

Abstract
Summary: The extent to which a substance in the circulation gains access to the CNS needs to be determined for potential neuropharmaceuticals as well as for drug candidates with primary targets in the periphery. Characteristics of the in vivo methods, ranging from classical pharmacokinetic techniques (intravenous administration and tissue sampling) over brain perfusions to microdialysis and imaging techniques, are highlighted. In vivo measurements remain unmatched with respect to sensitivity and for the characterization of carrier-mediated uptake, receptor-mediated transport, and active efflux. Isolated microvessels are valuable tools for molecular characterization of transporters. Endothelial cell culture models of the blood-brain barrier (BBB) are pursued as in vitro systems suitable for screening procedures. Recent applications of conditionally immortalized cell lines indicate that a particular weakness of culture models because of downregulation of BBB-specific transporter systems can be overcome. In silico approaches are being developed with the goal of predicting brain uptake from molecular structure at early stages of drug development. Currently, the predictive capability is limited to passive, diffusional uptake and predominantly relies on few molecular descriptors related to lipophilicity, hydrogen bonding capacity, charge, and molecular weight. A caveat with most present strategies is their reliance on surrogates of BBB transport, like CNS activity/inactivity or brain-to-blood partitioning rather than actual BBB permeability data.
Keywords: Blood-brain barrier, endothelial cell culture, pharmacokinetics, microdialysis, lipophilicity
INTRODUCTION
Valid information about permeability of drug candidates at the blood-brain barrier (BBB) is needed to decrease the attrition rate in drug discovery/development, as poor pharmacokinetics has been recognized as one of the leading causes of failure.1, 2
With the exceptions of diseases affecting primarily the brain surface, such as meningitis or cerebral manifestations of leukemia, the brain tissue concentration, and its temporal relation to blood or plasma concentrations is most relevant, as compared with concentrations in CSF. Brain tissue concentration needs to be further distinguished into the cell membrane bound and intracellular fractions, and the fraction in brain interstitial space, which amounts to about 19% of the tissue volume.3 Depending on the drug target and mechanism of action of a given substance, either whole tissue concentration or extracellular concentration may correlate to effect.
Figure 1 shows a schematic diagram of the brain and its barrier systems. The BBB and the blood-CSF barrier (B-CSF-B) differ in location, size, morphology, and function. The BBB is an endothelial barrier, where tight junctions between the endothelial cells seal off the vascular lumen from the abluminal side. In contrast, the B-CSF-B is formed by the plexus epithelial cells, which are connected by tight junctions, whereas the capillaries are fenestrated. The available experimental tools to study transport include in vivo techniques, in vitro models, and computational approaches. An understanding of the different parameters used in these methods for the description of brain uptake is required to make valid comparisons. Table 1 gives a synopsis of the techniques discussed in the following sections, highlighting the main advantages and limitations.
FIG. 1.

The interfaces between CNS and systemic blood: BBB is formed by microvascular endothelial cells. B-CSF-B is formed by the choroid plexus epithelial cells, which also secrete CSF. Transport processes at both barrier systems include (a) passive exchange via transcellular or paracellular diffusion, (b) uptake by facilitative or active carriers and receptor-mediated transport, (c) active efflux. In (d) there is exchange by diffusion and bulk flow between brain interstitial fluid (ISF) and CSF, which are not separated by a tight cellular barrier. In vivo methods for measurement of brain uptake differ in the sampling compartment: 1) whole tissue samples include vascular content, vasculature, brain cells and interstitial fluid, 2) microdialysis methods sample interstitial fluid, and 3) catheters in ventricles or subarachnoid space collect CSF.
TABLE 1.
Characteristics of the Techniques Used to Estimate Brain Uptake of Drugs
| Technique | Measured/Estimated Parameter | Advantages | Disadvantages/Caveats |
|---|---|---|---|
| In vivo methods | |||
| Intravenous injection/brain sampling | Influx; Influx/efflux | Most physiological approach; highest sensitivity; low technical difficulty | May require good analytical tools to exclude metabolite uptake and careful pharmacokinetic analysis to discriminate unidirectional uptake versus bidirectional transfer |
| Brain uptake index | Influx | Fast procedure; moderate technical difficulty; permits wide range of modifications of injectate composition; artifacts by metabolism largely excluded | Relatively insensitive (compared with intravenous injection and brain perfusion) |
| Brain perfusion | Influx | Higher sensitivity compared with BUI; permits modification of both perfusate composition and flow rates; artifacts by peripheral metabolism excluded | Technically more difficult than intravenous experiments and BUI |
| Quantitative autoradiography | Influx | Excellent spatial resolution | Time-consuming evaluation; no proof of integrity of tracer |
| External registration: MRI, SPECT, PET | Influx/efflux | Noninvasive and applicable in humans; allows time course measurements in individual subjects | Expensive equipment (MRI, PET) and tracers (PET); limited sensitivity (MRI) and availability of labeled tracers (MRI, PET); poor spatial resolution for small animals (SPECT) |
| Microdialysis | Influx/efflux | Allows time course measurements in individual subjects; samples well suited for subsequent analytical procedures | Technically involved; in vivo probe calibration required for valid quantitative evaluation;local damage to BBB integrity |
| CSF sampling | Influx/efflux | Readily accessible for sampling; applicable to humans | Reflects permeability of B-CSF-B and CSF fluid dynamics rather than BBB |
| In vitro methods | |||
| Fresh isolated brain microvessels | Binding, uptake, efflux | Representing the in vivo expression of transporters and efflux systems at the BBB | Transcellular passage cannot be measured |
| EC membrane vesicles | Carrier-mediated transport | Allows distinction of luminal versus abluminal transport activity | Large amounts of source material required, laborious preparation |
| Endothelial cell culture | |||
| Primary cultures, cell lines | Receptor binding; uptake; luminal to abluminal transfer (and opposite direction) | Permeability screening experiments (feasible with primary EC from bovine/porcine sources); effect of culture conditions on BBB transport properties may be studied (e.g., astroglial factors, serum effects, inflammatory stimuli, hypoxia/aglycemia) | No system yet able to represent in vivo condition with respect to barrier tightness and BBB specific transporter expression; multitude of models makes comparison of results between studies difficult |
| In silico models | CNS active (+/−); Log BB; Log PS | Screening of large compound libraries (depending on model selection and computational resources); screening of virtual libraries | Many current models based on data, which may not represent BBB permeability as such (log BB; CNS activity); still very limited data bases for BBB transport (log PS models) |
| Rules of thumb | |||
| Classification models | |||
| QSAR |
IN VIVO MEASUREMENTS
Intravenous experiments and pharmacokinetic principles
The intravenous technique remains the reference for brain uptake studies because it represents fully physiological conditions. Moreover, it offers the potentially highest sensitivity, as tissue uptake in intravenous experiments can be measured over long periods (i.e., multiple passages through the brain capillary bed). In addition, information on systemic pharmacokinetics of a compound may be gained simultaneously. However, awareness of potential pitfalls is required to avoid erroneous evaluation and data interpretation. It is salient that specific and sensitive analytical methods must be applied, especially with metabolically labile substances, to measure the intact fraction in blood and tissue. Two pharmacokinetic scenarios need to be distinguished with respect to brain uptake: 1) Unidirectional uptake from plasma into brain tissue, or 2) reversible uptake, i.e., influx and efflux are present simultaneously. Unless there is irreversible binding of the test substance in brain tissue, the second case will apply to any compound at sufficiently late time points.
Evaluation of initial rate experiments
The goal of such studies is to capture the unidirectional uptake phase. In that case no assumptions are required about distribution phenomena taking place after a substance has entered brain tissue (cellular binding, cellular uptake, metabolic degradation, efflux), which are often not readily experimentally accessible.
