

Abstract
Summary: The blood-brain barrier (BBB) is formed by the brain capillary endothelium and excludes from the brain ∼100% of large-molecule neurotherapeutics and more than 98% of all small-molecule drugs. Despite the importance of the BBB to the neurotherapeutics mission, the BBB receives insufficient attention in either academic neuroscience or industry programs. The combination of so little effort in developing solutions to the BBB problem, and the minimal BBB transport of the majority of all potential CNS drugs, leads predictably to the present situation in neurotherapeutics, which is that there are few effective treatments for the majority of CNS disorders. This situation can be reversed by an accelerated effort to develop a knowledge base in the fundamental transport properties of the BBB, and the molecular and cellular biology of the brain capillary endothelium. This provides the platform for CNS drug delivery programs, which should be developed in parallel with traditional CNS drug discovery efforts in the molecular neurosciences.
Keywords: Blood-brain barrier, endothelium, drug targeting, biological transport, neurotherapeutics
INTRODUCTION
The blood-brain barrier (BBB) is the bottleneck in brain drug development and is the single most important factor limiting the future growth of neurotherapeutics.1 The BBB problem is illustrated in Figure 1, which is a whole body autoradiogram of a mouse sacrificed 30 min after intravenous injection of radiolabeled histamine, a small molecule of only ∼100 Da in molecular mass. Histamine readily crosses the porous capillaries perfusing all peripheral tissues but is excluded from entry into the brain or spinal cord by the BBB.
FIG. 1.

Whole body autoradiogram of an adult mouse sacrificed 30 min after intravenous injection of radiolabeled histamine, a small molecule that readily enters all organs of the body, except for the brain and spinal cord.
The histamine example in Figure 1 refutes a common misconception that most small molecules readily cross the BBB. As discussed below, the transport of small molecules across the BBB is the exception rather than the rule, and 98% of all small molecules do not cross the BBB (FIG. 1). Moreover, all large-molecule products of biotechnology, such as monoclonal antibodies (mAbs), recombinant proteins, antisense, or gene therapeutics, do not cross the BBB (FIG. 1). Despite the large number of patients with disorders of the CNS and despite the fact that so few large- or small-molecule therapeutics cross the BBB, there are few pharmaceutical companies in the world today that have built a BBB drug targeting program (FIG. 1). However, even if a pharmaceutical company decided to develop a BBB program, there would be few BBB-trained scientists to hire because less than 1% of U.S. academic neuroscience programs emphasize BBB transport biology.
Because most drugs do not cross the BBB, and because the industry is not providing solutions to the BBB problem, it is not surprising that most disorders of the CNS could benefit from improved drug therapy (FIG. 2). For a small-molecule drug to cross the BBB in pharmacologically significant amounts, the molecule must have the dual molecular characteristics of: 1) molecular mass under a 400- to 500-Da threshold, and 2) high lipid solubility.1 There are only four categories of CNS disorders that consistently respond to such molecules, and these include affective disorders, chronic pain, and epilepsy (FIG. 2). Migraine headache may be a CNS disorder and could also be included in this category. In contrast, most CNS disorders such as those listed in Figure 2 have few treatment options. Parkinson’s disease patients are given l-dihydroxyphenylalanine (l-DOPA) for dopamine replacement therapy.2 As discussed below in the section on BBB carrier-mediated transport, l-DOPA is an example of a BBB drug targeting strategy. However, there is no neurotherapeutic that stops the neurodegeneration of Parkinson’s disease. Similarly, there is no therapy for other neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease, and amytrophic lateral sclerosis (ALS). Patients with multiple sclerosis (MS) are treated with cytokines that work on the peripheral immune system, but which do not permanently stop the progression of MS.3 The human immunodeficiency virus (HIV) infects the brain early in the course of acquired immune deficiency syndrome (AIDS).4 HIV in the periphery has been significantly reduced with highly active antiretroviral therapy (HAART) comprised of multiple small-molecule therapeutics. However, HAART drugs such as azidothymidine, 3TC, or protease inhibitors are substrates for BBB active efflux transporters, which are reviewed below, and HAART drugs have minimal penetration into brain parenchyma. Consequently, the brain remains a sanctuary for HIV in AIDS even with HAART.4,5 Brain cancer, stroke, and brain or spinal cord trauma are all examples of serious CNS disorders for which there is no effective drug therapy. The childhood disorders including autism, lysosomal storage disorders, fragile X syndrome, the ataxis, and blindness, are serious disorders where there is little effective treatment. In many of these cases, the gene underlying the disease is known, but BBB delivery is the rate-limiting problem in gene therapy or enzyme replacement therapy, and no therapeutics have been developed. Many of the disorders listed in the right-hand column in Figure 2 could be treated with drugs, enzymes, or genes already discovered. However, these drugs do not cross the BBB and cannot enter into brain drug development because no BBB solutions have been developed by industry. Given the absence of effective BBB drug targeting technology, CNS drug developers are left with the traditional approaches to solving the brain drug delivery problem: small molecules, trans-cranial brain drug delivery, and BBB disruption. A review of these approaches shows that none provide solutions to the BBB problem that could be practically implemented in large numbers of patients.
FIG. 2.

A review of the Comprehensive Medicinal Chemistry database shows that, of more than 7000 small-molecule drugs, only 5% treat the CNS, and these drugs only treat four disorders: depression, schizophrenia, chronic pain, and epilepsy.6,7 There are few effective small- or large-molecule drugs for the majority of CNS disorders, with the exception of Parkinson’s disease, e.g., l-DOPA, and multiple sclerosis, e.g., cytokines.
SMALL MOLECULES
Most small-molecule drugs do not cross the BBB. Of over 7000 drugs in the comprehensive medicinal chemistry (CMC) database, only 5% of all drugs treat the CNS, and these CNS active drugs only treat depression, schizophrenia, and insomnia.6 The average molecular mass of the CNS active drug is 357 Da. In another study, only 12% of drugs were active in the CNS, but only 1% of all drugs were active in the CNS for diseases other than affective disorders.7
BBB transport of small molecules is limited
Small molecules generally cross the BBB in pharmacologically significant amounts if 1) the molecular mass of the drug is less than 400-500 Da, and 2) the drug forms less than 8-10 hydrogen bonds with solvent water.1
The permeation of the drug across the BBB does not increase in proportion to lipid solubility when the molecular weight of the drug is increased. BBB permeation decreases 100-fold as the surface area of the drug is increased from 52 Angstroms2 (e.g., a drug with molecular mass of 200 Da) to 105 Angstroms2 (e.g., a drug of 450 Da).8 Drug diffusion through a biological membrane is not analogous to drug diffusion through solvent water. In contrast to water, diffusion of a drug through a biological membrane is dependent on the volume of the drug. The classical Overton rules that relate membrane permeation to solute lipid solubility do not predict the molecular weight threshold effect. As noted by Leib and Stein nearly 20 years ago,9 the molecular weight threshold effect is best predicted by the “hole-jumping” model of Trauble,10 which posits that solutes undergo a form of molecular “hitch hiking” across a biological membrane by moving through small holes in the membrane formed by kinking of the mobile unsaturated fatty acyl side chains in the phospholipid bilayer.
