

Abstract
The glucocorticoid receptor interacting protein (GRIP1) belongs to the p160 steroid receptor coactivator family that plays essential roles in nuclear receptor-dependent transcriptional regulation. Previously, we reported that the cAMP-dependent protein kinase (PKA) induces ubiquitination leading to degradation of GRIP1. Here we show that the cAMP response element-binding protein (CREB) downregulates GRIP1 and is necessary for the PKA-stimulated degradation of GRIP1, which leads to changes in the expression of a subset of genes regulated by estrogen receptor-α in MCF-7 breast cancer cells. Our data of domain-mapping and ubiquitination analyses suggest that CREB promotes the proteasomal breakdown of ubiquitinated GRIP1 through 2 functionally independent protein domains containing amino acids 347 to 758 and 1121 to 1462. We provide evidence that CREB interacts directly with GRIP1 and that CREB Ser-133 phosphorylation or transcriptional activity is not required for GRIP1 interaction and degradation. The basic leucine zipper domain (bZIP) of CREB is important for the interaction with GRIP1, and deletion of this domain led to an inability to downregulate GRIP1. We propose that CREB mediates the PKA-stimulated degradation of GRIP1 through protein-protein interaction and stimulation of proteasomal degradation of ubiquitinated GRIP1.
Nuclear receptors (NRs) comprise a superfamily of transcription factors that are essential for many physiological functions, and alterations in their transcriptional regulation are associated with a variety of disorders. NRs regulate gene transcription through coordinated recruitment of coactivator and corepressor complexes to the promoters of responsive genes (1). The glucocorticoid receptor-interacting protein 1 [GRIP1, also known as steroid receptor coactivator (SRC)-2] is a member of the SRC family (2, 3). As for the other 2 SRCs (SRC-1 and SRC-3), GRIP1 binds directly to NRs, including estrogen receptor α (ERα) (4), and potentiates NR-mediated gene transcription by serving as a core adaptor for recruitment of histone-modifying proteins and chromatin-remodeling complexes as well as mediating the assembly of the transcription initiation machinery on NR target genes (5). The SRCs have a common functional structure, which includes a conserved N-terminal basic helix-loop-helix Per-Arnt-Sim (bHLH-PAS) domain, a centrally located NR-interaction domain (NID) composed of 3 LXXLL motifs (NR-boxes) and C-terminal intrinsic activation domains (AD1 and AD2) (6)
Genetic ablation studies in mice have revealed important biological functions of GRIP1 in reproduction, energy homeostasis, and bone formation. Mice lacking GRIP1 exhibit reduced fertility due to placental hypoplasia in females and abnormal spermatogenesis in males (7, 8). Moreover, GRIP1−/− mice are resistant to develop high-fat–induced obesity (9). Ablation of GRIP1 in mice leads to changes in gene expression of several key regulatory enzymes in energy metabolism (10, 11), and loss of GRIP1 in bone results in a disruption of the transcription complex at target genes of the peroxisome proliferator activated receptor γ (PPARγ) and a subsequent increased bone mass in mice (12). SRC-1 and SRC-2 have been suggested as mediators of endocrine resistance in breast cancer (13, 14), and their expression levels are increased in normal and malignant breast tissue after tamoxifen treatment (15). Although it has been shown that GRIP1 is able to stimulate breast cancer cell proliferation (16–18), its exact role in breast carcinogenesis is not clear.
Despite the apparent essential biological functions of GRIP1, knowledge about the molecular mechanisms regulating GRIP1 protein level and availability is limited. It has been demonstrated that GRIP1-mediated coactivation of ERα- and androgen receptor (AR) target genes is stimulated by the ERK and p38 of the MAPK family due to GRIP1 phosphorylation at Ser736 (19–21). Recently, the energy-depletion-sensing kinase AMPK has been reported to phosphorylate and activate GRIP1 (22), whereas another study demonstrated that glucocorticoid induces phosphorylation of GRIP1 in a receptor-bound–dependent manner through casein kinase-2 and cyclin-dependent kinase-9 (23). GRIP1 colocalization with and coactivation of AR is also modulated by GRIP1 sumoylation at the NID (24). We have previously reported that the cAMP-dependent protein kinase (PKA) downregulates GRIP1 coactivator function by stimulating GRIP1 turnover through ubiquitination and proteasomal degradation (25, 26) and that PKA regulates the recruitment of GRIP1 to an ERα-responsive target gene (27). Activation of PKA has been shown to inhibit breast cancer cell proliferation (28, 29), but the knowledge of the underlying mechanisms is limited.
In this study, we sought to elucidate the molecular components involved in the PKA-mediated regulation of GRIP1. The role of cAMP-response element-binding protein (CREB), which is a transcription factor of the basic leucine zipper (bZIP) family and a key mediator of the cAMP/PKA signaling pathway (30), was examined. CREB is structurally and functionally characterized with a conserved N-terminal glutamine-rich domain (Q1), a kinase-inducible domain (KID), another glutamine-rich domain (Q2), and a C-terminal bZIP domain (31). Activation of PKA phosphorylates CREB at the serine 133 (Ser-133) residue, which promotes CREB interaction with the CREB-binding protein (CBP), leading to enhanced transcriptional activity of CREB (30, 32). The CREB-regulated transcription coactivators (CRTCs) also modulate CREB-dependent gene expression (33).