Single time-point analysis4 requires the least number of test subjects but is contingent on the presence of unidirectional transport. It involves arterial blood sampling at suitable intervals for analysis of the time curve of blood or plasma concentration, Cp, and obtaining a single brain tissue sample at the terminal time point. Provided the condition of unidirectional uptake holds, the brain tissue concentration, Cbr, accumulated at time T can be calculated as
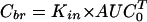 |
(1) |
where Kin is an organ clearance, the unidirectional influx constant from plasma to brain, and AUC0T (area under the curve) is the integral of plasma concentrations from time = 0 to time = T. Cbr in that equation should represent tissue concentration after correction for intravascular content. A brief washing procedure with buffer via the ascending aorta can be performed to clear intravascular content after sacrificing the animal. Alternatively, the intravascular volume of brain, V0, is experimentally determined by coadministration of a vascular marker (a substance not significantly accumulating in brain tissue during the time course of the experiment, e.g., radiolabeled serum albumin, inulin, or sucrose). The corrected brain concentration, Cbr, is then obtained according to
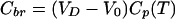 |
(2) |
where VD represents the apparent volume of distribution of the test substance in total brain at the sampling time T: VD = Cbr(total)/Cp.
Therefore, Kin, with dimensions [volume mass−1 time−1], is obtained from the measurable quantities as:
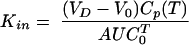 |
(3) |
The permeability of the BBB may then be derived from the Renkin-Crone equation of capillary transport, 5, 6 which provides a relation between Kin and PS, (the product of permeability P and capillary surface area S) and flow F:
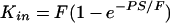 |
(4) |
Two boundary conditions can be identified. Kin will be equivalent to PS (with an error ≤ 10%) if BBB permeability is low relative to flow (PS ≤ 0.2 F).7 In the case of high PS products, Kin will approach cerebral blood flow (PS ≥ 2.3 F, with an error ≤ 10%) and is fully flow limited. Hence, for the valid calculation of PS products in the range of PS > 0.2 F, simultaneous measurement of Kin and of cerebral blood flow is required. Substances with high permeability suitable as flow markers include diazepam and butanol.
Multiple-time uptake data
For brain tissue concentrations measured at different sampling times, Patlak formulated a method to estimate Kin from a graphical representation of the experimental data.8 VD in brain is plotted versus the ratio AUC0T/Cp(T), also called the “exposure time.” This plot yields a straight line for the period of unidirectional uptake, and linear regression analysis can be applied to fit the data to the equation used in this approach:
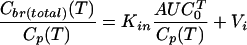 |
(5) |
The fitting provides Kin as the slope of the regression line. Vi is the ordinate intercept and represents the initial volume of distribution, which comprises intravascular space, V0, and any brain compartments in rapid equilibration with plasma.9 Theoretically, a vascular marker is not required, in contrast to the single time point analysis. However, without additional experimental information the assignment of a physiological meaning to Vi values exceeding the typical intravascular volume of about 10-12 μl/g is not straightforward. In addition, if an apparent linearity of the uptake phase is observed in the presence of large y-intercepts (= high Vi values), the assumption of unidirectional uptake is likely no longer justified.10
Bidirectional transfer across the BBB after systemic drug administration is included in multicompartmental models, such as the distributed model, 11 which distinguish concentrations in brain intracellular fluid, extracellular fluid and CSF, but the practical application requires considerable experimental input. Direct experimental measurements of efflux can be performed (see brain efflux index as discussed in this issue of NeuroRx® by Terasaki and Ohtsuki).12
For cases where whole brain tissue concentrations Cbr are measured and passive distribution across the BBB occurs by diffusional transport, the relationship to the plasma concentration-time course after an intravenous bolus administration can be described by an equation of the form:
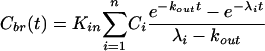 |
(6) |
where Ci and λi are the parameters of an exponential disposition function from plasma.13 The single rate constant of elimination from brain tissue, kout, may comprise loss by efflux and/or local metabolism. Equation (6) is equivalent to equation (1) for the special case, kout = 0.
The evaluations outlined above assume linear pharmacokinetics. Saturable processes for brain uptake or systemic clearance can be accounted for by applying Michaelis-Menten kinetics (see brain perfusion studies described below).
Brain uptake index and isolated brain perfusion
These methods complement the intravenous analysis and offer some unique advantages. Both techniques permit to manipulate the composition of the intravascular fluid in the brain microcirculation within wide limits, which is not possible with intravenous administration. In addition, metabolism in other organs is excluded, facilitating analysis and avoidance of artifactual uptake measurements.
The brain uptake index (BUI), as originally described and refined by Oldendorf, 14, 15 is a single pass method. It greatly facilitated transport studies because of its technical simplicity and versatility compared with the indicator diffusion method, a single passage technique, which requires sampling of venous cerebral outflow.5 The BUI employs a rapid bolus injection of radiolabeled test and reference substances into the common carotid artery of anesthetized animals (e.g., in the rat 0.2 ml in less than 0.5 s). The option to choose freely the composition of the injectate buffer allows to analyze the effects of test solute concentration, electrolytes and pH, osmotic pressure, as well as the influence of protein binding. After the 1- to 2-s passage time through the brain capillaries, brain uptake is measured as the single pass extraction, E, by brain tissue sampling after decapitation, which is performed within 5-15 s. The short time interval excludes systemic recirculation and also minimizes potential wash-out.15 A second reference substance is coinjected, which does not penetrate the BBB, to correct for the fraction of the bolus remaining in the vascular lumen at the time of brain sampling (about 2%). Suitable combinations of radioisotopes must be available to allow for double or triple isotope counting of the tracers in injectate and brain tissue, respectively. The calculation of BUI (as percentage) is defined by the equation:
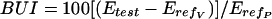 |
(7) |
where Etest is the brain extraction of the unknown test substance, ErefV is the apparent extraction of a vascular marker (nonpermeant reference) and ErefP is the extraction of the permeant reference. The BUI represents a relative measure, but it may be converted to a true extraction value, provided the absolute extraction fraction, Eref, of the permeant reference, is known from independent experiments.15 Further, the Renkin-Crone equation gives the relation to PS product and cerebral blood flow, F:
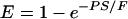 |
(8) |
With widespread use of the carotid single injection method over the years, BUI data of a host of endogenous and exogenous compounds accumulated, including the characterization of BBB nutrient transport systems and drug transport.16 BUI measurements are particularly suitable for substances with moderate to high BBB permeabilities, whereas the single capillary transit implies that absolute extraction values of poorly permeant solutes (at E values of about 0.05 or less) cannot be determined reliably. As an example, the classical CNS drug morphine with a BUI of 2.6 is barely different from vascular markers with a BUI value of 2.15
In addition to the low sensitivity, other limitations have been realized. Mixing of endogenous plasma with the injectate solution has been estimated at 5%, 17 which is relevant in cases of saturable transport with low Michaelis-Menten constant (Km) and high plasma concentrations of endogenous substrates (e.g., neutral amino acids and the LAT1 transporter) and for drugs with high plasma protein binding. Because of the short measurement time, it is also not possible to study slower uptake processes, such as receptor-mediated endocytosis.