Hydrogen bonding
BBB permeation decreases exponentially with the addition of each pair of hydrogen bonds added to the drug structure.11 It does not matter whether the functional group is a hydrogen bond donor or a hydrogen bond acceptor because each hydrogen bond carries equal weight. Hydrogen bond donor groups such as hydroxyls form two hydrogen bonds because a hydroxyl group acts as both a hydrogen bond donor and hydrogen bond acceptor, whereas a carbonyl group only acts as a hydrogen bond acceptor. Once the total number of hydrogen bonds on the drug exceeds a threshold of 8-10, there is minimal transport of the drug across the BBB in pharmacologically active amounts. Both the hydrogen bonding and the molecular weight of drugs currently emanating from CNS drug discovery programs generally are higher than drugs discovered 20 years ago.7 This is because CNS drug discovery programs now rely extensively on receptor-based high-throughput screening (HTS) programs. HTS-based drug screening invariably selects for drugs that have higher molecular weights and higher hydrogen bonding because these factors enable higher affinity drug binding to the target receptor.
HTS-based CNS drug discovery
Current CNS drug discovery programs are generally broken down into four major areas: 1) receptor target identification, 2) drug “hit” identification, 3) “lead” identification, and 4) drug lead optimization. After screening several hundred thousand small-molecule drugs with a given target, several hundred hits may be found, leading to a score of potential drug leads. The HTS drug lead compounds must then be optimized with respect to distribution, metabolism, and pharmacokinetics (DMPK).12 However, the drugs generally require so much medicinal chemistry to block polar functional groups that the original high receptor affinity is lost in an attempt to produce a drug with acceptable DMPK properties. The difficulty in using medicinal chemistry to increase the lipid solubility of a drug is illustrated by considering that there is not a single drug currently in CNS clinical practice that is an example of a water soluble drug that was made lipid soluble with medicinal chemistry optimization such that the drug then became pharmacologically active in the brain in vivo.
The pharmacokinetic rule
When medicinal chemistry is used to increase the lipid solubility of the drug, this may increase penetration across the BBB, but it also increases penetration across all biological membranes in vivo. Therefore, the lipidized form of the drug is rapidly removed from the blood, and in pharmacokinetic terms, the plasma area under the concentration curve (AUC) is substantially decreased for the lipidized form of the drug. Drug action in brain is a function of drug uptake, expressed as percent of injected dose (ID) per gram brain, and the % ID/g is equally dependent on two factors, the BBB permeability-surface area (PS) product and the plasma AUC:
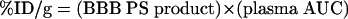 |
(Eq. 1) |
Although an increase in lipid solubility of the drug may increase the BBB PS product, there is a proportional decrease in the plasma AUC with lipidization. The increased BBB PS product and the decreased plasmid AUC have offsetting effects, which minimizes the increase in brain uptake caused by lipidization.1
Medicinal chemistry and brain drug lead optimization
The use of medicinal chemistry to increase the lipid solubility of drug to solve the BBB drug delivery problem is problematical for the reasons listed above. However, a new approach to the use of medicinal chemistry to solve the BBB drug delivery problem is discussed below. Medicinal chemistry can be used to alter the structure of a lead drug candidate to make that drug transportable on one of several carrier-mediated transport (CMT) systems within the BBB. However, redirection of the use of medicinal chemistry to increase the carrier-mediated transport of a drug, as oppose to the lipid-mediated transport of the drug, requires knowledge on the structural characteristics of a drug that enable CMT across the BBB. Therefore, a knowledge base in BBB CMT must be developed before the use of medicinal chemistry to increase drug penetration to the brain via endogenous BBB carriers.
TRANS-CRANIAL BRAIN DRUG DELIVERY
Trans-cranial brain drug delivery approaches attempt to bypass the BBB using one of three neurosurgical-based delivery approaches: intracerebral implantation, intracerebroventricular (ICV) infusion, and convection enhanced diffusion (CED). The factor limiting either the intracerebral or ICV infusion approach is that either method relies on diffusion for drug penetration into the brain from the depot site. Solute diffusion decreases with the square of the diffusion distance.1 Therefore, the concentration of drug decreases logarithmically with each millimeter of brain tissue that is removed from the injection site, in the case of intracerebral implantation, or from the ependymal surface of the brain, in the case of ICV infusion. The concentration of a small molecule is decreased by 90% at a distance of only 0.5 mm from the intracerebral implantation site in rat brain.13 The logarithmic decrease in drug concentration from the ependymal surface following an ICV infusion was shown in the 1970s in adult Rhesus monkeys; after ICV drug injection, the concentration of small molecules in brain parenchyma removed only 1-2 mm from the ependymal surface is only about 1-2% of the concentration in the CSF compartment.14 The limited diffusion of drug from an intracerebral implant is shown in Figure 3, which is an autoradiogram of rat brain taken 2 days after the intracerebral implantation of a wafer embedded with radiolabeled NGF.15 The size of the wafer is approximately equal to the magnification bar in the figure, which indicates that there has been minimal penetration of NGF into brain parenchyma from the implant site. The limited diffusion of BDNF into brain parenchyma following injection into a lateral ventricle (LV)16 is shown in Figure 3. The BDNF is sequestered by the ependymal surface but does not significantly diffuse into brain parenchyma. This limited diffusion of BDNF into brain parenchyma is not due to the fact that BDNF is a cationic protein, as a similar logarithmic decrease in brain penetration is found for any drug following ICV injection.14 This slow rate of drug diffusion into brain parenchyma is to be contrasted with the rapid rate of bulk flow of CSF through the ventricular compartments. CSF is then rapidly absorped into the peripheral bloodstream at the superior sagittal sinus. The ICV injection of drug should be regarded as a slow intravenous infusion rather than a direct administration of drug into the brain.17 The rapid rate of cytokine distribution into blood, but minimal penetration into brain, following an ICV injection has been demonstrated in adult rhesus monkeys.18
FIG. 3.

Trans-cranial drug delivery to the brain. A: Autoradiogram of rat brain 48 h after an intracerebral implantation of a polymer carrying radiolabeled NGF.15 The size of the polymer approximates the magnification bar, indicating the NGF has not significantly diffused from the implantation site. B: Autoradiogram of rat brain 24 h after an intracerebroventricular injection of BDNF into an LV.16 The BDNF distributes to the ependymal surface of the ipsilateral LV and the third ventricle (3V), but not into brain parenchyma. C: Convection enhanced diffusion in the primate brain forces fluid through the brain tissue. The direction of fluid flow, principally via white matter tracts,19 can be traced with immunocytochemistry using an antibody to GFAP, which shows an astrogliotic reaction in the path of fluid flow.20 The hole in the brain left by the catheter is noted by the asterisk. The fluid moved from the catheter in the putamen (Pu) via the internal capsule (ic) white matter to the caudate (Cd).