Here we demonstrate that CREB downregulates GRIP1 coactivator function by reducing GRIP1 protein level, mediated through a proteasomal degradation pathway. CREB is required for cAMP/PKA-stimulated degradation of GRIP1, and its inhibitory effect on GRIP1 leads to changes in expression of several ERα target genes. Through mutational analysis, we mapped the PKA/CREB-targeted GRIP1 domains to the protein regions containing amino acids (aa) 347 to 758 and 1121 to 1462. These regions of GRIP1 are highly subjected to ubiquitination, and proteasomal degradation of their ubiquitinated products is stimulated by CREB, independently of each other. Interestingly, CREB interacts directly with GRIP1, and the interaction does not require the Ser-133 phosphorylation or transcriptional activity of CREB. We show that the CREB bZIP domain is essential for GRIP1 interaction, and deletion of this domain blocks CREB from degrading GRIP1. Our data suggest that CREB promotes the proteasomal breakdown of ubiquitinated GRIP1 independently of CREB Ser-133 phosphorylation, and we hypothesize that CREB mediates GRIP1 degradation through protein-protein interaction at the bZIP domain.
Materials and Methods
Plasmid constructs
GRIP1 wild type (wt) and mutants with N-terminal hemagglutinin (HA) epitope tags and GAL4-GRIP1 were expressed from vector pSG5 (34) and pM-GRIP1 (35), respectively. The following plasmids were used for expression of CREB in cells: RSV-CREB (R. H. Goodman, Portland, Oregon), pCMV5-CREB (wt, S133A, and KCREB; BD Biosciences, San Jose, California), GAL4-CREB (36), VP16-CREB, and pFlag-CMV2-CREB (wt, ΔQ1, ΔQ2, ΔKID, ΔbZIP, and bZIP). In vitro expression of CREB was achieved using the T7 promoter-carrying expression vector pcDNA3.1 (Invitrogen, Carlsbad, California). Details on construction of the plasmids are provided in the Supplemental Materials and Methods (published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). The expression plasmids encoding human ERα (pSG5-hERα), human GR (pSG5-hGR), the catalytic subunit of PKA (pCMV5-Cα), and the luciferase reporter constructs pG5-luc and ERE-TATA-luc were described previously (26, 27). The empty vectors pM encoding GAL4-DBD (GAL4), pSG5, and the expression plasmid encoding p53 protein fused with GAL4-DBD (GAL4-p53) were purchased from Promega (Madison, Wisconsin), Stratagene (La Jolla, California), and Clontech (Mountain View, California), respectively. pRc/RSV-CBP-HA encoding mouse CBP was provided by R. H. Goodman (Vollum Institute, Portland, Oregon). Expression of recombinant GRIP1 protein was achieved by employing pFastBac-HTa-GRIP1, which was created by cloning GRIP1 cDNA into the pFastBac-HTa vector (Invitrogen) at an EcoRI site. pcDNA-His-Ubiquitin encoding wt ubiquitin tagged to a histidine sequence at the N terminus [His-tagged ubiquitin (His-Ub)] was kindly supplied by Dr E. Treuter (Karolinska Institute, Stockholm, Sweden).
Cell culture, transfections, and introduction of intracellular antibodies
COS-1 and MCF-7 cells were cultured and transiently transfected with plasmid DNAs as described previously (27). For experiments using 17β-estradiol and for MCF-7 cells, phenol red-free DMEM (Dulbecco's modified Eagle's medium) supplemented with 5% charcoal-stripped fetal bovine serum (FBS) was used for 1 (COS-1) to 3 (MCF-7) days before the experiment. Transient transfections of cells with small interference RNA (siRNA) duplexes were performed 3 hours before cotransfection with plasmid DNAs, both siRNAs targeting against CREB mRNA (CREB siRNA) and nonspecific negative control (NSC siRNA) (Upstate, Lake Placid, New York) were used. Introduction of antibodies into the cells was achieved through protein transfection using Chariot (ActiveMotif, La Hulpe, Belgium) 3 hours after transfection with expression plasmids. Rabbit anti-CREB antibody and normal rabbit serum IgG (normal IgG; Upstate) were used. Luciferase assays were performed using the luciferase assay kit (BIOThema AB, Haninge, Sweden) or the dual-luciferase reporter assay system (Promega), as described previously (26).
Generation of stable GRIP1 knockdown MCF-7 cells
Knockdown of GRIP1 in MCF-7 cells was obtained by transduction of the cells with lentivirus carrying GRIP1 short hairpin RNA (shRNA) inserted into a pLKO.1 puro vector (Mission; Sigma). A control MCF-7 cell line was also generated using lentivirus carrying a corresponding empty vector (control shRNA). Transduced cells with stable expression of shRNA (control or GRIP1 shRNA) were selected by treatment with puromycin (1 μg/ml) for 3 weeks. Lysates of the puromycin-selected cells were analyzed by Western blotting for the expression and knockdown of GRIP1.
RNA isolation, microarray analyses, and real-time quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini kit (QIAGEN, Valencia, California). The shRNA-expressing MCF-7 cells were grown for 3 days in phenol red-free DMEM with 5% charcoal-stripped FBS and 10nM 17β-estradiol. Cells were subsequently left untreated or were treated with 150μM 8-4-chlorophenylthio-cAMP, 50μM 3-isobutyl-1-methylxanthine, and 10μM forskolin (cAMP) for 24 hours. Sample preparations were balanced and randomized in each step of lysate collection, RNA extraction, labeling, and microarray hybridization. Microarray was performed using the Illumina HumanRef-8 version 3.0 Expression BeadChips. Data analyses were achieved using J-Express Pro version 2. Significant changes in gene expression were detected through the significance analysis of microarray and defined by a fold change of at least 1.5, false discovery rate = 0, and q-value = 0.