These shortfalls of the BUI have been addressed by the development of brain perfusion methods.18–20 The Takasato method uses a retrograde catheterization of the external carotid artery in the anesthetized rat, and ligation of all branches of the external and internal carotid artery.18 The perfusion with an oxygenated physiological buffer containing the test substance is started after ligating the common carotid artery. Recent simplifications of the technique21 include direct catheterization of the common carotid artery, and stopping endogenous blood flow by severing the heart ventricles before start of the perfusion to minimize mixing of perfusate and endogenous blood. In addition to the manipulation of perfusate composition, control of flow rate and duration is possible with the perfusion technique. Because of the constant perfusate concentration during the perfusion time, T, equation (3) for calculating Kin simplifies to:
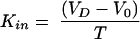 |
(9) |
In accordance with the longer exposure of the brain capillary bed to the test substance compared to the single pass technique, the sensitivity of the perfusion method is enhanced by about 2 orders of magnitude. Perfusion times with buffer can be extended up to 10 min, 22 and up to 1 h by addition of oxygen carriers (washed erythrocytes).21
Receptor-mediated transport at the BBB via transcytosis has been shown for peptides and proteins, 23 e.g., hormones like insulin and leptin, transport proteins (transferrin, apolipoproteins), and various cytokines. The relatively slow transport process, operating on a time scale of minutes, as opposed to the rapid carrier mediated transport, makes brain perfusion the method of choice to characterize receptor-mediated uptake. The BUI method is too insensitive, whereas the intravenous technique is prone to artifacts, especially with respect to the metabolic lability of peptide hormones.
It is crucial in uptake experiments to distinguish between the fraction of a test substance, which has passed through the endothelium and entered brain extracellular space or brain cells, and the fraction associated with the endothelial cells. Such an analysis is possible with the capillary depletion method.22 A density centrifugation step of brain homogenate in dextran separates the vascular component from the rest of the tissue. The power of the method was demonstrated in studies revealing the vascular sequestration of adenosine after carrier mediated uptake and endothelial metabolism, 24 and by receptor-mediated endocytosis without exocytosis on the abluminal side22 for acetylated low-density lipoprotein (LDL). However, it is important to be cognizant of the limitations of the approach. Because vascular depletion is not an instantaneous process, redistribution may produce artifacts, e.g., because of dissociation from low affinity or nonspecific binding sites at the endothelial cell plasma membrane, as observed with a synthetic dermorphin tetrapeptide.25
Analysis of saturable uptake
A modified Michaelis-Menten equation is employed to characterize BBB permeability by saturable transport systems under different concentrations of test solute in the perfusate, Cpf. To account for the diffusional component of uptake, the following equation is used:
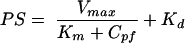 |
(10) |
where Vmax, the maximal rate of the saturable component of transport, Km, the perfusate concentration at which half maximal transport occurs, and Kd, the nonsaturable, diffusion uptake clearance. The parameters of Km and Vmax can be estimated by regression analysis after plotting BBB PS products versus perfusate concentrations. This type of analysis has been extensively used with both the BUI and perfusion techniques to study carrier mediated uptake of glucose, amino acids, and other metabolites.26–28
Quantitative autoradiography
Quantitative autoradiography (QAR) methods were originally developed by Sokoloff’s group29, 30 for measurement of local glucose metabolism with [14C] 2-deoxyglucose and for blood flow with iodo[14C]antipyrine. The strength of the technique lies in the high spatial resolution in the micrometer range. Preferred applications include studies in which regional differentiation is crucial, as is the case for brain tumors31 and ischemia.32 The radioactive tracers, typically labeled with 14C, are administered intravenously, and arterial blood samples are drawn to measure the concentration time course. Pharmacokinetic evaluations correspond to the procedures outlined for intravenous experiments above, with the difference that brain activity measurements are performed on cryostate sections of the harvested organ, which are dried and exposed to x-ray film or phosphor imaging screens along with radioactive standards. Quantification of V0 values to correct for intravascular volume is done in separate experimental series after injection of a marker with low permeability (125I-labeled albumin, [14C] sucrose). To calculate regional PS values of a test substance, separate measurement of regional blood flow must be performed.33 Employing suitable combinations of isotopes, dual label autoradiographic methods are possible. QAR methods serve as the reference for developing external detection techniques described below.
External detection methods
Because of their noninvasive character, external detection techniques can be applied in humans and permit to measure individually the time course of uptake together with plasma pharmacokinetics. Spatial resolution of single photon computed tomography (SPECT) is in the centimeter range, whereas magnetic resonance imaging (MRI) and positron emission tomography (PET) approach autoradiographic resolutions. In the case of the radioisotope techniques (SPECT, PET) no signal discriminating intact tracer and metabolites is obtained, which limits the time course of experiments if tracers are chemically unstable.
Three main types of PET tracers are in use: 1) nutrient analogs like 18F-deoxy-glucose and several amino acid analogs (e.g., 11C-methionine, 18F-DOPA) for measurement of metabolism, 2) ligands for neurotransmitter receptors or transporters, and 3) tracers for detection of BBB damage (e.g., 68Ga-EDTA). The primary focus in studies with the first two classes is on brain metabolism and receptor binding, respectively. Quantitative measurements of PS products at the BBB use dynamic scanning procedures and require the arterial input function to be measured, either by blood sampling or scanning of large vessels. Evaluations are performed graphically, or with compartmental pharmacokinetic models.34, 35
Recent efforts are directed at the development of radiotracers, which are substrates of the membrane efflux transporter, p-glycoprotein (MDR). For example, 11C-verapamil and 11C-carvedilol have been introduced as PET tracers for this purpose and may be beneficial for screening the pharmacokinetic behavior of other P-gp substrates in inhibition experiments.36
MRI is clinically used for detection of BBB defects in diseases like multiple sclerosis, stroke, and brain tumors. Routinely the focal BBB breakdown associated with these conditions is qualitatively measured after injection of suitable paramagnetic MR contrast media, e.g., gadolinium-DTPA (Gd-DTPA). Quantitative measurement of BBB PS products is feasible with this tracer, 37 which behaves as a vascular marker under physiological conditions, showing a PS product at the intact BBB below the value of sucrose.38 Dynamic MRI measurements follow the concentration changes of the contrast agent in brain tissue after an intravenous bolus injection. The evaluation of concentration time curves for determination of PS products either employs multicompartmental kinetic models or the graphical Patlak analysis.38 Gd-DTPA may be useful to follow the effect of drug treatment on restoring BBB integrity. The measurement of drug uptake itself through the BBB by MRI is limited by the inherently low sensitivity of MRI, which requires high local concentrations to generate detectable MR contrast.39 As an illustration, typical systemic doses of the BBB leakage probe, Gd-DTPA, are on the order of 0.1 mmol/kg.
Brain microdialysis
The microdialysis technique has gained increasing popularity over the last decade for studying drug distribution to the CNS.40, 41 This method requires stereotaxic probe implantation under anesthesia, which is an invasive procedure causing acute traumatic injury.42 Once the probe is implanted, microdialysis offers the advantage of allowing repeated or continuous sampling in freely moving animals. The method is applicable in humans, and the concentration-time course (and the AUC) of a test substance can be measured in individual subjects. Moreover, the dialysate is readily available for chromatographic analysis.
Valid quantitative interpretation of microdialysis data requires the individual calibration of the probes to obtain correction factors, which relate the dialysate concentration to the brain extracellular fluid (ECF) concentration. Recovery rates of dialysis probes for certain test substances can be low, e.g., in the range of 3% with small peptides. In addition to the physicochemical properties of the solute, the probe recovery as measured in vitro is dependent on factors such as probe geometry, membrane material, and dialysate flow rate. Notably, theoretical models clearly demonstrated that in vitro and in vivo recovery rates differ, because the latter is modulated by tissue-specific factors.43 Experimentally, several techniques are in use to account for this fact and determine the in vivo probe recovery. These include the retrodialysis method, the zero-net-flux approaches, 44 and the reference method.40 Moreover, the underlying pharmacokinetic principles need to be considered in the interpretation when microdialysis is used to analyze BBB permeability.45 Because the samples reflect unbound test solute concentration in brain extracellular fluid, microdialysate data are not only dependent on the uptake rate and efflux rate through the BBB, but also on the rates of transfer between brain intra- and extracellular compartments. When drug concentrations are measured under steady-state conditions, a ratio of unity is expected for AUCbrain ECF/AUCplasma unbound. A significantly lower ratio is interpreted as evidence of local metabolism or the presence of efflux transport at the BBB.46
For correct estimates of BBB influx clearances or PS products, the apparent brain volume of distribution needs to be known, which requires the separate measurement of total tissue concentrations.40, 47 Erroneous conclusions based on isolated microdialysis data may be drawn, as a controversy around the BBB permeability of morphine-6-glucoronide (M6G) may illustrate. The postulate of good BBB permeability of M6G was based on the AUC in brain dialysate of this pharmacologically active morphine metabolite being only four-fold lower compared with its parent compound after equivalent systemic doses.48 However, determining BBB permeability of M6G with intravenous injection and whole tissue measurement revealed a PS product 32- to 57-fold lower than morphine.49, 50 The discrepancy was reconciled by studies, in which both brain dialysate and whole tissue concentrations were taken into account.51, 52 These comparisons showed that M6G is largely confined to brain extracellular space. Despite a much lower uptake rate through the BBB concentrations approach those of morphine, which accumulates predominantly in the intracellular compartment.