The effective penetration of drug into brain can be increased to a treatment radius of a few millimeters when bulk flow is used to deliver drug into brain parenchyma, and this is possible by forcing fluid through the brain with CED. However, the brain has no lymphatic system and is not designed for a significant intraparenchymal volume flow. CED in humans with glioblastoma multiform causes a preferential flow of the forced fluid along white matter tracts.19 CED in the adult Rhesus monkey brain with glial-derived neurotrophic factor involved the infusion of relatively small volumes of ∼0.1 ml/day over a 4-week period.20 This led to diffuse white matter astrogliosis, which was visualized by immunocytochemistry of the autopsy primate brain, and immunostaining with an antibody to GFAP as shown in Figure 3. In addition, there was a microglial response and demyelination around the catheter, with extension of the astrogliotic reaction from the catheter in the putamen (Pu) through the internal capsule (ic) to the caudate (cd) (FIG. 3). These findings of an intense astrogliotic reaction along white matter tracts after CED in the primate brain raise concerns about the long-term effects of this delivery approach for humans.
BLOOD-BRAIN BARRIER DISRUPTION
In parallel with trans-cranial brain drug delivery strategies, there has been a significant effort in delivering drugs to the brain with BBB disruption after the intracarotid arterial infusion of vasoactive agents such as those listed in Table 1. The intracarotid arterial infusion of 2 m concentrations of poorly diffusible solutes such as mannitol causes disruption of the BBB owing to osmotic shrinkage of the endothelial cells.21 This is associated with severe vasculopathy22 and chronic neuropathologic changes in rodent models23 and is also associated with seizures in either animal models24 or humans.25 Plasma proteins such as albumin are toxic to brain cells,26 and BBB disruption allows for the uptake of plasma into the brain.
TABLE 1.
BBB Disruption after Intracarotid Arterial Infusion of Noxious Agents
| Method | Comments (References) |
|---|---|
| Hyperosmolar | Leads to chronic neuropathologic changes and vasculopathy in the brain and seizures21–25 |
| Vasoactive agents | Examples are bradykinin, histamine, and multiple other vasoactive compounds; opens BBB in brain tumor to greater extent than normal brain72 |
| Solvents | BBB is solubilized with high dose ethanol, DMSO, SDS, Tween 80 (polysorbate-80)27–30 |
| Alkylating agents | Examples are etoposide and melphalan; may alkylate key sulfhydryl residues similar to mercury73,74 |
| Immune adjuvants | Freunds adjuvant opens BBB to IgG for weeks; enable IgG uptake into brain in rodent vaccine models, such as Alzheimer’s disease32 |
| Ultrasound | The combination of administration of high-dose air bubbles (2–4 μm) and high-dose ultrasound (10–1000 watt/cm2) can induce BBB disruption75 |
| Cytokines | Intracerebral interleukin-1β or CXC chemokines can attract white cells from blood and cause BBB disruption76,77 |
| Miscellaneous | Intracarotid acid pH, cold temperatures, or high-dose free fatty acid all cause BBB disruption78–80 |
Solvent/adjuvant-mediated BBB disruption
The BBB, like cell membranes in general, is subject to solvent-mediated disruption with chemicals such as ethanol, dimethylsulfoxide (DMSO), or detergents such as SDS, or Tween 80 also known as polysorbate-80.27–30 There are numerous examples in the literature where the peripheral administration of a drug, which normally should not cross the BBB, is followed by pharmacological activity in the brain. Such an observation could arise because the drug is transported across the BBB via an endogenous transport system. However, an alternative explanation is that the drug is injected in a diluent that is membrane destabilizing, and causes BBB disruption. Often the drug is solubilized in solvents such as ethanol or DMSO, or surfactants such as SDS, a Tween detergent, or other surfactants, such as polyethyleneglycol hydroxy stearate. Doses of solvents such as ethanol or DMSO at a level of 1-4 g/kg may cause solvent-mediated disruption of the BBB.27,28 This dose of DMSO or ethanol is given to animal models with surprising frequency, particularly small rodent models such as mice, which weigh only 20-30 g. The administration of just 50 μl of 50% DMSO to a 20-g mouse is equivalent to 1.25 g/kg DMSO, and there are examples in the literature of pharmacologic effects achieved in brain following systemic administration of drugs that normally do not cross the BBB. These drugs are administered in solvents such as ethanol or DMSO and the dose of solvent is such that BBB disruption may be caused by administration of the drug/solvent mixture. Tween 80, also known as polysorbate-80, is frequently administered in CNS drug formulations. A dose of polysorbate-80 of 3-30 mg/kg will cause BBB disruption in mice.30 Analgesia with kyotorphin, a oligopeptide that normally does not cross the BBB, is possible following the peripheral administration of the peptide, providing Tween 80 is coadministered.31 Low doses of another surfactant, SDS, are frequently included in CNS drug diluents. However, doses of SDS as low as 1.0 μg/kg can cause disruption of the BBB for short periods. Immune adjuvants such as Freund’s complete or incomplete adjuvant cause disruption of the BBB to circulating IgG that can persist for weeks.32 This is relevant to rodent vaccine models where active immunization is attempted as a new therapy for the treatment of brain diseases. The vaccine for Alzheimer’s disease was based on the administration of the Aβ peptide mixed in Freund’s adjuvant to transgenic mice with brain amyloid.33 The adjuvant has two effects. First, it recruits the immune system to the injection site so that antibodies are made to the target peptide, in this case the Aβ. Second, the immune adjuvant causes an inflammatory response that results in opening of the BBB. This latter property allows the circulating anti-Aβ antibodies to enter the brain. In the absence of BBB disruption, the circulating IgG cannot enter the brain. In either active or passive immunization approaches to brain disorders, the circulating IgG must be enabled to cross the BBB and enter brain to cause the intended pharmacological effect. IgG molecules do not cross the BBB, in the absence of specific transport mechanisms. It is unlikely that active or passive immunization will be effective in humans, if the BBB is not disrupted.
If a CNS drug is formulated in a vehicle other than a physiological buffer, then the amounts of any solvent, surfactant, or adjuvant, that are included in the formulation should be evaluated critically as to whether drug treatment is associated with solvent-mediated BBB disruption. In this setting, there is a high likelihood that chronic drug administration will have toxic side effects.