The real-time quantitative RT-PCR (qRT-PCR) analyses were carried out using the LightCycler RNA Master SYBR Green I kit in a LightCycler rapid thermal cycler system (Roche, Basel, Switzerland). mRNA expression levels of target genes were quantified relative to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or TATA box binding protein. The primer sequences are provided in the Supplemental Materials and Methods.
Western blotting and coimmunoprecipitation
Western blotting was performed as described previously (26). The primary antibodies used for detection of GRIP1 were rat monoclonal anti-HA-peroxidase (Roche, Mannheim, Germany) and mouse monoclonal anti-TIF2 (transcriptional intermediary factor 2) (BD Biosciences). Other antibodies included rabbit polyclonal anti-ERα and anti-CBP (Santa Cruz Biotechnology, Santa Cruz, California), mouse monoclonal anti-GAPDH (Chemicon International, Temecula, California), and rabbit polyclonal anti-CREB (Upstate). Coimmunoprecipitation was performed using anti-CREB (Upstate) or anti-nuclear receptor coactivator 2 (GRIP1) (Bethyl, Montgomery, Texas) antibodies as described previously (27). Immunoprecipitated proteins were resolved through SDS-PAGE and Western blotting using anti-HA-peroxidase (Zymed Laboratories Inc, San Francisco, California), anti-TIF2 (BD Biosciences), and anti-CREB (Upstate) antibodies. Densitometry was performed using ImageJ software. Relative protein levels were calculated by dividing the band density at a particular experimental condition to the control.
In vitro protein-protein interaction
Full-length and mutant CREB proteins were expressed in vitro using the TNT reticulocyte lysate system (Promega) in the presence of [35S]methionine (Amersham Biosciences, Uppsala, Sweden). The recombinant 6xHis-GRIP1 protein was expressed and purified from the insect sf21 cell line (Supplemental Materials and Methods). An equivalent amount (6 μg) of recombinant 6xHis-GRIP1, 6xHis-β actin (Abcam, Cambridge, United Kingdom) or 6xHis-peptide (Abbiotec, San Diego, California) was incubated with the [35S]methionine-labeled CREB protein in an interaction buffer (Supplemental Materials and Methods) for 1.5 hours at room temperature. The interaction mixture was subsequently incubated and pulled down using Ni-Sepharose beads (Amersham Biosciences), followed by extensive washings with the interaction buffer. Bound proteins were eluted in 2× sodium dodecyl sulfate (SDS) sample buffer and resolved by SDS-PAGE followed by autoradiography or Western blotting using anti-TIF2 (BD Biosciences).
His-tag pulldown ubiquitination assays
Transfected COS-1 cells were lysed in 1× SDS sample buffer (for input) or in a 6M guanidine-HCl–containing lysis buffer for pulldown. The cell lysates were cleared by centrifugation at 13 000g for 5 minutes and subsequently incubated with Ni-NTA agarose beads (QIAGEN) for 4 hours. After the incubation, the beads were washed sequentially in 4 buffers varied in guanidine, Triton X-100 concentration, and/or pH (Supplemental Materials ad Methods). Proteins bound to Ni-nitrilotriacetic acid beads were eluted in an elution buffer containing 250mM imidazole, 150mM Tris-HCl (pH 6.7), 30% glycerol, 20mM β-mercaptoethanol, and 5% SDS (pH 6.7) through shaking and resolved by SDS-PAGE and Western blotting.
Statistics
Student's t test was used to evaluate the significance of differential luciferase activity or mRNA expression assessed by qRT-PCR. The significance levels were defined as P ≤ .05 and P ≤ .01.
Results
CREB downregulates protein level of GRIP1 and its coactivator function
Previously, we demonstrated that PKA enhances GRIP1 ubiquitination and targets it to degradation (26). This prompted us to investigate the possibility that this may be mediated by CREB. First we used a GAL4-based luciferase reporter gene assay. We observed that overexpression of CREB resulted in a dose-dependent decrease in GAL4-GRIP1–mediated transcription (Figure 1A), and a similar effect on the GRIP1 protein level was seen (Figure 1B). In contrast, overexpression of GAL4-CREB together with an increasing amount of GRIP1 did not reduce the reporter activity, and there was no dose-dependent downregulation of the transcriptional activity of GAL4-p53 by CREB (Supplemental Figure 1, A and B), suggesting there was no general inhibiting effect on transcriptional activity by overexpressing CREB. GAL4-GRIP1 activates transcription of the GAL4-responsive reporter gene by binding to the gene via the GAL4 DNA-binding domain and by recruitment of other coactivators including primarily CBP/p300 and secondarily coactivator-associated arginine methyltransferase 1 (CARM1) through the GRIP1 activation domains (34, 37–39). As expected, coexpression of CBP enhanced GAL4-GRIP1–dependent transcriptional activity (Supplemental Figure 1C). However, overexpression of CREB did not affect this enhancing effect of CBP on GRIP1. Importantly, the protein level of CBP was not affected by CREB overexpression (Figure 1B). The results suggest that the inhibition of GRIP1 activity by CREB was mediated through downregulation of GRIP1. This was confirmed in ERα-responsive reporter gene (ERE-TATA-luc) assays, where we observed that GRIP1-mediated coactivation of ERα was significantly decreased by the coexpression of CREB, both in the absence and presence of 17β-estradiol (Figure 1C). Of note, ERα protein level was not reduced upon CREB overexpression in COS-1 cells (data not shown), and overexpression of ERα and glucocorticoid receptor (GR) did not lead to reduced levels of GRIP1; neither did ERα and GR affect the CREB-induced reduction of GRIP1 (Supplemental Figure 2). It appeared that ERα even stabilized the GRIP1 protein level. Taken together, these data suggest that CREB decreases GRIP1 coactivator function and protein level.