Brain microdialysis appears to perform best for drugs with intermediate permeability. For substances with high permeability, which also tend to show high intracellular accumulation, concentrations in dialysate may be immeasurably low. An example is the highly lipophilic benzopyranone analog KA-672, which reached concentrations in brain homogenate three times higher than in plasma, whereas microdialysate concentrations were below the detection limit.53 At the other end of the permeability spectrum, microdialysis likely overestimates the permeability of poorly permeable compounds (e.g., sucrose, AIB, inulin, urea) because of subtle, yet persistent BBB damage. For example, an increase of the BBB PS product of sucrose by a factor between 5 and 20 was measured, when compared with the intravenous administration/single time point brain sampling method and QAR.54, 55
Limitations of CSF sampling
Especially in human pharmacokinetic studies the measurement of CSF concentrations is often taken as a substitute for brain interstitial fluid concentrations. However, as Collins and Dedrick11 already derived theoretically for the distributed model, drug delivery to brain and CSF must not be considered identical. Concentration gradients between CSF and brain tissue exist, with the magnitude being determined by the relations of CSF bulk flow, distance from ependymal surface, and brain capillary permeability. This holds even in the absence of any active transport processes. However, many drugs are affected by active influx or efflux systems at the CNS barriers, or by local metabolism.56 Quantitative and qualitative differences in the expression of transporters between BBB and B-CSF-B will result in unequal concentrations in CSF and brain after systemic drug administration. Well documented examples are the quinolone antibiotics, which reach much lower concentrations in brain extracellular fluid compared to CSF because of highly efficient efflux transport at the BBB.57, 58 The reverse transcriptase inhibitor azidothymidine readily reaches CSF as a nucleoside analog via a thymidine carrier system at the choroid plexus. On the other hand, azidothymidine shows negligible brain uptake, consistent with efflux at the BBB.59
IN VITRO MODELS OF BRAIN UPTAKE
Isolated brain microvessels
The ability to isolate viable microvessels from brain tissue of various species, including human autopsy brain, spurred multiple in vitro approaches.60, 61 The isolated capillaries remain metabolically active, although the ATP content is significantly depleted.62 The BBB-specific gene expression of endothelial receptors and carrier proteins reflects the in vivo situation. Hence, microvessel preparations can be successfully used to perform binding and uptake assays and to study BBB transport systems for nutrients and peptides at the mRNA and protein level.61 Examples of binding/uptake assays conducted with isolated microvessels to characterize peptide receptors at the BBB, include those for transferrin, insulin and insulin like growth factors, leptin and tumor necrosis factor (TNF) α.28, 63 Because both plasma membrane surfaces are accessible to ligand in a capillary suspension, no differentiation between binding and uptake by the luminal versus the abluminal side is obtained. However, after incubation of fresh capillaries with fluorescence-labeled ligands, confocal microscopy can be used to analyze the spatial distribution. This approach allowed the demonstration of luminal and abluminal localization of transferrin receptors with a monoclonal antibody attached to fluorescent labeled liposomes.64 Recently, Miller et al.65 used isolated brain microvessels to establish a semiquantitative assay for drug efflux transport at the luminal endothelial plasma membrane. These authors used confocal microscopy to measure concentrations of fluorescent P-glycoprotein substrates. Microvessels were loaded with fluorescent drugs (e.g., daunomycin, fluorescent derivatives of cyclosporin A and ivermectin) and then fluorescence intensity accumulating inside the lumen of a defined isolated brain capillary was dynamically quantified. Nonfluorescent drugs may also be analyzed, if they inhibit efflux of fluorescent substrates (e.g., inhibition of P-gp mediated transport by verapamil). Efflux mediated by ABC transporters is known to be ATP dependent, and the fact that it could be suppressed by the inhibitor NaCN lends further support to the metabolic viability of isolated microvessels.65
Another option to separately analyze transport processes at the luminal and abluminal plasma membrane is provided by the isolation of membrane vesicles derived from these membrane domains with ultracentrifugation in Ficoll gradients.66 However, large amounts of brain microvessels are required as a source for the membrane preparation, which need to be isolated from a typical starting material on the order of 100 bovine brains.67 The membrane vesicles have been used in initial rate experiments and in equilibrium studies to analyze kinetics and distribution of transport systems. To date, nutrient transport (amino acids, glucose) and electrolyte transport have been characterized, but the technique should also be applicable to study carrier-mediated drug transport.
Endothelial cell culture models
Much effort is dedicated to the development of brain microvessel endothelial cell (BMEC) culture systems, which could be used for quantitative permeability studies and as screen in drug discovery.68, 69 A large diversity of models exists with respect to species of origin, primary culture or cell lines, and culture conditions.
Typical set-ups for transport studies have been compared.70 A vertical system for transport studies utilizes the Transwell insert system, where an endothelial cell monolayer is grown to confluence on collagen/fibronectin coated, removable polycarbonate filter inserts separating an upper (donor) and lower (acceptor) chamber. This system is popular primarily because of the convenient handling. Its disadvantages include the difficulty of a tight temperature control and poor agitation of the solutions, which does not guarantee well-stirred conditions. This presents a significant difference to the in vivo condition, as there is no significant unstirred water layer at the BBB.71 In addition, the construction makes a small hydrostatic pressure gradient unavoidable.
Alternative technical solutions place the filter with the endothelial cell layer between horizontal diffusion chambers, and continuous mixing of donor and acceptor fluid is created either by gas-lift (Snapwell system; Corning, Acton, MA) or by magnetic stir bars (Side-bi-Side system; Crown Glass, Somerville, NJ). Both horizontal systems avoid hydrostatic pressure gradients and provide temperature stability using a water jacket.
The actual transport experiments and the evaluations are straightforward. After adding the test substance to the donor compartment, repeated samples are taken from the donor compartment over the desired time course (typically 30-60 min). The concentration measured in these samples and the known volumes of the compartments (Vdonor and Vacceptor) are used to calculate the incremental clearance volumes ΔVCl for each time point:
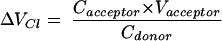 |
(11) |
As long as the concentration in the acceptor compartment is small and ΔVCl increases in linear manner, the slope of the line can be interpreted as the PS product for unidirectional transfer. With the known exchange surface, S, (= filter area) the permeability may be obtained as P = PS/S. Finally, a correction needs to be made for the permeability of the cell-free filter:
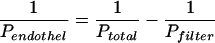 |
(12) |
Primary endothelial cell cultures
Accepted criteria for monitoring the quality of monolayers in transport studies include transendothelial electrical resistance (TEER) and permeability to hydrophilic markers like 14C-sucrose, which reflect the degree of tight junction formation. Using these criteria, none of the primary endothelial cell culture models matches yet in vivo conditions (TEER in the range >2 kΩcm2 up to 8 kΩcm2, 72 and sucrose permeability of about 0.3 × 10−7 cm s−1).4 Although primary cultures can be established from rat, mouse, bovine, porcine, primate, or human brain, only bovine or porcine sources provide sufficient quantities for permeability screening of drugs.