TRANS-NASAL DRUG DELIVERY TO THE BRAIN
The delivery of drugs after intranasal administration is based on the rationale that drugs can exit the submucous space of the nose and cross the arachnoid membrane, and enter into olfactory CSF. It is posited that drug may then enter the brain from the CSF flow tracts following intranasal administration of drug. There are two points to consider when evaluating the potential efficacy of trans-nasal drug delivery to the brain. First, any drug that enters into olfactory CSF will exit the CSF flow tracts and enter the peripheral bloodstream like any other ICV route of administration. The second consideration is that the arachnoid membrane, which separates olfactory CSF from the submucous spaces of the nose, has high resistance tight junctions, just like the capillary endothelium that forms the BBB.34 Therefore, only lipid-soluble small molecules may cross the arachnoid membrane and enter into olfactory CSF in the absence of arachnoid membrane disruption. Conversely, if the arachnoid membrane and other membranes in the nose are physically or chemically disrupted, then drug may enter the CSF from the nose. The human nasal cavity can only receive about 100 μl per nostril without local injury.35 The volume of drug administered into the nose is invariably ≫200 μl. Melanocyte-stimulating hormone, a seven-amino acid neuropeptide, entered CSF following intranasal instillation in humans after these subjects ingested 20 consecutive puffs of drug via an atomizer into each nares.36 When drug is administered to the nose via volumes that are not injurious to the nose, then no distribution into CSF is found for a water-soluble drug such as vitamin B12 or a relatively lipid soluble drug such as melatonin.35 In the absence of local injury, distribution of neuropeptides to olfactory CSF is nil, unless the protein has access to a specialized transport system that enables movement across the arachnoid membrane. This was demonstrated in the case of a conjugate of HRP and wheat germ agglutinin (WGA). The latter is a glycoprotein that crosses membranes via absorptive mediated endocytosis, based on binding to membrane lectin sites.37 Whereas the HRP alone cannot penetrate the olfactory CSF, the HRP-WGA conjugate can cross plasma membranes via absorptive-mediated endocytosis.
TRANSVASCULAR DRUG DELIVERY TO THE BRAIN VIA ENDOGENOUS BBB TRANSPORTERS
The complexity of the vascular tree in the cortex of rat brain is shown with the India ink38 study in Figure 4. The vascular density in the human brain is even more complex. In the human brain, there are over 100 billion capillaries. The distance between capillaries is ∼50 μm. Therefore, the maximum diffusion distance in brain parenchyma following transvascular delivery is only 25 μm. Even a molecule as large as albumin, 68,000 Da molecular mass, will diffuse 25 μm in less than 1 s.1 Because the intercapillary distance in brain is so small, every neuron is virtually perfused by its own blood vessel. The length of capillaries in human brain is ∼400 miles, and the surface area of the brain capillary endothelium in the human brain is ∼20 m2. However, the volume of the intraendothelial space is only 1 μl for adult rat brain and is only 5 ml for the human brain. Therefore, the brain capillary endothelial surface, which forms the BBB in vivo, forms a very broad but thin barrier system. The thickness of the endothelial cell is only ∼200 nm, which is less than 5% of the thickness of most cells.
FIG. 4.

India ink study shows vascular density in the cortex of adult rat brain. Reprinted with permission from Bar. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol 59:I–VI,1–62. Copyright © 1980, Springer-Verlag.38 All rights reserved.
Transport across the BBB involves movement across two membranes in series: the luminal and abluminal membranes of the capillary endothelium, separated by the 200 nm of endothelial cytoplasm. The microvascular endothelium in brain is completely invested by a basement membrane, but the basement membrane constitutes no diffusion barrier. Approximately 90% of the brain side of the capillary is covered by astrocyte foot processes,39 although these astrocyte foot processes similarly constitute no diffusion barrier. Therefore, solutes freely and instantaneously distribute throughout the entire brain extravascular volume after transport across the limiting membrane, which is the capillary endothelial membrane. The BBB has a very high resistance owing to the tight junctions, which cement adjacent endothelial cells together. Due to the presence of the tight junctions, there is no para-cellular pathway for solute distribution into brain interstitial fluid from blood. Circulating molecules can only gain access to brain interstitium via a trans-cellular route through the brain capillary endothelial membranes. If a molecule is lipid soluble and has a molecular mass less than 400 Da and is not avidly bound by plasma proteins or is a substrate for an active efflux transport system at the BBB, then the circulating molecule may gain access to brain by lipid-mediated free diffusion. In the absence of the lipid-mediated pathway, circulating molecules may gain access to brain only via transport on certain endogenous transport systems within the brain capillary endothelium. These endogenous transporters have an affinity for both small molecules and large molecules and can be broadly classified into three categories: 1) CMT; 2) active efflux transport, or AET; and 3) receptor-mediated transport, or RMT.
CMT
CMT systems for hexoses, monocarboxylic acids such as lactic acid, neutral amino acids such as phenylalanine, basic amino acids such as arginine, quaternary ammonium molecules such as choline, purine nucleosides such as adenosine, and purine bases such as adenine, are shown in Figure 5, which represents the luminal membrane of the brain capillary endothelium. The individual endogenous nutrients shown in Figure 5 are representative substrates because each carrier system transports a group of nutrients of common structure. The CMT systems shown in Figure 5 are all members of the Solute Carrier (SLC) gene family (Table 2). The BBB glucose carrier is GLUT1 (glucose transporter type 1), which is a member of the SLC2 family; the BBB monocarboxylic acid transporter is MCT1, which is a member of the SLC16 family; the BBB large neutral amino acid and cationic amino acid transporters are LAT1 and CAT1, respectively, which are members of the SLC7 family; LAT1 and CAT1 are the light chains of heterodimeric proteins, and the heavy chain of the dimer is 4F2hc, which is a member of the SLC3 family; the BBB adenosine transporter is CNT2, which is a member of the SLC28 family (Table 2). Each of the SLC families shown in Table 2 represent many common genes of overlapping nucleotide identity and some of the SLC families are comprised of over 100 different genes.
FIG. 5.

BBB CMT systems are shown for seven different classes of nutrients, and the genes for five of these systems has been identified. GLUT1 = glucose transporter type 1; MCT1 = monocarboxylic acid transporter type 1; LAT1 = large neutral amino acid transporter type 1; CAT1 = cationic amino acid transporter type 1; CNT2 = concentrative nucleoside transporter type 2.
TABLE 2.