Figure 1.
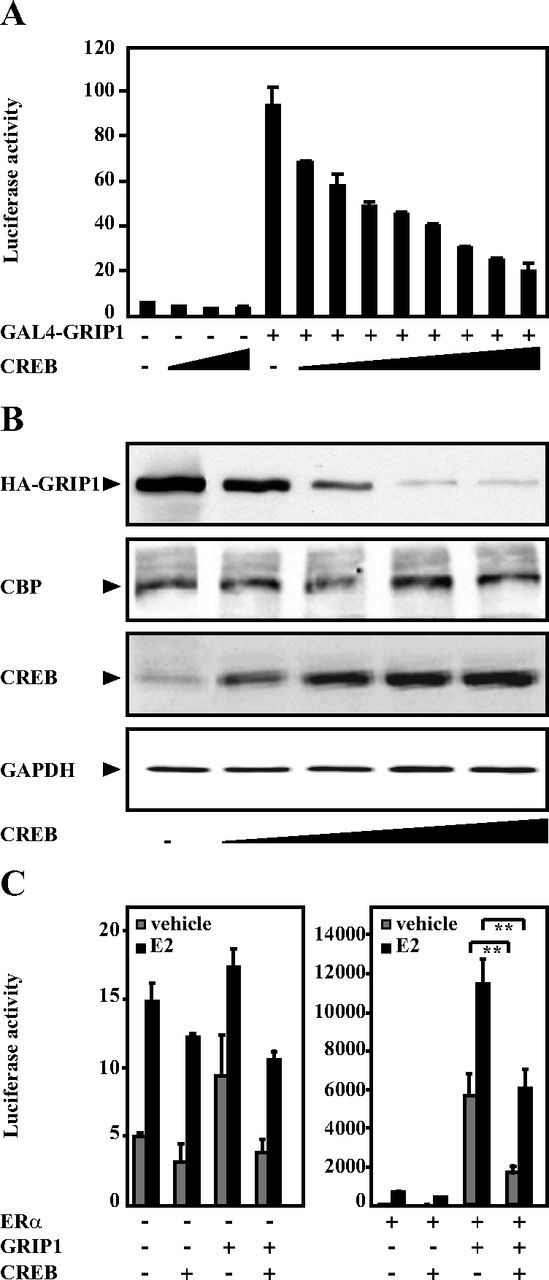
CREB inhibits GRIP1-stimulated activation of the GAL4-reporter gene and reduces GRIP1 protein level. A, COS-1 cells were transiently transfected with a GAL4-responsive luciferase reporter construct (GAL4-luc) (1.1 μg) together with expression plasmids encoding GAL4-GRIP1 (1.0 μg) and CREB (0.01, 0.02, 0.05, 0.1, 0.25, 0.5, 1.0, and 2.0 μg) for 48 hours. Luciferase activities were measured as described in Materials and Methods. The figures show the mean ± SD of triplicate transfections from at least 3 independent experiments. B, COS-1 cells were transfected with expression plasmids encoding HA-GRIP1 (2.0 μg) and CREB (0.5, 1.0, 1.5, and 2.0 μg). Forty-eight hours after transfection, cell were lysed and analyzed by Western blotting using anti-HA (GRIP1), anti-CBP, anti-CREB, and anti-GAPDH antibodies. The results presented are representative of at least 3 independent experiments. C, COS-1 cells seeded in phenol red-free DMEM containing charcoal-stripped FBS were transfected with expression plasmids encoding human ERα (0.1 μg), HA-GRIP1 (1.0 μg), and CREB (0.1 μg) together with the ERE-TATA-luc reporter construct (1.1 μg). The cells were treated with vehicle or 17β-estradiol (E2, 0.1μM) for 24 hours. Luciferase activities were measured 48 hours after transfection. The figure shows the mean ± SD of triplicate transfections (**, P < .01, Student's t test) and is representative of 3 independent experiments.
CREB reduces GRIP1 protein level through proteasomal protein degradation
GRIP1 is regulated posttranslationally through coupled ubiquitination and proteasomal protein degradation (26, 40, 41). As shown in Figure 2A, CREB induced a decrease in GRIP1-mediated coactivation of ERα-dependent transcriptional activity [gray bars, dimethylsulfoxide (DMSO)], and the effect was diminished when cells were treated with the proteasome inhibitor MG132 (black bars). Of note, treatment with MG132 resulted in lower luciferase activities, which have also been observed previously and are believed to be due to a general deleterious effect of MG132 on the luciferase enzyme activity (26, 42, 43). Furthermore, Western blot analyses showed that MG132 inhibited CREB-induced reduction of both endogenous (Figure 2B) as well as overexpressed (Figure 2C) GRIP1 protein in COS-1 cells, despite a stabilization of CREB upon treatment with MG132 (Figure 2, B and C). Similar to the results observed in Figure 1, overexpression of CREB did not decrease the protein level of CBP and ERα, despite that treatment with MG132 also resulted in elevated levels of CBP, ERα, and CREB (Figure 2, A and B). Meanwhile, PKA and CREB did not modulate GRIP1 mRNA level in cells (data not shown). Overall, our data suggest that CREB reduces GRIP1 protein level through the proteasomal protein degradation pathway.