Bovine systems in several variants are widely used. For example, Audus and Borchardt’s labs68, 70 developed a monoculture system of endothelial cells grown out from microvessels, which are obtained by Percoll centrifugation after several steps of enzymatic digestion of brain tissue (dispase/collagenase). These cells form monolayers with TEER values of 160-200 Ωcm2 and sucrose permeabilities of 1-2.4 × 10−5 cm s−1 after 9-10 days in culture.68
One intervention to improve barrier tightness and BBB specific gene expression is to mimic the astroglial influence present in vivo by adding astrocyte conditioned culture medium or by coculture with astrocytes. For example, Cecchelli and co-workers73 use rat astrocytes grown at the bottom of the culture well, with bovine BMEC growing on the membrane inset. In this case, the endothelial cells are derived from clones sprouting out of capillary fragments, and can be passaged up to seven times. TEER is reported to increase significantly compared to monocultures, up to 800 Ωcm2. Some BBB characteristics also improved, as concluded from the stimulated expression of transferrin receptors, LDL receptors and P-glycoprotein compared with endothelial cells in monoculture without astrocytes.
A different approach toward an improved BMEC model has been introduced by Galla’s group.74 These investigators grow primary cultures from porcine BMEC under serum-free conditions, without astrocytes, but with hydrocortisone in the media. The tightness of this model appears remarkably high, with TEER of 600-1000 Ωcm2, and sucrose permeability as low as 3 × 10−7 cm s−1.
To overcome the disadvantages of primary culture systems with respect to labor intensity and batch-to-batch reproducibility, a number of immortalized cell lines have been established, mostly generated by transformation with viral proteins. A recent review listed 18 brain derived endothelial cell lines.69 Typically, these cell lines do not form tight monolayers and are unsuitable for transport experiments. However, rat (TR-BBB) and murine cell lines (-BBB), which carry a temperature-sensitive simian virus 40 large T antigen, show high potential as in vitro models of BBB transport, specifically when active influx or efflux carrier systems are involved. These cells grow at a permissive temperature of 33°C, and stop proliferating at 37°C. The analysis of nutrient transporters, neurotransmitter transporters, and ABC transporters revealed that many of the systems known to be active in vivo are also highly expressed by these cell lines.75 For example, the transport activity of GLUT1 glucose transporter in TR-BBB cells reached 20% of the in vivo value, whereas this carrier is downregulated in primary BMEC by 100-fold.76
Computational prediction of brain uptake
The in silico models evolved from experimental work on the correlation between brain uptake and two principal molecular properties: lipophilicity and molecular weight.77, 78 Modeling efforts concentrate on passive permeability at the BBB, because the quantitative structural information necessary for predicting active or facilitative transport processes is currently insufficient.
Classification models assign drugs to categories based on their pharmacological activity, such as CNS active and CNS inactive.79 Predictive models for assignment of a test substance to these classes have been built based on one- and two-dimensional molecular descriptors, or on substructure analysis.80 The data sets used in these models are large, ranging from hundreds to ten thousands of compounds. With respect to a correct prediction of BBB permeability, the pitfalls of the approach are well recognized.79, 81 In particular, CNS-inactive drugs may actually be able to penetrate the BBB but lack a drug target (receptor) inside brain tissue. Prediction accuracies for test sets of about 80% for CNS-active drugs are reported with various algorithms, whereas accuracies for correct prediction of CNS-inactive drugs are typically 10-20% lower.81
A different category of models, which represents the bulk of current computational Quantitative Structure Activity Relationship (QSAR) approaches, is based on a pharmacokinetic parameter: the brain/blood partitioning at steady state, expressed as log BB [= log (Cbrain/ Cblood)]. All the strategies using log BB share caveats, which jeopardize the validity of predictions.82–84 First, valid log BB values need to be determined at steady state. This prerequisite is probably often violated in the existing data sets. Taking a single pair of blood and tissue samples before the steady state of tissue distribution is reached, will distort the estimate of brain uptake. Second, whole brain tissue concentrations are measured, which may not have good correlations with free concentrations and occupancy of specific binding sites in brain. Third, log BB reflects processes other than BBB permeability, including plasma protein binding, tissue binding, local metabolism, and efflux across the BBB or clearance by interstitial fluid flow and CSF reabsorption.
Regarding details of the models, the number of descriptors necessary to build QSAR models of brain uptake appears limited. As argued by Lipinski, the chemical space is characterized by few descriptors (five to six) related to molecular size, lipophilicity, polarity, H-bonding status and charge with regard to Absorption Distribution Metabolism Excretion (ADME) properties.85
The 1-octanol/water partition coefficient, log Poctanol/water, has been in long standing experimental use as a descriptor of lipophilicity, and early predictive models were based on that parameter.86, 87 One reason for its popularity are computer algorithms providing calculated log P values, like ClogP86 and MlogP, 88 which often closely match (within about 0.3 log units) experimentally measured values.89 For ionizable molecules, the distribution coefficients, i.e., log P values at a defined pH (typically the physiological plasma pH of 7.4) are used. If log P and pKa is known, log D may be derived using the Henderson-Hasselbalch equation.
Solvation equations and linear free energy relationships were investigated extensively by Abraham’s group, culminating recently in a version of their regression model based on the descriptors molar refraction R2, polarizability π2H, hydrogen bond acidity α2H and basicity β2H, McGowan molecular volume Vχ, and an indicator variable for the presence or absence of a carboxyl group.90 The regression analysis was based on a data set of 157 drug and non-drug-like substances. After omitting nine outliers, multivariate linear regression resulted in the following statistics:
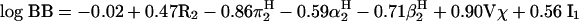 |
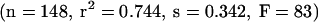 |
Hence, regression analysis indicated that higher polarity and hydrogen bonding decreased brain accumulation, whereas higher molecular volume was positively correlated. Alternative thermodynamic approaches (e.g., Keseru and Molnar91) with faster computational algorithms yielded similar statistical qualities.
In a quest to increase calculation speed by simplified modeling with a reduced number of descriptors, a group of QSAR models evolved, which are based on polar surface area (PSA) as the only, or as the dominant physico-chemical descriptor.92–95 PSA values can be calculated from the chemical structure, and these are highly correlated to the hydrogen bonding capacity of a molecule.92 By analyzing 125 drugs classified as CNS active or nonactive, the constraints of CNS active drugs were defined by van de Waterbeemd et al.2 as displaying a polar surface area less than 90 Å, and molecular weight less than 450. Another, less stringent descriptor in that study was molecular shape, expressed as the ratio of a molecule’s principal axes, which should be <5.92 Based on a different data set, Kelder et al.94 estimated a lower value (<60-70 Å2) as the upper limit for PSA of CNS-active drugs.
Despite a wide spectrum of molecular descriptors used, the recent computational approaches seem to achieve similar predictive quality for brain partitioning, when judged by correlation coefficients (r2 in the range of 0.7–0.9) and standard deviation (about 0.4 log units).81, 96 From classification and QSAR studies with log BB data described above, a few additional rules of thumb have been derived. Norinder and Haeberlein96 noted a linear relation between PSA and the sum of N + O atoms, and postulated that (N + O) ≤ 5 predicts good brain uptake. A second rule was derived from a model by Clark, 93 which uses both PSA and logP as descriptors: If log [P –(N + O)] > 0, then also log BB >0.