Solute Carrier (SLC) Gene Families of Small-Molecule Transporters
| Family | Substrate Specificity | Abbreviations |
|---|---|---|
| SLC1 | Acidic amino acid transporter | EEAT |
| ASC small neutral amino acid transporter | ASCT | |
| SLC2 | Glucose transporter | GLUT |
| H+-myo-inositol transporter | HMIT | |
| SLC3 | Heavy chain of heterodimeric amino acid transporters | 4F2hc |
| SLC4 | Bicarbonate/carbonate exchangers and Na+ coupled transporters | AE, NBC |
| SLC5 | Sodium/substrate cotransporters (glucose, choline) | SGLT, CHT |
| SLC6 | Neurotransmitter transporters (GABA, glycine, taurine, monoamines, creatine) | GAT, TAUT |
| SLC7 | Cationic amino acid transporter | CAT |
| Light chain of amino acid transporters | LAT | |
| SLC8 | Sodium/calcium exchanger | NCX |
| SLC9 | Sodium/proton exchanger | NHE |
| SLC10 | Sodium/bile salt cotransporter | NTCP, ASBT |
| SLC11 | Natural resistance-associated macrophage protein | NRAMP |
| Divalent metal-ion transporter | DMT | |
| SLC12 | Potassium/chloride cotransporter | KCC |
| SLC13 | Sodium/sulphate cotransporter | NaS |
| Sodium/dicarboxylate transporter | NaDC | |
| SLC14 | Urea transporter | UT |
| SLC15 | Proton peptide transporter | PEPT |
| SLC16 | Monocarboxylic acid transporter (lactate, pyruvate, ketone bodies) | MCT |
| SLC17 | Vesicular glutamic acid transporter | VGLUT |
| SLC18 | Vesicular amine transporter | VAT |
| SLC19 | Vitamin transporters (folic acid, thiamine) | THTR |
| SLC20 | Sodium-phosphate cotransporters | Pit |
| SLC21 | Organic anion transporters | OATP |
| SLC22 | Organic cation transporters | OCTN, OAT |
| SLC23 | Sodium/ascorbic acid transporter | SVCT |
| SLC24 | Sodium/calcium-potassium exchanger | NCKX |
| SLC25 | Mitochondrial carriers | MC |
| SLC26 | Anion exchangers | CFTR |
| SLC27 | Fatty acid transport proteins | FATP |
| SLC28 | Sodium dependent nucleoside transporters | CNT |
| SLC29 | Equilibrative nucleoside transporters | ENT |
| SLC30 | Zinc efflux transporters | ZNT |
| SLC31 | Copper efflux transporters | CTR |
| SLC32 | Vesicular neurotransmitter transporters | VIAAT, VGAT |
| SLC33 | Acetyl-CoA transporters | AT |
| SLC34 | Sodium/phosphate cotransporters | NaPi |
| SLC35 | Nucleotide sugar transporters | UGT |
| SLC36 | Lysosomal amino acid transporters | LYAAT |
| SLC37 | Glucose-6-phosphate transporter | G6PT |
| SLC38 | Sodium coupled neutral amino acid transporters | SNAT |
| SLC39 | Metal ion transporters | ZIP |
| SLC40 | Iron efflux transporter | MTP |
BBB GLUT1 transports glucose, 2-deoxyglucose, 3-O-methyl-glucose, galactose, and mannose, but not l-glucose.40 BBB MCT1 transports lactate, pyruvate, ketone bodies, and monocarboxylic acids.41 BBB LAT1 transports the neutral amino acids with preferential affinity for the large neutral amino acids.42 BBB CAT1 transports arginine, lysine, ornithine.43 The BBB choline transporter transports choline, and perhaps other quaternary ammonium molecules.44 To date, the BBB choline transporter has not been cloned. CHT1 is a sodium-dependent choline transporter member of the SLC5 family (Table 2), which corresponds to the sodium-dependent synaptosomal choline carrier. However, the BBB choline transporter is sodium independent45 and is likely a member of a different SLC gene family. The BBB adenosine carrier transports adenosine, guanosine, and certain pyrimidine nucleosides such as uridine,46 and is derived from the CNT2 gene,47 where CNT = concentrative nucleoside transporter. Purine nucleosides are also transported by sodium independent or equilibrative nucleoside transporters (ENT), which are members of the SLC29 gene family (Table 2). However, BBB transport in vivo on the blood side of the endothelium is sodium dependent,48 which excludes the role of an ENT carrier in mediating uptake of circulating adenosine. Pyrimidine nucleosides are primarily transported by CNT1, and, to date, there is no evidence that the BBB expresses CNT1. Purine bases such as adenine and guanine are transported by a nucleobase transporter (NBT)46 but, to date, no eukaryotic NBT transporter gene has been cloned.
In addition to the CMT systems shown in Figure 5, there are many other CMT genes expressed at the BBB, which enable the BBB transport of water-soluble vitamins, thyroid hormones, and other compounds. All of these CMT systems at the BBB, which may number in the dozens, are potential portals of entry of drugs to the brain. The CMT systems comprise highly stereospecific pore-based transporters, and there are significant structural requirements for transporter affinity. Therefore, it is unlikely that a drug, which is normally not transported across the BBB, would be made transportable by simply coupling to the drug to another molecule that undergoes CMT across the BBB. Rather, the structure of the pharmaceutical should be altered with medicinal chemistry so that it takes on the structure of a pseudo-nutrient and thus is able to undergo transport across the BBB via one of the CMT systems. For example, the α-carboxylation of dopamine results in the formation of l-DOPA, and DOPA, a large neutral amino acid, is a substrate for the BBB LAT1. Once across the BBB, the l-DOPA is decarboxylated back to dopamine via aromatic amino acid decarboxylase. l-DOPA is the primary example of a pro-drug that traverses biological membranes, not via lipid mediation, but via carrier mediation.
AET
P-glycoprotein is the prototypic AET system at the BBB, and accounts for the active efflux of molecules in the brain to blood direction. P-glycoprotein, which is a product of the ABC-B1 gene (FIG. 6), is just one of many members of the ATP binding cassette (ABC) gene family of transporters. There are several multidrug resistance protein (MRP) transporters, which also belong to the ABC gene family. The excessive focus on p-glycoprotein, also called the multidrug resistance (MDR) gene product, overlooks the fact that P-glycoprotein is just one member of a large gene family, and many members of the ABC gene family may participate in BBB AET. A second consideration is that active efflux in the brain to blood direction requires the concerted actions of two different types of transporters: an energy requiring transporter at one membrane of the endothelium, and an energy-independent transporter, or exchanger, at the opposite membrane of the capillary endothelium. Examples of energy-independent exchangers are members of the solute carrier (SLC) transporter gene family and include the organic anion transporter (OAT) gene family or the organic anion transporter polypeptide (OATP) gene family (FIG. 6). OATP and OAT are members of the SLC21 and SLC22 gene families, respectively (Table 2).
FIG. 6.

BBB AET systems are comprised of an energy-dependent system at one side of the brain capillary endothelium and an energy-independent system at the opposite endothelial membrane. As a hypothetical example, members of the ABC gene family are shown at the luminal endothelial membrane, and members of the SLC gene family are shown at the abluminal endothelial membrane.
Certain drugs are excluded from penetration into brain because these drugs are substrates for BBB AET systems. One strategy for increasing brain penetration of such drugs is the development of “co-drugs” that inhibit BBB AET systems and thereby allow increased brain penetration of the therapeutic drug. The development of pro-drugs to increase brain penetration of therapeutics might focus on MRP, OATP, or OAT transporters at the BBB in addition to p-glycoprotein.
RMT
Certain large-molecule peptides or proteins undergo transport from brain to blood via RMT across the BBB. There are at least three different types of BBB receptor systems as depicted in Figure 7. The transferrin receptor (TfR) is an example of a bidirectional RMT system that causes both the receptor-mediated transcytosis of holo-transferrin in the blood to brain direction, and the reverse transcytosis of apo-transferrin in the brain to blood direction.49,50 The neonatal Fc receptor (FcRn) is an example of a reverse RMT system that functions only to mediate the reverse transcytosis of IgG in the brain to blood direction, but not in the blood to brain direction.51,52 The type 1 scavenger receptor (SR-VI) is an example of a receptor-mediated endocytosis system that mediates the uptake of modified low-density lipoprotein (LDL) from the blood compartment into the intraendothelial compartment, and this endocytosis is not followed by exocytosis into brain interstitial fluid.53
FIG. 7.