Figure 2.
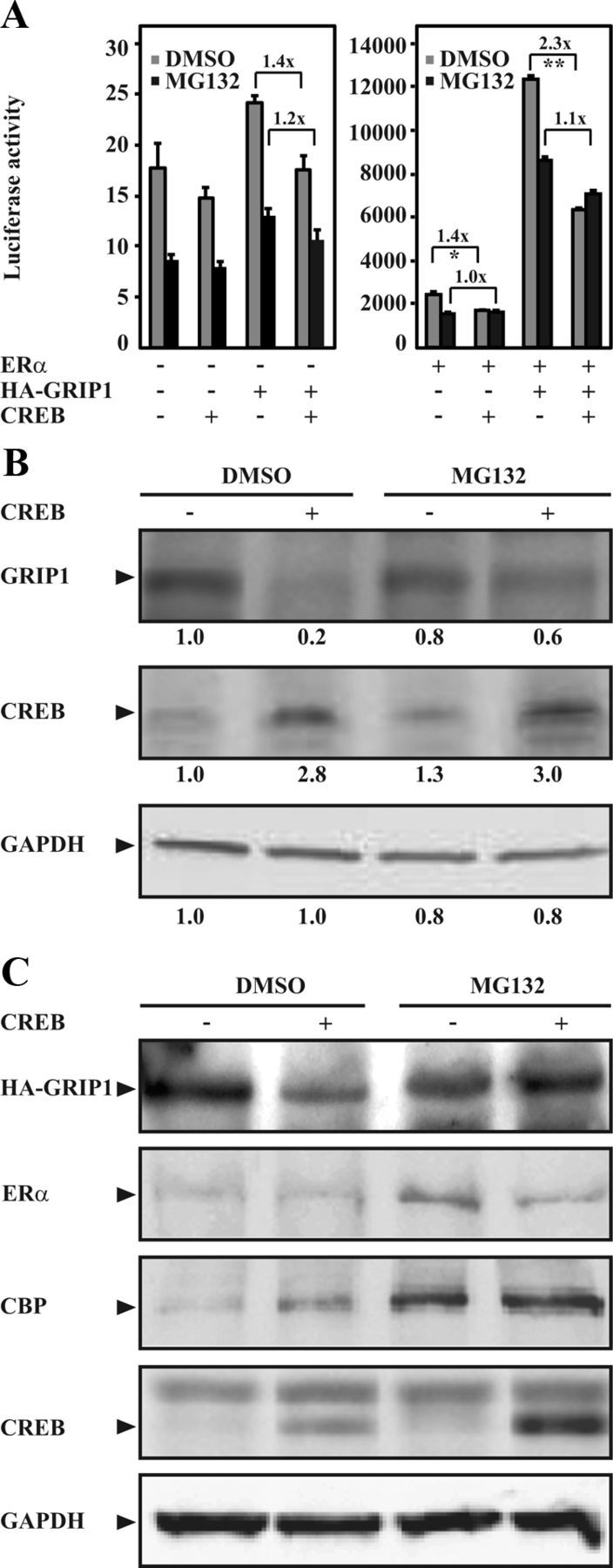
CREB reduces GRIP1 protein level through proteasomal protein degradation. A, COS-1 cells seeded in phenol red-free DMEM supplemented with charcoal-stripped FBS (5%) were transfected with expression plasmids encoding human ERα (0.1 μg), HA-GRIP1 (1.0 μg), and CREB (0.1 μg) together with the ERE-TATA-luc reporter construct (1.1 μg). Twenty-four hours after transfection, cells were treated with 17β-estradiol (E2, 0.1μM). Transfected cells were also treated with vehicle (DMSO) or MG132 (1μM) for 16 hours as indicated in the figure. Luciferase assays were performed 48 hours after transfections. The results are presented as mean ± SD of triplicate transfections from 3 independent experiments (**P < .01; *P < .05, Student's t test). Fold reduction is also presented. B and C, COS-1 cells were transfected with expression plasmid encoding CREB (0.1 μg) alone (B) or together with expression plasmids encoding human ERα (0.1 μg) and HA-GRIP1 (1.0 μg, C) and treated with DMSO or MG132 as in A. Cell lysates were analyzed by Western blotting using anti-TIF2 or anti-HA antibodies for detection of endogenous GRIP1 (B) or overexpressed GRIP1 (HA-GRIP1, C), respectively. The levels of CREB, ERα, CBP, and GAPDH were also examined. Relative protein levels of GRIP1, CREB, and GAPDH (B) were assessed as described in Materials and Methods and indicated below each respective panel.
CREB is required for the PKA-mediated degradation of GRIP1
It has been established that CREB is a central mediator of the cAMP/PKA signaling pathway in many biological processes (31). We therefore investigated whether PKA-induced degradation of GRIP1 was dependent on CREB. The endogenous level of CREB in COS-1 cells was reduced by siRNA with simultaneous transfection of a GAL4-luc reporter construct and a GAL4-GRIP1 expression plasmid to study GRIP1 transcriptional activity or with the expression plasmid encoding HA-GRIP1 to examine protein level. As shown in Figure 3, A–C, elevation of the cAMP level resulted in decreased GRIP1 activity and protein level, as reported previously (26). Importantly, knockdown of CREB prevented reduction of GRIP1 transcriptional activity and protein level after activation of the PKA signaling pathway (Figure 3, A–C). We also employed an alternative method whereby anti-CREB antibodies were introduced to block endogenous CREB protein from its normal functions (Figure 3D). In line with the other results, interference of CREB function by anti-CREB antibody terminated the cAMP-induced inhibition of GRIP1 coactivator function, whereas introduction of normal IgG had no such effect (Figure 3D). Taken together, these results suggest that CREB is required for the cAMP/PKA-stimulated downregulation of GRIP1.
