. doi: 10.1128/JVI.79.6.3595-3605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCown MF, Pekosz A. 2006. Distinct domains of the influenza A virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J Virol 80:8178–8189. doi: 10.1128/JVI.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossman JS, Lamb RA. 2010. Swine-origin influenza virus and the 2009 pandemic. Am J Respir Crit Care Med 181:295–296. doi: 10.1164/rccm.200912-1876ED. [DOI] [PubMed] [Google Scholar]
- 28.Roberts KL, Leser GP, Ma C, Lamb RA. 2013. The amphipathic helix of influenza A virus M2 protein is required for filamentous bud formation and scission of filamentous and spherical particles. J Virol 87:9973–9982. doi: 10.1128/JVI.01363-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zebedee SL, Lamb RA. 1989. Growth restriction of influenza A virus by M2 protein antibody is genetically linked to the M1 protein. Proc Natl Acad Sci U S A 86:1061–1065. doi: 10.1073/pnas.86.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder C, Heider H, Moncke-Buchner E, Lin TI. 2005. The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein. Eur Biophys J 34:52–66. doi: 10.1007/s00249-004-0424-1. [DOI] [PubMed] [Google Scholar]
- 31.Hess ST, Kumar M, Verma A, Farrington J, Kenworthy A, Zimmerberg J. 2005. Quantitative electron microscopy and fluorescence spectroscopy of the membrane distribution of influenza hemagglutinin. J Cell Biol 169:965–976. doi: 10.1083/jcb.200412058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leser GP, Lamb RA. 2005. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 342:215–227. doi: 10.1016/j.virol.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 33.Scheiffele P, Rietveld A, Wilk T, Simons K. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J Biol Chem 274:2038–2044. doi: 10.1074/jbc.274.4.2038. [DOI] [PubMed] [Google Scholar]
- 34.Takeda M, Leser GP, Russell CJ, Lamb RA. 2003. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc Natl Acad Sci U S A 100:14610–14617. doi: 10.1073/pnas.2235620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Leser GP, Pekosz A, Lamb RA. 2000. The cytoplasmic tails of the influenza virus spike glycoproteins are required for normal genome packaging. Virology 269:325–334. doi: 10.1006/viro.2000.0228. [DOI] [PubMed] [Google Scholar]
- 36.Jin H, Leser G, Lamb RA. 1994. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J 13:5504–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin H, Leser GP, Zhang J, Lamb RA. 1997. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J 16:1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitnaul LJ, Castrucci MR, Murti KG, Kawaoka Y. 1996. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J Virol 70:873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Pekosz A, Lamb RA. 2000. Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J Virol 74:4634–4644. doi: 10.1128/JVI.74.10.4634-4644.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enami M, Enami K. 1996. Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol 70:6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali A, Avalos RT, Ponimaskin E, Nayak DP. 2000. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74:8709–8719. doi: 10.1128/JVI.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kretzschmar E, Bui M, Rose JK. 1996. Membrane association of influenza virus matrix protein does not require specific hydrophobic domains or the viral glycoproteins. Virology 220:37–45. doi: 10.1006/viro.1996.0283. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Lamb RA. 1996. Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology 225:255–266. doi: 10.1006/viro.1996.0599. [DOI] [PubMed] [Google Scholar]
- 44.Barman S, Ali A, Hui EK, Adhikary L, Nayak DP. 2001. Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res 77:61–69. doi: 10.1016/S0168-1702(01)00266-0. [DOI] [PubMed] [Google Scholar]
- 45.Mora R, Rodriguez-Boulan E, Palese P, Garcia-Sastre A. 2002. Apical budding of a recombinant influenza A virus expressing a hemagglutinin protein with a basolateral localization signal. J Virol 76:3544–3453. doi: 10.1128/JVI.76.7.3544-3553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez-Puertas P, Albo C, Perez-Pastrana E, Vivo A, Portela A. 2000. Influenza virus matrix protein is the major driving force in virus budding. J Virol 74:11538–11547. doi: 10.1128/JVI.74.24.11538-11547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latham T, Galarza JM. 2001. Formation of wild-type and chimeric influenza virus-like particles following simultaneous expression of only four structural proteins. J Virol 75:6154–6165. doi: 10.1128/JVI.75.13.6154-6165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen BJ, Leser GP, Morita E, Lamb RA. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81:7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanan DA, Anderson RGW. 1991. Simultaneous visualization of LDL receptor distribution and clathrin lattices on membranes torn from the upper surface of cultured cells. J Histochem Cytochem 39:1017–1024. doi: 10.1177/39.8.1906908. [DOI] [PubMed] [Google Scholar]