BBB RMT systems are shown for three classes of systems. An example of a bidirectional RMT system is the endothelial transferrin receptor (TfR), which mediates the transport of holo-transferrin (Tf) in the blood to brain direction, and the transport of apo-Tf in the brain to blood direction. A reverse RMT system such as the neonatal Fc receptor (FcRn) transports IgG in the brain to blood direction only. An endocytosis system is illustrated by the type I scavenger receptor (SR-BI), which mediates the endocytosis of acetylated low-density lipoprotein into the endothelial compartment without transcytosis across the BBB.
Molecular Trojan horses and BBB RMT
Certain endogenous ligands or peptidomimetic mAbs that bind exofacial epitopes on BBB RMT systems and that are endocytosing antibodies can act as molecular Trojan horses to ferry drugs, proteins, and nonviral gene medicines across the BBB using the endogenous RMT systems. This BBB molecular Trojan horse technology has been reduced to practice in vivo in the following systems:
Vasoactive intestinal peptide (VIP) causes a 60% increase in cerebral blood flow after intravenous injection in conscious rats.54
BDNF causes 100% normalization of the pyramidal cell density in the CA1 sector of the hippocampus in adult rats subjected to transient forebrain ischemia after delayed intravenous administration.55
BDNF reduces stroke volume 65-70% in adult rats with either permanent or reversible middle cerebral artery occlusion (MCAO) after delayed intravenous administration.56,57
FGF-2 causes an 80% reduction in stroke volume in a permanent MCAO model in adult rats after delayed intravenous administration.58
Epidermal growth factor (EGF) can be used as a peptide radiopharmaceutical to enable early detection of brain cancer that overexpresses the EGF receptor.59
Aβ1-40 can be used as a peptide radiopharmaceutical for the early detection of brain amyloid in Alzheimer’s disease.60
Sequence-specific peptide nucleic acids (PNA) can be used as antisense radiopharmaceuticals for the in vivo imaging of gene expression in brain, in either transgenic mouse models or adult rats with experimental brain cancer.61,62
In all of these studies, the peptide or antisense agent was ineffective in the brain in vivo after intravenous administration owing to the lack of transport of the molecule across the BBB. However, the intended CNS pharmacologic effect in vivo was achieved after intravenous administration, owing to conjugation of the peptide or antisense therapeutic to a BBB molecular Trojan horse. Molecular Trojan horses can also target liposomes63 and nanoparticles64 across the BBB. Nonviral plasmid DNA is encapsulated in pegylated liposomes, which are then targeted across the BBB and the brain cell membrane with peptidomimetic monoclonal antibodies that function as molecular Trojan horses.65 The pegylated immunoliposome (PIL) nonviral gene transfer technology has enabled 100% normalization of striatal tyrosine hydroxylase activity in experimental Parkinson’s,66 and a 100% increase in survival time of adult mice with experimental brain cancer.67 After intravenous administration of PILs carrying an exogenous reporter gene, the exogenous gene was globally expressed in all regions of the brain of the adult Rhesus monkey after intravenous injection of a nonviral formulation.68 Plasmid DNA that produces short hairpin RNA for the purposes of silencing genes through a mechanism of RNA interference (RNAi) can be delivered across the BBB with the PIL gene targeting technology.69 This resulted in an 88% increase in survival time in adult mice with experimental human brain cancer that were treated with DNA-based RNAi therapeutics directed against the human EGF receptor.70
CONCLUSIONS
The development of new drugs for brain disorders is a formidable challenge, and there is no effective treatment for the majority of brain diseases (FIG. 2). The inability to treat most brain diseases is incongruous with the tremendous progress made in the molecular neurosciences. The brain drug discovery sciences have, in fact, been highly successful, and many new therapeutics have been discovered, which could potentially be used to treat the brain, if the BBB problem was solved. However, if the drugs cannot be delivered across the BBB, then there is no translation from the lab to the clinic. Step number 1 in CNS drug development is providing solutions to the BBB problem (FIG. 8). If no BBB delivery solutions are in place, which is the standard in the pharmaceutical industry, then the number of drugs that can be developed as new neurotherapeutics is less than 2% of small molecules and is ∼0% of large molecules. The few small molecules that do cross the BBB are those drugs that have high lipid solubility and molecular mass less than 400 Da, and these drugs generally only treat certain CNS disorders, such as epilepsy, affective disorders, and chronic pain (FIG. 8). In the absence of an effective BBB technology, the pharmaceutical industry cannot provide therapeutics for the majority of patients with brain disorders. It is estimated that the global CNS pharmaceutical market would have to grow by more than 500% just to equal the cardiovascular market,71 and there are more patients with CNS disorders than there are with cardiovascular disease. If BBB delivery solutions were in place for either small or large molecules, then almost any pharmaceutical could enter clinical drug development programs and therapies could be developed for most CNS disorders (FIG. 8).
FIG. 8.

Step 1 in CNS drug development is the availability of effective BBB drug or gene targeting technology. In the absence of a BBB technology, then the CNS drug developer is limited to lipid-soluble low molecular weight drugs, and only a few CNS diseases consistently respond to this class of molecule.
Acknowledgments
This work was supported by National Institutes of Health Grant NS34698.
REFERENCES
- 1.Pardridge WM. Brain drug targeting: the future of brain drug development. Cambridge, UK: Cambridge University Press, 2001.