Figure 3.

CREB is required for cAMP/PKA-mediated degradation of GRIP1. A, COS-1 cells were grown in 24-well plates and transfected with the GAL4-GRIP1 expression plasmid (0.7 μg) and the GAL4-luc reporter construct (0.6 μg) as well as siRNA specific (CREB-siRNA, 50 pmol) or nonspecific (NSC-siRNA, 50 pmol) to CREB. Cells were treated with forskolin (10μM), 3-isobutyl-1-methylxanthine (50μM), and 8-4-chlorophenylthio-cAMP (500μM) (cAMP, as indicated) for 24 hours before lysis. Luciferase assays were performed as described in Materials and Methods. The figure shows the mean ± SD of triplicate transfections from 3 independent experiments (**P < .01, Student's t test). B, COS-1 cells were grown in 12-well plates and transfected with the HA-GRIP1 expression plasmid (1.0 μg) and the siRNAs as described above (150 pmol each). Cells were treated with forskolin, IBMX, and 8-CPT-cAMP as described in A. Cell lysates were subjected to Western blotting using anti-HA, anti-CREB, and anti-GAPDH antibodies. C, Protein expression levels of GRIP1 and CREB in COS-1 cells transfected as described in B are presented as relative to GAPDH. Calculations were performed based on the band density using ImageJ software. The results are the mean ± SD of 3 independent experiments (*P < .05, Student's t test). D, COS-1 cells were grown in 24-well plates and transfected with the GAL4-GRIP1 expression plasmid (0.7 μg) and the GAL4-luc reporter (0.6 μg) as well as anti-CREB antibodies (α-CREB) or normal IgG (0.4–1.0 μg) as indicated. Treatment of the cells with forskolin, IBMX, and 8-CPT-cAMP was carried out as in A. Luciferase assays were performed 48 hours after transfection. The figure shows the mean ± SE of triplicate transfections (**P < .01, Student's t test).
CREB-mediated downregulation of GRIP1 modulates the expression of ERα target genes
We then wanted to study in more detail the effects of overexpressed CREB on endogenous levels of GRIP1 and ERα target genes in MCF-7 cells. Similar to our findings in COS-1 cells, overexpression of CREB in MCF-7 cells resulted in reduced GRIP1 protein level, whereas the ERα protein level increased slightly, an effect that may be associated with the PKA-induced increase of ERα protein observed in other studies (44) (Figure 4A). In another study, we performed a microarray analysis on MCF-7 cells to compare the global gene expression after knockdown of GRIP1 with elevation of the cAMP level, respectively (unpublished). ERα target genes that showed similar significant changes in expression in response to reduction of GRIP1 caused by either GRIP1 shRNA or treatment with cAMP-elevating agents were selected for further analyses in the present study, including MYB (45), BCAS1 (46, 47), and RET (48) as well as EGR1 (49, 50). The protein level of GRIP1 in MCF-7 cells was significantly reduced after transduction with GRIP1 shRNA compared with control shRNA (Figure 4B). Microarray analyses showed that MYB and EGR1 were significantly downregulated (approximate fold change = 1.9 and 1.5, respectively), whereas BCAS1 and RET were upregulated (fold change = 2.1 and 1.8, respectively) in MCF-7 cells with GRIP1 knockdown (Supplemental Table 1). The effects of GRIP1 knockdown on the gene expression of MYB, EGR1, BCAS1, and RET were verified by qRT-PCR (Figure 4C and data not shown). Overexpression of CREB in control cells (control shRNA) induced similar effects as when GRIP1 protein level was reduced (GRIP1 shRNA). Because CREB did not have any additional effects on the gene expression after GRIP1 knockdown, our data suggest that CREB cooperates with GRIP1 to regulate the expression of these genes (Figure 4C). The results indicate that downregulation of GRIP1 by PKA/CREB leads to changes in the transcription of ERα target genes in MCF-7 breast cancer cells.
Figure 4.

CREB-mediated reduction of GRIP1 modulates the expression of ERα target genes. A, MCF-7 cells grown in phenol red-free DMEM supplemented with charcoal-stripped FBS (5%) and 17β-estradiol (E2, 10nM) were transfected with the expression plasmid encoding CREB (0.3 μg) for 48 hours. Lysates from the transfected cells were analyzed by Western blotting using anti-TIF2 (GRIP1), anti-ERα, anti-CREB, and anti-GAPDH antibodies. B, Western blot analyses of lysates from GRIP1 knockdown (GRIP1 shRNA) and control (control shRNA) MCF-7 cells were performed as described in Materials and Methods. GRIP1 and GAPDH were detected using anti-TIF2 (GRIP1) and anti-GAPDH antibodies, respectively. The results shown are representative of at least 3 independent experiments. The values below each panel represent the relative levels of each indicated protein, and were calculated as described in Materials and Methods. C, GRIP1 knockdown (GRIP1 shRNA) and control (control shRNA) MCF-7 cells were grown as described in A and transfected with CREB expression plasmid for 48 hours. The mRNA expression of the genes was quantitated by real-time qRT-PCR. The expression levels of each gene relative to the housekeeping gene TBP are shown (mean ± SD of triplicate samples). *P ≤ .05 (Student's t test). Calculated fold change = −1.82 ± 0.16 (EGR1), −1.63 ± 0.18 (MYB), 1.92 ± 0.28 (BCAS1), and 1.76 ± 0.21 (RET).