- 50.Wilson BS, Pfeiffer JR, Raymond-Stintz MA, Lidke D, Andrews N, Zhang J, Yin W, Steinberg S, Oliver JM. 2007. Exploring membrane domains using native membrane sheets and transmission electron microscopy. Methods Mol Biol 398:245–261. doi: 10.1007/978-1-59745-513-8_17. [DOI] [PubMed] [Google Scholar]
- 51.Andrews NL, Lidke KA, Pfeiffer JR, Burns AR, Wilson BS, Oliver JM, Lidke DS. 2008. Actin restricts FcεRI diffusion and facilitates antigen-induced receptor immobilization. Nat Cell Biol 10:955–963. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carroll-Portillo A, Spendier K, Pfeiffer J, Griffiths G, Li H, Lidke KA, Oliver JM, Lidke DS, Thomas JL, Wilson BS, Timlin JA. 2010. Formation of a mast cell synapse: Fc epsilon RI membrane dynamics upon binding mobile or immobilized ligands on surfaces. J Immunol 184:1328–1338. doi: 10.4049/jimmunol.0903071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Signoret N, Hewlett L, Wavre S, Pelchen-Matthews A, Oppermann M, Marsh M. 2005. Agonist-induced endocytosis of CC chemokine receptor 5 is clathrin dependent. Mol Biol Cell 16:902–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. 2003. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson BS, Pfeiffer JR, Oliver JM. 2000. Observing FcepsilonRI signaling from the inside of the mast cell membrane. J Cell Biol 149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson BS, Steinberg SL, Liederman K, Pfeiffer JR, Surviladze Z, Zhang J, Samelson LE, Yang LH, Kotula PG, Oliver JM. 2004. Markers for detergent-resistant lipid rafts occupy distinct and dynamic domains in native membranes. Mol Biol Cell 15:2580–2592. doi: 10.1091/mbc.E03-08-0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prior IA, Muncke C, Parton RG, Hancock JF. 2003. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol 160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ripley BD. 1977. Modelling spatial patterns. J R Stat Soc Series B 39:172–212. [Google Scholar]
- 60.Ripley BD. 1979. Tests of ‘randomness’ for spatial point patterns. J R Stat Soc Series B 41:368–374. [Google Scholar]
- 61.Lillemeier BF, Pfeiffer JR, Surviladze Z, Wilson BS, Davis MM. 2006. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc Natl Acad Sci U S A 103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bourmakina SV, Garcia-Sastre A. 2003. Reverse genetics studies on the filamentous morphology of influenza A virus. J Gen Virol 84:517–527. doi: 10.1099/vir.0.18803-0. [DOI] [PubMed] [Google Scholar]
- 63.Lindwasser OW, Resh MD. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J Virol 75:7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dittmann M, Hoffmann HH, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, Wilson SJ, Crotta S, Yu Y, Flatley B, Xiao JW, Casanova JL, Wack A, Bieniasz PD, Rice CM. 2015. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell 160:631–643. doi: 10.1016/j.cell.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheiffele P, Roth MG, Simons K. 1997. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J 16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barman S, Nayak DP. 2000. Analysis of the transmembrane domain of influenza virus neuraminidase, a type II transmembrane glycoprotein, for apical sorting and raft association. J Virol 74:6538–6545. doi: 10.1128/JVI.74.14.6538-6545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohkura T, Momose F, Ichikawa R, Takeuchi K, Morikawa Y. 2014. Influenza A virus hemagglutinin and neuraminidase mutually accelerate their apical targeting through clustering of lipid rafts. J Virol 88:10039–10055. doi: 10.1128/JVI.00586-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pralle A, Keller P, Florin E-L, Simons K, Horber JKH. 2000. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J Cell Biol 148:997–1007. doi: 10.1083/jcb.148.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma P, Varma R, Sarasij RC, Gousset KI, Krishnamoorthy G, Rao M, Mayor S. 2004. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 116:577–589. doi: 10.1016/S0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- 70.