It can be argued that taking into consideration the experimental error of log BB values of about 0.3 log units, the performance of current log BB/QSAR models may have approached its limit.81 Furthermore, the question has been raised whether log BB predictions are relevant to the discovery of clinically useful drugs.82, 83 One way to address this concern is expanding the neglected, alternative approach, which builds predictive models from BBB permeability data, as demonstrated early by Levin’s work.77 He measured permeability coefficients using an intravenous method, and concluded from a series of 22 compounds that BBB transfer can be described by a regression between log (permeability) and the product of log Poctanol times square root of molecular weight. The regression held for compounds up to 400-600 Da. Substances falling several orders of magnitude above or below the line are substrates of carrier mediated uptake or active efflux, respectively.
Among the few QSAR models using BBB uptake rates, a study by Gratton et al.97 applied Abraham’s Linear Gibbs free-energy relationship (LFER) approach to 18 compounds covering 7 log orders in measured PS products. The regression equation obtained for log PS was qualitatively and quantitatively distinct from Platts’ results on log BB.90
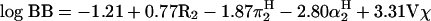 |
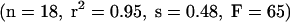 |
Interestingly, for the same data set a good correlation of log PS and log Poctanol/water was found, in contrast to poor correlations of log BB and Poctanol/water described earlier.98 These comparisons underscore the notion that models based on log BB describe properties other than BBB permeability alone. On the other hand, there was a positive correlation of log PS and molecular size, as seen for the log BB model as well.90 This finding is at odds with most other studies, which defined the molecular mass threshold of 400-600 Da mentioned above. The presence of such a threshold is supported by a physical model of phospholipid bilayers, which postulates the formation of temporary pores of corresponding size, because of kinks in the lipid tails caused by unsaturated fatty acids.99 Although the majority of substances in Gratton’s data set represented small organic nondrug molecules, Liu et al.100 recently included 23 drug-like compounds in a log PS model. Using stepwise multivariate regression analysis, these authors settled on three descriptors (log D, van der Waals area of basic atoms, topological van der Waals polar surface area) and achieved statistics (r2 = 0.74, with SD = 0.50) superior to log P- or log D-based models, and better than Gratton’s LFER model. This shows the persisting problem with cross-validation of models using different data sets. Clearly, many more experimental data are required as a basis to improve the validity of PS-based models.
REFERENCES
- 1.Kennedy T. Managing the drug discovery/development interface. Drug Discovery Today 2: 436–444, 1997. [Google Scholar]
- 2.van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov 2: 192–204, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Levin VA, Fenstermacher JD, Patlak CS. Sucrose and inulin space measurements of cerebral cortex in four mammalian species. Am J Physiol (Lond) 219: 1528–1533, 1970. [DOI] [PubMed] [Google Scholar]
- 4.Ohno K, Pettigrew KD, Rapoport SI. Lower limits of cerebrovascular permeability to nonelectrolytes in the conscious rat. Am J Physiol (Lond) 235: H299–H307, 1978. [DOI] [PubMed] [Google Scholar]
- 5.Crone C. The permeability of capillaries in various organs as determined by use of the ’indicator diffusion’ method. Acta Physiol Scand 58: 292–305, 1963. [DOI] [PubMed] [Google Scholar]
- 6.Renkin EM. Transport of potassium-42 from blood to tissue in isolated mammalian skeletal muscles. Am J Physiol (Lond) 197: 1205–1210, 1959. [DOI] [PubMed] [Google Scholar]
- 7.Fenstermacher JD, Blasberg RG, Patlak CS. Methods for quantifying the transport of drugs across brain barrier systems. Pharmacol Ther 14: 217–248, 1981. [DOI] [PubMed] [Google Scholar]
- 8.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 3: 1–7, 1983. [DOI] [PubMed] [Google Scholar]
- 9.Blasberg RG, Fenstermacher JD, Patlak CS. Transport of α-aminoisobutyric acid across brain capillary and cellular membranes. J Cereb Blood Flow Metab 3: 8–32, 1983. [DOI] [PubMed] [Google Scholar]
- 10.Smith QR. A review of blood-brain barrier transport techniques. Methods Mol Med 89: 193–208, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Collins JM, Dedrick RL. Distributed model for drug delivery to CSF and brain tissue. Am J Physiol (Lond) 245: R303–R310, 1983. [DOI] [PubMed] [Google Scholar]
- 12.Tetsuya T, Ohtsuki S. Brain-to-blood transporters for endogenous substrates and xenobiotics at the blood-brain barrier: an overview of biology and methodology. NeuroRx 2: 63–71, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bickel U, Thomsen T, Fischer JP, Weber W, Kewitz H. Galanthamine: pharmacokinetics, tissue distribution and cholinesterase inhibition in brain of mice. Neuropharmacology 30: 447–454, 1991. [DOI] [PubMed] [Google Scholar]
- 14.Oldendorf WH. Measurement of brain uptake of radiolabeled substances using a tritiated water internal standard. Brain Res 24: 372–376, 1970. [DOI] [PubMed] [Google Scholar]
- 15.Oldendorf WH. Clearance of radiolabeled substances by brain after arterial injection using a diffusible internal standard. In: Research methods in neurochemistry (Marks N, Rodnight R, eds.). New York: Plenum Publishing Co., 91–112, 1981.
- 16.Cornford EM. The carotid artery single injection technique. In: Introduction to the blood-brain barrier: methodology, biology and pathology (Pardridge WM, ed), pp 11–23. Cambridge, UK: Cambridge University Press, 1998.
- 17.Pardridge WM, Landaw EM, Miller LP, Braun LD, Oldendorf WH. Carotid artery injection technique: bounds for bolus mixing by plasma and by brain. J Cereb Blood Flow Metab 5: 576–583, 1985. [DOI] [PubMed] [Google Scholar]
- 18.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol (Lond) 247: H484–H493, 1984. [DOI] [PubMed] [Google Scholar]
- 19.Zlokovic BV, Begley DJ, Djuricic BM, Mitrovic DM. Measurement of solute transport across the blood-brain barrier in the perfused guinea pig brain: method and application to N-methyl-α-aminoisobutyric acid. J Neurochem 46: 1444–1451, 1986. [DOI] [PubMed] [Google Scholar]
- 20.Dagenais C, Rousselle C, Pollack GM, Scherrmann JM. Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab 20: 381–386, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Smith QR, Allen DD. In situ brain perfusion technique. Methods Mol Med 89: 209–218, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem 54: 1882–1888, 1990. [DOI] [PubMed] [Google Scholar]
- 23.Pardridge WM. Receptor-mediated transcytosis of peptides. In: Brain drug targeting—the future of brain drug development. Cambridge, UK: Cambridge University Press, 82–125, 2001.
- 24.Pardridge WM, Yoshikawa T, Kang YS, Miller LP. Blood-brain barrier transport and brain metabolism of adenosine and adenosine analogs. J Pharmacol Exp Ther 268: 14–18, 1994. [PubMed] [Google Scholar]
- 25.Samii A, Bickel U, Stroth U, Pardridge WM. Blood-brain barrier transport of neuropeptides: analysis with a metabolically stable dermorphin analogue. Am J Physiol (Lond) 267: E124–E131, 1994. [DOI] [PubMed] [Google Scholar]
- 26.Pardridge WM, Oldendorf WH. Kinetics of blood-brain transport of hexoses. Biochim Biophys Acta 382: 377–392, 1975. [DOI] [PubMed] [Google Scholar]
- 27.Smith QR, Takasato Y. Kinetics of amino acid transport at the blood-brain barrier studied using an in situ brain perfusion technique. Ann NY Acad Sci 481: 186–201, 1986. [DOI] [PubMed] [Google Scholar]
- 28.Pardridge WM. Blood-brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res 23: 635–644, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28: 897–916, 1977. [DOI] [PubMed] [Google Scholar]
- 30.Sakurada O, Kennedy C, Jehle J, Brown JD, Carbin GL, Sokoloff L. Measurement of local cerebral blood flow with iodo [14C] antipyrine. Am J Physiol (Lond) 234: H59–H66, 1978. [DOI] [PubMed] [Google Scholar]
- 31.Asotra K, Ningaraj N, Black KL. Measurement of blood-brain and blood-tumor barrier permeabilities with [14C]-labeled tracers. Methods Mol Med 89: 177–190, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Belayev L, Zhao W, Busto R, Ginsberg MD. Transient middle cerebral artery occlusion by intraluminal suture: I. Three-dimensional autoradiographic image-analysis of local cerebral glucose metabolism-blood flow interrelationships during ischemia and early recirculation. J Cereb Blood Flow Metab 17: 1266–1280, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Fenstermacher JD, Wei L. Measuring local cerebral capillary permeability-surface area products by quantitative autoradiography. In: Introduction to the blood-brain barrier: methodology, biology and pathology (Pardridge WM, ed), pp 122–132. Cambridge, UK: Cambridge University Press, 1998.