GRIP1 domain in the protein regions containing aa 347 to 758 and 1121 to 1462 are targeted for PKA/CREB-induced downregulation
GRIP1 contains 1462 aa and several functional domains, including the bHLH-PAS (aa 1-368), NID (aa 638-753), AD1 (aa 1056-1121), and AD2 (aa 1121-1462) (Figure 5A). To identify which aa region of GRIP1 is involved in the PKA- and CREB-mediated regulation, we examined protein levels of different GRIP1 deletion mutants (Figure 5A) in response to PKA-Cα or CREB (Figure 5B). In line with the previous results, overexpression of PKA-Cα and CREB induced a significant reduction in the protein level of GRIP1 wt (Figure 5Bi). GRIP1 mutants lacking the C-terminal region spanning the AD1 and AD2 domains (ΔAD1–2) or even lacking the sequence from aa 759 to 1462 (5–758) were also subjected to downregulation by PKA and CREB (Figure 5B, ii and iii). In contrast, the mutant containing only the N-terminal aa 5-345 was resistant to degradation (Figure 5Biv), suggesting that the aa 346 to 758 region is important for the PKA/CREB-induced downregulation of GRIP1, which was confirmed by overexpression of the GRIP1 aa 347 to 758 mutant (Figure 5Bv). This region of GRIP1 harbors the NID, which is composed of the 3 LXXLL motifs. However, mutations in these 3 LXXLL motifs (NRB123) did not affect the ability of PKA and CREB to reduce the level of GRIP1 (Figure 5Bvi), and we also observed a PKA/CREB-induced reduction of a GRIP1 N-terminal mutant that lacks the NID (5–637) (Figure 5Bvii). These results suggest that the NID is not important for the PKA/CREB-mediated regulation of GRIP1 and confirm our previous observation that GRIP1 is regulated by PKA independently of its association with NRs (26). In summary, the results suggest that PKA and CREB regulate the GRIP1 protein level through the region of aa 346 to 637. However, GRIP1 mutants lacking this region (Δ 346–637 and 638–1462) were still downregulated by PKA and CREB (Figure 5B, viii and ix, respectively). Thus, it appears that more than one GRIP1 region may serve as target for PKA/CREB. The resistance of GRIP1 aa 638 to 1056 and 730 to 1121 mutants to PKA- and CREB-stimulated degradation (Figure 5B, x and xi) suggest that the GRIP1 aa 1121 to 1462 region also serves as a target for PKA/CREB-mediated downregulation. This was confirmed by the observation that the protein level of the GRIP1 aa 1121 to 1462 fragment was also reduced (Figure 5Bxii). Furthermore, deletion of both the aa 346 to 637 and 1121 to 1462 regions (Δ 346–637 and 1121–1462) led to an attenuation of PKA/CREB-stimulated reduction of GRIP1 (Figure 5Bxiii). To ensure that the GRIP1 mutants resistant to PKA/CREB-mediated downregulation were localized in the nuclear compartment where CREB is normally localized, we performed Western blot analyses of nuclear and cytoplasmic extracts from transfected COS-1. All mutants were detected in the nuclear and also in the cytoplasmic extracts (data not shown). In conclusion, our data suggest that the PKA/CREB-stimulated downregulation of GRIP1 is mediated through 2 GRIP1 domains that are located within aa 347 to 758 and aa 1121 to 1462, respectively, and that they function independently of each other.
Figure 5.

The aa 347-758 and 1121-1462 regions of GRIP1 are targets for degradation by PKA and CREB. A, Schematic illustrations of the functional domains of full-length GRIP1 (wt) and the GRIP1 truncated or deletion mutants studied in this report. The numbers represent the spanning or deleted (Δ) aa region of each truncated or deletion mutant, respectively. Q-rich indicates a glutamine-rich region; NRB123 indicates mutations in the 3 LXXLL motifs of the NID. B, Western blot analyses of lysates from COS-1 cells transfected with expression plasmids encoding HA-GRIP1 wt or mutant protein (2 μg), CREB (0.5 μg), and PKA-Cα (0.2 μg), where indicated, were performed using anti-HA and anti-GAPDH antibodies. Relative GRIP1 levels shown to the right were calculated from densitometry analyses (ImageJ) of GRIP1 and normalized to GAPDH. Observed downregulation (+) or no change (−) of GRIP1 protein level by PKA/CREB overexpression is also indicated. The results are representative of at least 3 independent experiments.