- 2.Lloyd K, Hornykiewicz O. Parkinson’s disease activity of l-dopa decarboxylase in discrete brain regions. Science 170: 1212–1213, 1970. [DOI] [PubMed] [Google Scholar]
- 3.Zivadinov R, Zorzon M, Tommasi MA, Nasuelli D, Bernardi M, Monti-Bragadin L, et al. A longitudinal study of quality of life and side effects in patients with multiple sclerosis treated with interferon β-1a. J Neurol Sci 216: 113–118, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Antinori A, Cingolani A, Giancola ML, Forbici F, De Luca A, Perno CF. Clinical implications of HIV-1 drug resistance in the neurological compartment. Scand J Infect Dis Suppl [35 Suppl] 106: 41–44, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Kandanearatchi A, Williams B, Everall IP. Assessing the efficacy of highly active antiretroviral therapy in the brain. Brain Pathol 13: 104–110, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1: 55–68, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44: 235–249, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Fischer H, Gottschlich R, Seelig A. Blood-brain barrier permeation molecular parameters governing passive diffusion. J Membr Biol 165: 201–211, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Lieb WR, Stein WD. NonStokesian nature of transverse diffusion within human red cell membranes. J Membr Biol 92: 111–119, 1986. [DOI] [PubMed] [Google Scholar]
- 10.Trauble H. The movement of molecules across lipid membranes: a molecular theory. J Membrane Biol 4: 193–208, 1971. [DOI] [PubMed] [Google Scholar]
- 11.Pardridge WM, Mietus LJ. Transport of steroid hormones through the rat blood-brain barrier. Primary role of albumin-bound hormone. J Clin Invest 64: 145–154, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundquist S, Renftel M. The use of in vitro cell culture models for mechanistic studies and as permeability screens for the blood–brain barrier in the pharmaceutical industry-background and current status in the drug discovery process. Vasc Pharmacol 38: 355–364, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Fung LK, Shin M, Tyler B, Brem H, Saltzman WM. Chemotherapeutic drugs released from polymers distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res 13: 671–682, 1996. [DOI] [PubMed] [Google Scholar]
- 14.Blasberg RG, Patlak C, Fenstermacher JD. Intrathecal chemotherapy brain tissue profiles after ventriculocisternal perfusion. J Pharmacol Exp Ther 195: 73–83, 1975. [PubMed] [Google Scholar]
- 15.Krewson CE, Klarman ML, Saltzman WM. Distribution of nerve growth factor following direct delivery to brain interstitium. Brain Res 680: 196–206, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Yan Q, Matheson C, Sun J, Radeke MJ, Feinstein SC, Miller JA. Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. Exp Neurol 127: 23–36, 1994. [DOI] [PubMed] [Google Scholar]
- 17.Christy NP, Fishman RA. Studies of the blood-cerebrospinal fluid barrier to cortisol in the dog. J Clin Invest 40: 1997–2006, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Billiau A, Heremans H, Ververken D, van Damme J, Carton H, de Somer P. Tissue distribution of human interferons after exogenous administration in rabbits, monkeys, and mice. Arch Virol 68: 19–25, 1981. [DOI] [PubMed] [Google Scholar]
- 19.Voges J, Reszka R, Gossmann A, Dittmar C, Richter R, Garlip G, et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol 54: 479-487, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Ai Y, Markesbery W, Zhang Z, Grondin R, Elseberry D, Gerhardt GA, et al. Intraputamenal infusion of GDNF in aged rhesus monkeys distribution and dopaminergic effects. J Comp Neurol 461: 250–261, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Zunkeler B, Carson RE, Olson J, Blasberg RG, DeVroom H, Lutz RJ, et al. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg 85: 1056–1065, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Lossinsky AS, Vorbrodt AW, Wisniewski HM. Scanning and transmission electron microscopic studies of microvascular pathology in the osmotically impaired blood-brain barrier. J Neurocytol 24: 795–806, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Salahuddin TS, Johansson BB, Kalimo H, Olsson Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. Acta Neuropathol 77: 5–13 [DOI] [PubMed]
- 24.Neuwelt EA, Rapoport SI. Modification of the blood-brain barrier in the chemotherapy of malignant brain tumors. Fed Proc 43: 214–219, 1984. [PubMed] [Google Scholar]
- 25.Doolittle ND, Petrillo A, Bell S, Cummings P, Eriksen S. Blood-brain barrier disruption for the treatment of malignant brain tumors The National Program. J Neurosci Nurs 30: 81–90, 1998. [DOI] [PubMed] [Google Scholar]
- 26.Nadal A, Fuentes E, Pastor J, McNaughton PA. Plasma albumin is a potent trigger of calcium signals and DNA synthesis in astrocytes. Proc Natl Acad Sci USA 92: 1426–1430, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanig JP, Morrison JM Jr, Krop S. Ethanol enhancement of blood-brain barrier permeability to catecholamines in chicks. Eur J Pharmacol 18: 79–82, 1972. [DOI] [PubMed] [Google Scholar]
- 28.Broadwell RD, Salcman M, Kaplan RS. Morphologic effect of dimethyl sulfoxide on the blood-brain barrier. Science 217: 164–166, 1982. [DOI] [PubMed] [Google Scholar]
- 29.Saija A, Princi P, Trombetta D, Lanza M, De Pasquale A. Changes in the permeability of the blood-brain barrier following sodium dodecyl sulphate administration in the rat. Exp Brain Res 115: 546–551, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Azmin MN, Stuart JF, Florence AT. The distribution and elimination of methotrexate in mouse blood and brain after concurrent administration of polysorbate 80. Cancer Chemother Pharmacol 14: 238–242, 1985. [DOI] [PubMed] [Google Scholar]
- 31.Sakane T, Tanaka C, Yamamoto A, Hashida M, Sezaki H, Ueda H, et al. The effect of polysorbate 80 on brain uptake and analgesic effect of D-kyotorphin. Int J Pharm 57: 77–83, 1989. [Google Scholar]
- 32.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund’s adjuvant in mice provoke leakage of serum proteins through the blood-brain barrier without inducing reactive gliosis. Brain Res 832: 84–96, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173–177, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Kristensson K, Olsson Y. Uptake of exogenous proteins in mouse olfactory cells. Acta Neuropathol (Berl) 19: 145–154, 1971. [DOI] [PubMed] [Google Scholar]
- 35.Merkus P, Guchelaar HJ, Bosch DA, Merkus FW. Direct access of drugs to the human brain after intranasal drug administration? Neurology 60: 1669–1671, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL. Sniffing neuropeptides a transnasal approach to the human brain. Nat Neurosci 5: 514–516, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Thorne RG, Emory CR, Ala TA, Frey WH 2nd. Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res 692: 278–282, 1995. [DOI] [PubMed] [Google Scholar]
- 38.Bar T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol 59: I–VI, 1–62, 1980. [DOI] [PubMed] [Google Scholar]
- 39.Jaeger CB, Blight AR. Spinal cord compression injury in guinea pigs structural changes of endothelium and its perivascular cell associations after blood-brain barrier breakdown and repair. Exp Neurol 144: 381–399, 1997. [DOI] [PubMed] [Google Scholar]
- 40.Pardridge WM, Oldendorf WH. Kinetics of blood-brain barrier transport of hexoses. Biochim Biophys Acta 382: 377–392, 1975. [DOI] [PubMed] [Google Scholar]
- 41.Enerson BE, Drewes LR. Molecular features, regulation, and function of monocarboxylate transporters implications for drug delivery. J Pharm Sci 92: 1531–1544, 2003. [DOI] [PubMed] [Google Scholar]
- 42.Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci USA 96: 12079–12084, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith QR, Stoll J. Blood-brain barrier amino acid transport. Introduction to the blood-brain barrier: methodology and pathology. Cambridge, UK: Cambridge University Press, 1998.