- 34.Huang SC, Hoh C, Barrio JR, Phelps ME. Measurement of blood-brain barrier permeability in humans with positron emission tomography. In: Introduction to the blood-brain barrier: methodology, biology and pathology (Pardridge WM, ed), pp 122–146. Cambridge, UK: Cambridge University Press, 1998.
- 35.Schmidt KC, Turkheimer FE. Kinetic modeling in positron emission tomography. Q J Nucl Med 46: 70–85, 2002. [PubMed] [Google Scholar]
- 36.Elsinga PH, Hendrikse NH, Bart J, Vaalburg W, van Waarde A. PET studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS. Curr Pharm Des 10: 1493–1503, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Tofts PS, Kermode AG. Measurement of the blood-brain barrier permeability and leakage space using dynamic MR imaging. 1. Fundamental concepts. Magn Reson Med 17: 357–367, 1991. [DOI] [PubMed] [Google Scholar]
- 38.Ewing JR, Knight RA, Nagaraja TN, Yee JS, Nagesh V, Whitton PA, et al. Patlak plots of Gd-DTPA MRI data yield blood-brain transfer constants concordant with those of 14C-sucrose in areas of blood-brain opening. Magn Reson Med 50: 283–292, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Artemov D. Molecular magnetic resonance imaging with targeted contrast agents. J Cell Biochem 90: 518–524, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Deguchi Y, Morimoto K. Application of an in vivo brain microdialysis technique to studies of drug transport across the blood-brain barrier. Curr Drug Metab 2: 411–423, 2001. [DOI] [PubMed] [Google Scholar]
- 41.de Lange EC, de Boer AG, Breimer DD. Methodological issues in microdialysis sampling for pharmacokinetic studies. Adv Drug Deliv Rev 45: 125–148, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Westergren I, Nystrom B, Hamberger A, Johansson BB. Intracerebral dialysis and the blood-brain barrier. J Neurochem 64: 229–234, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Bungay PM, Morrison PF, Dedrick RL. Steady-state theory for quantitative microdialysis of solutes and water in vivo and in vitro. Life Sci 46: 105–119, 1990. [DOI] [PubMed] [Google Scholar]
- 44.Elmquist WF, Sawchuk RJ. Application of microdialysis in pharmacokinetic studies. Pharm Res 14: 267–288, 1997. [DOI] [PubMed] [Google Scholar]
- 45.Hammarlund-Udenaes M. The use of microdialysis in CNS drug delivery studies. Pharmacokinetic perspectives and results with analgesics and antiepileptics. Adv Drug Deliv Rev 45: 283–294, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Hammarlund-Udenaes M, Paalzow LK, de Lange EC. Drug equilibration across the blood-brain barrier—pharmacokinetic considerations based on the microdialysis method. Pharm Res 14: 128–134, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Welty DF. The simultaneous estimation of the influx and efflux blood-brain barrier permeabilities of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res 13: 398–403, 1996. [DOI] [PubMed] [Google Scholar]
- 48.Aasmundstad TA, Morland J, Paulsen RE. Distribution of morphine 6-glucuronide and morphine across the blood-brain barrier in awake, freely moving rats investigated by in vivo microdialysis sampling. J Pharmacol Exp Ther 275: 435–441, 1995. [PubMed] [Google Scholar]
- 49.Wu D, Kang YS, Bickel U, Pardridge WM. Blood-brain barrier permeability to morphine-6-glucuronide is markedly reduced compared with morphine. Drug Metab Dispos 25: 768–771, 1997. [PubMed] [Google Scholar]
- 50.Bickel U, Schumacher OP, Kang YS, Voigt K. Poor permeability of morphine 3-glucuronide and morphine 6-glucuronide through the blood-brain barrier in the rat. J Pharmacol Exp Ther 278: 107–113, 1996. [PubMed] [Google Scholar]
- 51.Bouw MR, Xie R, Tunblad K, Hammarlund-Udenaes M. Blood-brain barrier transport and brain distribution of morphine-6-glucuronide in relation to the antinociceptive effect in rats—pharmacokinetic/pharmacodynamic modelling. Br J Pharmacol 134: 1796–1804, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stain-Texier F, Boschi G, Sandouk P, Scherrmann JM. Elevated concentrations of morphine 6-β-D-glucuronide in brain extracellular fluid despite low blood-brain barrier permeability. Br J Pharmacol 128: 917–924, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilgert M, Noldner M, Chatterjee SS, Klein J. KA-672 inhibits rat brain acetylcholinesterase in vitro but not in vivo. Neurosci Lett 263: 193.–196, 1999. [DOI] [PubMed] [Google Scholar]
- 54.Groothuis DR, Ward S, Schlageter KE, Itskovich AC, Schwerin SC, Allen CV, et al. Changes in blood-brain barrier permeability associated with insertion of brain cannulas and microdialysis probes. Brain Res 803: 218–230, 1998. [DOI] [PubMed] [Google Scholar]
- 55.Morgan ME, Singhal D, Anderson BD. Quantitative assessment of blood-brain barrier damage during microdialysis. J Pharmacol Exp Ther 277: 1167–1176, 1996. [PubMed] [Google Scholar]
- 56.de Lange EC, Danhof M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet 41: 691–703, 2002. [DOI] [PubMed] [Google Scholar]
- 57.Ooie T, Terasaki T, Suzuki H, Sugiyama Y. Quantitative brain microdialysis study on the mechanism of quinolones distribution in the central nervous system. Drug Metab Dispos 25: 784–789, 1997. [PubMed] [Google Scholar]
- 58.Ooie T, Terasaki T, Suzuki H, Sugiyama Y. Kinetic evidence for active efflux transport across the blood-brain barrier of quinolone antibiotics. J Pharmacol Exp Ther 283: 293–304, 1997. [PubMed] [Google Scholar]
- 59.Wu D, Clement JG, Pardridge WM. Low blood-brain barrier permeability to azidothymidine (AZT), 3TC, and thymidine in the rat. Brain Res 791: 313–316, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Joo F. The blood-brain barrier in vitro: the second decade. Neurochem Int 23: 499–521, 1993. [DOI] [PubMed] [Google Scholar]
- 61.Pardridge WM. Isolated brain capillaries: and in vitro model of blood-barin barrier research. In: Introduction to the blood-brain barrier: methodology, biology and pathology (Pardridge WM, ed), pp 49–61. Cambridge, UK: Cambridge University Press, 1998.