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol 84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang T, Hong M. 2015. Investigation of the curvature induction and membrane localization of the influenza virus M2 protein using static and off-magic-angle spinning solid-state nuclear magnetic resonance of oriented bicelles. Biochemistry 54:2214–2226. doi: 10.1021/acs.biochem.5b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidt NW, Mishra A, Wang J, DeGrado WF, Wong GC. 2013. Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission. J Am Chem Soc 135:13710–13719. doi: 10.1021/ja400146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ho CS, Khadka NK, She F, Cai J, Pan J. 2016. Influenza M2 transmembrane domain senses membrane heterogeneity and enhances membrane curvature. Langmuir 32:6730–6738. doi: 10.1021/acs.langmuir.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. 2011. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J Virol 85:6117–6126. doi: 10.1128/JVI.00378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He J, Sun E, Bujny MV, Kim D, Davidson MW, Zhuang X. 2013. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog 9:e1003701. doi: 10.1371/journal.ppat.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yanguez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, Konig R, Stertz S, Garcia-Sastre A, Chanda SK. 2015. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thaa B, Levental I, Herrmann A, Veit M. 2011. Intrinsic membrane association of the cytoplasmic tail of influenza virus M2 protein and lateral membrane sorting regulated by cholesterol binding and palmitoylation. Biochem J 437:389–397. doi: 10.1042/BJ20110706. [DOI] [PubMed] [Google Scholar]
- 78.Tian C, Gao PF, Pinto LH, Lamb RA, Cross TA. 2003. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci 12:2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stewart SM, Wu WH, Lalime EN, Pekosz A. 2010. The cholesterol recognition/interaction amino acid consensus motif of the influenza A virus M2 protein is not required for virus replication but contributes to virulence. Virology 405:530–538. doi: 10.1016/j.virol.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leser GP, Martin TE. 1987. Changes in heterogeneous nuclear RNP core polypeptide complements during the cell cycle. J Cell Biol 105:2083–2094. doi: 10.1083/jcb.105.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC. 2006. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A 103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hilsch M, Goldenbogen B, Sieben C, Hofer CT, Rabe JP, Klipp E, Herrmann A, Chiantia S. 2014. Influenza A matrix protein M1 multimerizes upon binding to lipid membranes. Biophys J 107:912–923. doi: 10.1016/j.bpj.2014.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baudin F, Petit I, Weissenhorn W, Ruigrok RW. 2001. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology 281:102–108. doi: 10.1006/viro.2000.0804. [DOI] [PubMed] [Google Scholar]
- 84.Zhirnov OP, Manykin AA, Rossman JS, Klenk HD. 2016. Intravirion cohesion of matrix protein M1 with ribonucleocapsid is a prerequisite of influenza virus infectivity. Virology 492:187–196. doi: 10.1016/j.virol.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 85.Iwatsuki-Horimoto K, Horimoto T, Noda T, Kiso M, Maeda J, Watanabe S, Muramoto Y, Fujii K, Kawaoka Y. 2006. The cytoplasmic tail of the influenza A virus M2 protein plays a role in viral assembly. J Virol 80:5233–5240. doi: 10.1128/JVI.00049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paterson RG, Lamb RA. 1993. The molecular biology of influenza viruses and paramyxoviruses, p 35–73. In Davidson A, Elliott RM (ed), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 87.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants by a novel eukaryotic vector. Gene 108:193–200. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 88.Zhang J. 2000. Role of the influenza virus glycoprotein cytoplasmic tails and the matrix protein in virus assembly. PhD thesis Northwestern University, Evanston, IL. [Google Scholar]
- 89.Burns AR, Oliver JM, Pfeiffer JR, Wilson BS. 2008. Visualizing clathrin-mediated IgE receptor internalization by electron and atomic force microscopy. Methods Mol Biol 440:235–245. doi: 10.1007/978-1-59745-178-9_18. [DOI] [PubMed] [Google Scholar]
- 90.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baddeley A, Turner R. 2005. Spatstat: an R package for analyzing spatial point patterns. J Stat Softw 12:1–42. [Google Scholar]