- 44.Cornford EM, Braun LD, Oldendorf WH. Carrier mediated blood-brain barrier transport of choline and certain choline analogs. J Neurochem 30: 299–308, 1978. [DOI] [PubMed] [Google Scholar]
- 45.Allen DD, Smith QR. Characterization of the blood-brain barrier choline transporter using the in situ rat brain perfusion technique. J Neurochem 76: 1032–1041, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Cornford EM, Oldendorf WH. Independent blood-brain barrier transport systems for nucleic acid precursors. Biochim Biophys Acta 394: 211–219, 1975. [DOI] [PubMed] [Google Scholar]
- 47.Li JY, Boado RJ, Pardridge WM. Cloned blood-brain barrier adenosine transporter is identical to the rat concentrative Na+ nucleoside cotransporter CNT2. J Cereb Blood Flow Metab 21: 929–936, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Pardridge WM, Yoshikawa T, Kang Y-L, Miller LP. Blood-brain barrier transport and brain metabolism of adenosine and adenosine analogs. J Pharmacol Exp Ther 268: 14–18, 1994. [PubMed] [Google Scholar]
- 49.Zhang Y, Pardridge WM. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J Neurochem 76: 1597–1600, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Skarlatos S, Yoshikawa T, Pardridge WM. Transport of [125I]transferrin through the rat blood-brain barrier. Brain Res 683: 164–171, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol 114: 168–172, 2001. [DOI] [PubMed] [Google Scholar]
- 52.Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J Neurochem 81: 203–206, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem 54: 1882–1888, 1990. [DOI] [PubMed] [Google Scholar]
- 54.Wu D, Pardridge WM. Central nervous system pharmacologic effect in conscious rats after intravenous injection of a biotinylated vasoactive intestinal peptide analog coupled to a blood-brain barrier drug delivery system. J Pharmacol Exp Ther 279: 77–83, 1996. [PubMed] [Google Scholar]
- 55.Wu D, Pardridge WM. Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc Natl Acad Sci USA 96: 254–259, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Pardridge WM. Conjugation of brain-derived neurotrophic factor to a blood-brain barrier drug targeting system enables neuroprotection in regional brain ischemia following intravenous injection of the neurotrophin. Brain Res 889: 49–56, 2001. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Pardridge WM. Neuroprotection in transient focal brain ischemia after delayed intravenous administration of brain-derived neurotrophic factor conjugated to a blood-brain barrier drug targeting system. Stroke 32: 1378–1384, 2001. [DOI] [PubMed] [Google Scholar]
- 58.Song BW, Vinters HV, Wu D, Pardridge WM. Enhanced neuroprotective effects of basic fibroblast growth factor in regional brain ischemia after conjugation to a blood-brain barrier delivery vector. J Pharmacol Exp Ther 301: 605–610, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Kurihara A, Pardridge WM. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood-brain barrier. Cancer Res 59: 6159–6163, 1999. [PubMed] [Google Scholar]
- 60.Lee HJ, Zhang Y, Zhu C, Duff K, Pardridge WM. Imaging brain amyloid of Alzheimer disease in vivo in transgenic mice with an Aβ peptide radiopharmaceutical. J Cereb Blood Flow Metab 22: 223–231, 2002. [DOI] [PubMed] [Google Scholar]
- 61.Lee HJ, Boado RJ, Braasch DA, Corey DR, Pardridge WM. Imaging gene expression in the brain in vivo in a transgenic mouse model of Huntington’s disease with an antisense radiopharmaceutical and drug-targeting technology. J Nucl Med 43: 948–956, 2002. [PubMed] [Google Scholar]
- 62.Suzuki T, Wu D, Schlachetzki F, Li JY, Boado, RJ, Pardridge WM. Imaging endogenous gene expression in brain cancer in vivo with 111In-peptide nucleic acid antisense radiopharmaceuticals and brain drug-targeting technology. J Nucl Med 10: 1766–1775, 2004. [PubMed] [Google Scholar]
- 63.Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci USA 93: 14164–14169, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olivier JC, Huertas R, Lee HJ, Calon F, Pardridge WM. Synthesis of pegylated immunonanoparticles. Pharm Res 19: 1137–1143, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Shi N, Zhang Y, Zhu C, Boado RJ, Pardridge WM. Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci USA 98: 12754–12759, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Schlachetzki F, Zhang YF, Boado RJ, Pardridge WM. Normalization of striatal tyrosine hydroxylase and reversal of motor impairment in experimental parkinsonism with intravenous nonviral gene therapy and a brain-specific promoter. Hum Gene Ther 15: 339–350, 2004. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Zhu C, Pardridge WM. Antisense gene therapy of brain cancer with an artificial virus gene delivery system. Mol Ther 6: 67–72, 2002. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Schlachetzki F, Pardridge WM. Global non-viral gene transfer to the primate brain following intravenous administration. Mol Ther 7: 11–18, 2003. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Boado RJ, Pardridge WM. In vivo knockdown of gene expression in brain cancer with intravenous RNAi in adult rats. J Gene Med 5: 1039–1045, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Bryant J, Zhang YF, Charles A, Boado RJ, Pardridge WM. Intravenous RNAi gene therapy targeting the human EGF receptor prolongs survival in intra-cranial brain cancer. Clin Cancer Res 10: 3667–3677. [DOI] [PubMed]
- 71.Pardridge WM. Why is the global CNS pharmaceutical market so under-penetrated? Drug Discov Today 7: 5–7, 2002. [DOI] [PubMed] [Google Scholar]
- 72.Matsukado K, Sugita M, Black KL. Intracarotid low dose bradykinin infusion selectively increases tumor permeability through activation of bradykinin B2 receptors in malignant gliomas. Brain Res 792: 10–15, 1998. [DOI] [PubMed] [Google Scholar]
- 73.Spigelman MK, Zappulla RA, Goldberg JD, Goldsmith SJ, Marotta D, Malis LI, et al. Effect of intracarotid etoposide on opening the blood-brain barrier. Cancer Drug Deliv 1: 207–211, 1984. [DOI] [PubMed] [Google Scholar]
- 74.Cornford EM, Young D, Paxton JW, Finlay GJ, Wilson WR, Pardridge WM. Melphalan penetration of the blood-brain barrier via the neutral amino acid transporter in tumor-bearing brain. Cancer Res 52: 138–143, 1992. [PubMed] [Google Scholar]
- 75.Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 220: 640–646, 2001. [DOI] [PubMed] [Google Scholar]
- 76.Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 86: 1245–1257, 1998. [DOI] [PubMed] [Google Scholar]
- 77.Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, et al. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol 8: 923–926, 1998. [DOI] [PubMed] [Google Scholar]
- 78.Oldendorf WH, Stoller BE, Tishler TA, Williams JL, Oldendorf SZ. Transient blood-brain barrier passage of polar compounds at low pH. Am J Physiol (Lond) 267: H2229–H2236, 1994. [DOI] [PubMed] [Google Scholar]
- 79.Oztas B, Kucuk M. Intracarotid hypothermic saline infusion a new method for reversible blood-brain barrier disruption in anesthetized rats. Neurosci Lett 190: 203–206, 1995. [DOI] [PubMed] [Google Scholar]
- 80.Sztriha L, Betz AL. Oleic acid reversibly opens the blood–brain barrier. Brain Res 550: 257–262, 1991. [DOI] [PubMed] [Google Scholar]