- 62.Lasbennes F, Gayet J Capacity for energy metabolism in microvessels isolated from rat brain. Neurochem Res 9: 1–10, 1984. [DOI] [PubMed] [Google Scholar]
- 63.Osburg B, Peiser C, Domling D, Schomburg L, Ko YT, Voigt K, et al. Effect of endotoxin on expression of TNF receptors and transport of TNF-α at the blood-brain barrier of the rat. Am J Physiol (Lond) Endocrinol Metab 283: E899–E908, 2002. [DOI] [PubMed] [Google Scholar]
- 64.Huwyler J, Pardridge WM. Examination of blood-brain barrier transferrin receptor by confocal fluorescent microscopy of unfixed isolated rat brain capillaries. J Neurochem 70: 883–886, 1998. [DOI] [PubMed] [Google Scholar]
- 65.Miller DS, Nobmann SN, Gutmann H, Toeroek M, Drewe J, Fricker G. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol Pharmacol 58: 1357–1367, 2000. [DOI] [PubMed] [Google Scholar]
- 66.Sanchez del Pino MM, Hawkins RA, Peterson DR. Neutral amino acid transport by the blood-brain barrier. Membrane vesicle studies. J Biol Chem 267: 25951–25957, 1992. [PubMed] [Google Scholar]
- 67.Peterson DR, Hawkins RA. Transport studies using membrane vesicles. Methods Mol Med 89: 233–247, 2003. [DOI] [PubMed] [Google Scholar]
- 68.Gumbleton M, Audus KL. Progress and limitations in the use of in vitro cell cultures to serve as a permeability screen for the blood-brain barrier. J Pharm Sci 90: 1681–1698, 2001. [DOI] [PubMed] [Google Scholar]
- 69.Reichel A, Begley DJ, Abbott NJ. An overview of in vitro techniques for blood-brain barrier studies. Methods Mol Med 89: 307–324, 2003. [DOI] [PubMed] [Google Scholar]
- 70.Audus KL, Rose JM, Wang W, Borchardt RT. Brain microvessel endothelia cell culture systems. In: Introduction to the blood-brain barrier: methodology, biology and pathology (Pardridge WM, ed), pp 86–93. Cambridge, UK: Cambridge University Press, 1998.
- 71.Patlak CS, Paulson OB. The role of unstirred layers for water exchange across the blood-brain barrier. Microvasc Res 21: 117–127, 1981. [DOI] [PubMed] [Google Scholar]
- 72.Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol (Lond) 429: 47–62, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cecchelli R, Dehouck B, Descamps L, Fenart L, Buee-Scherrer VV, Duhem C, et al. In vitro model for evaluating drug transport across the blood-brain barrier. Adv Drug Deliv Rev 36: 165–178, 1999. [DOI] [PubMed] [Google Scholar]
- 74.Hoheisel D, Nitz T, Franke H, Wegener J, Hakvoort A, Tilling T, Galla HJ. Hydrocortisone reinforces the blood-brain properties in a serum free cell culture system. Biochem Biophys Res Commun 247: 312–315, 1998. [PubMed] [Google Scholar]
- 75.Terasaki T, Ohtsuki S, Hori S, Takanaga H, Nakashima E, Hosoya K. New approaches to in vitro models of blood-brain barrier drug transport. Drug Discov Today 8: 944–954, 2003. [DOI] [PubMed] [Google Scholar]
- 76.Pardridge WM, Triguero D, Yang J, Cancilla PA. Comparison of in vitro and in vivo models of drug transcytosis through the blood-brain barrier. J Pharmacol Exp Ther 253: 884–891, 1990. [PubMed] [Google Scholar]
- 77.Levin VA. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem 23: 682–684, 1980. [DOI] [PubMed] [Google Scholar]
- 78.Oldendorf WH. Lipid solubility and drug penetration of the blood brain barrier. Proc Soc Exp Biol Med 147: 813–815, 1974. [DOI] [PubMed] [Google Scholar]
- 79.Ajay, Bemis GW, Murcko MA. Designing libraries with CNS activity. J Med Chem 42: 4942–4951, 1999. [DOI] [PubMed] [Google Scholar]
- 80.Engkvist O, Wrede P, Rester U. Prediction of CNS activity of compound libraries using substructure analysis. J Chem Inf Comput Sci 43: 155–160, 2003. [DOI] [PubMed] [Google Scholar]
- 81.Clark DE. In silico prediction of blood-brain barrier permeation. Drug Discov Today 8: 927–933, 2003. [DOI] [PubMed] [Google Scholar]
- 82.Martin I. Prediction of blood-brain barrier penetration: are we missing the point? Drug Discov Today 9: 161–162, 2004. [DOI] [PubMed] [Google Scholar]
- 83.Pardridge WM. Log(BB), PS products and in silico models of drug brain penetration. Drug Discov Today 9: 392–393, 2004. [DOI] [PubMed] [Google Scholar]
- 84.Habgood MD, Begley DJ, Abbott NJ. Determinants of passive drug entry into the central nervous system. Cell Mol Neurobiol 20: 231–253, 2000. [DOI] [PubMed] [Google Scholar]
- 85.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44: 235–249, 2000. [DOI] [PubMed] [Google Scholar]
- 86.Hansch C, Bjorkroth JP, Leo A. Hydrophobicity and central nervous system agents: on the principle of minimal hydrophobicity in drug design. J Pharm Sci 76: 663–687, 1987. [DOI] [PubMed] [Google Scholar]
- 87.Buchwald P, Bodor N. Octanol-water partition: searching for predictive models. Curr Med Chem 5: 353–380, 1998. [PubMed] [Google Scholar]
- 88.Moriguchi I, Hirono S, Liu Q, Nakagome Y, Matsushita Y. Simple method calculating octanol/water partition coefficient. Chem Pharm Bull 40: 127–130, 1992. [Google Scholar]
- 89.Hansch C, Leo A, Mekapati SB, Kurup A. QSAR and ADME. Bioorg Med Chem 12: 3391–3400, 2004. [DOI] [PubMed] [Google Scholar]
- 90.Platts JA, Abraham MH, Zhao YH, Hersey A, Ijaz L, Butina D. Correlation and prediction of a large blood-brain distribution data set—an LFER study. Eur J Med Chem 36: 719–730, 2001. [DOI] [PubMed] [Google Scholar]
- 91.Keseru GM, Molnar L. High-throughput prediction of blood-brain partitioning: a thermodynamic approach. J Chem Inf Comput Sci 41: 120–128, 2001. [DOI] [PubMed] [Google Scholar]
- 92.van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J Drug Target 6: 151–165, 1998. [DOI] [PubMed] [Google Scholar]
- 93.Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J Pharm Sci 88: 815–821, 1999. [DOI] [PubMed] [Google Scholar]
- 94.Kelder J, Grootenhuis PD, Bayada DM, Delbressine LP, Ploemen JP. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res 16: 1514–1519, 1999. [DOI] [PubMed] [Google Scholar]
- 95.Lobell M, Molnar L, Keseru GM. Recent advances in the prediction of blood-brain partitioning from molecular structure. J Pharm Sci 92: 360–370, 2003. [DOI] [PubMed] [Google Scholar]
- 96.Norinder U, Haeberlein M. Computational approaches to the prediction of the blood-brain distribution. Adv Drug Deliv Rev 54: 291–313, 2002. [DOI] [PubMed] [Google Scholar]
- 97.Gratton JA, Abraham MH, Bradbury MW, Chadha HS. Molecular factors influencing drug transfer across the blood-brain barrier. J Pharm Pharmacol 49: 1211–1216, 1997. [DOI] [PubMed] [Google Scholar]
- 98.Abraham MH, Chadha HS, Mitchell RC. Hydrogen bonding. 33. Factors that influence the distribution of solutes between blood and brain. J Pharm Sci 83: 1257–1268, 1994. [DOI] [PubMed] [Google Scholar]
- 99.Trauble H. The movement of molecules across lipid membranes: a molecular theory. J Membr Biol 4: 193–208, 1971. [DOI] [PubMed] [Google Scholar]
- 100.Liu X, Tu M, Kelly RS, Chen C, Smith BJ. Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos 32: 132–139, 2004. [DOI] [PubMed] [Google Scholar]