Ubiquitination and proteasomal degradation of GRIP1
Because GRIP1 is targeted to ubiquitination (26, 41), we next performed ubiquitination analyses of the GRIP1 aa 347 to 758 and aa 1121 to 1462 domains in response to CREB or PKA overexpression. The GRIP1 constructs were coexpressed with a His-tagged ubiquitin (His-Ub) in COS-1 cells with simultaneous treatment with MG132 or vehicle (DMSO). His-Ub-attached GRIP1 (ubiquitinated GRIP1) was pulled by taking advantage of the His tag through His-pulldown assays and resolved and detected with a specific antibody targeting GRIP1. As shown in Figure 6, ladders of specific higher molecular weight bands (arrows) representing ubiquitination from the GRIP1 aa 347 to 758 (Figure 6i, top panel) and 1121 to 1462 (Figure 6ii, top panel) were observed. The levels of ubiquitinated GRIP1 347 to 758 and 1121 to 1462 were reduced upon CREB and PKA-Cα overexpression in cells treated with the vehicle (lanes 1-3). After treatment with the proteasome inhibitor MG132, the reduction was blocked (lanes 4–6). Although we did not observe a marked increase in ubiquitination of the examined GRIP1 regions upon overexpression of CREB, our data suggest that GRIP1 is highly ubiquitinated at its PKA/CREB-targeted domains and that CREB stimulates the proteasomal degradation of ubiquitinated GRIP1.
Figure 6.
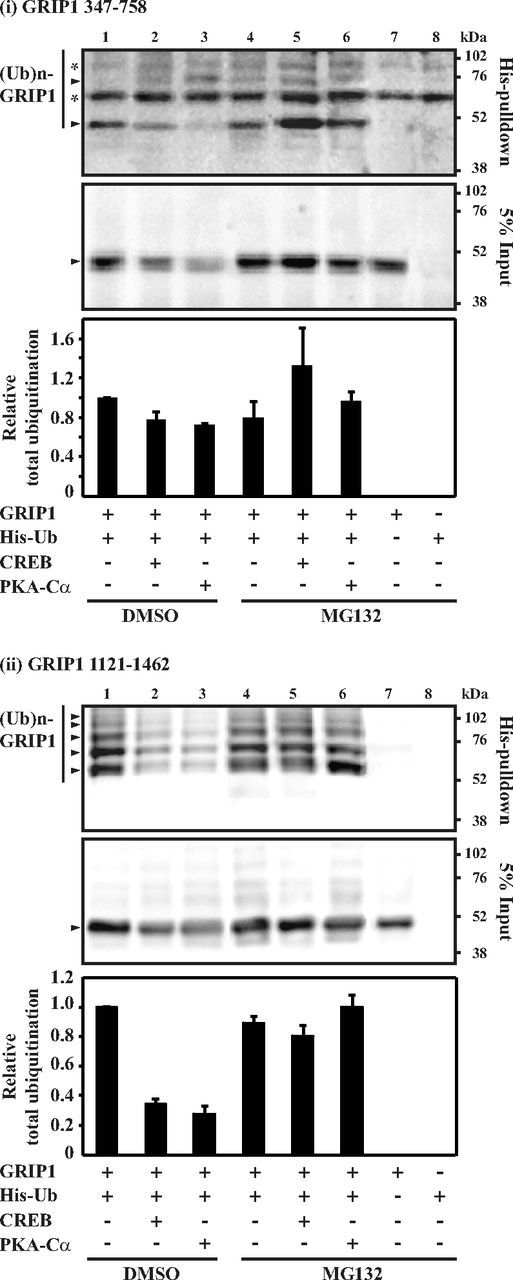
CREB promotes the proteasomal breakdown of ubiquitinated GRIP1. COS-1 cells were transfected with expression plasmids encoding HA-tagged GRIP1 347-758 (i) or 1121-1462 (ii) (4.0 μg), together with plasmids encoding His-Ub (4.0 μg), CREB (1.0 μg), and PKA-Cα (0.25 μg) as indicated, in the presence of vehicle (DMSO) or MG132. Cell lysates were subjected to His-pulldown assays as described in Materials and Methods. Pulldown samples (top panels, His-pulldown) as well as inputs (middle panels, 5% input) were analyzed by Western blotting using an anti-HA antibody (α-HA). (Ub)n-GRIP1 indicates ladders of ubiquitinated GRIP1. Arrows point to bands of His-tagged ubiquitinated GRIP1. *Bands of unspecifically bound proteins. The total ubiquitination level under each experimental condition for each GRIP1 fragment was calculated as sum (± SD) of specific ubiquitination after normalization for the unspecificity, using band densitometries, and presented as ratios over the control condition (bottom panels, relative total ubiquitination).
CREB interacts with GRIP1
To further elucidate the mechanism underlying the CREB-mediated downregulation of GRIP1, we examined whether the 2 proteins interact with each other. In vitro protein interaction assays were performed using a recombinant hexahistidine-tagged GRIP1 (6xHis-GRIP1) expressed and natively purified from a baculovirus system and a [35S]methionine-labeled and in vitro-translated CREB protein. For controls, a hexahistidine (6xHis)-peptide and another recombinant 6xHis-tagged protein (6xHis-β-actin) were used. As shown in Figure 7A, incubation of CREB with 6xHis-GRIP1 resulted in a strong binding signal as compared with the 6xHis-peptide and 6xHis-β-actin (top panel). The weak binding signals in the latter lane were due to background binding of CREB to Ni-Sepharose, because we also observed an equivalent binding intensity of CREB when CREB was incubated with only protein buffer (data not shown). Western blot analyses of the interaction complexes using an anti-GRIP1 antibody confirmed that 6xHis-GRIP1 was immobilized during pulldown of the interaction complexes using Ni-Sepharose (bottom panel).
Figure 7.

CREB interacts with GRIP1 in vitro and in vivo. A, The recombinant 6xHis-GRIP1 protein, 6xHis-β-actin, or 6xHis-peptide was incubated with [35S]methionine-labeled in vitro-translated CREB (35S-CREB) and subsequently pulled down by using Ni-Sepharose beads. Proteins bound to the Ni-Sepharose beads were subsequently resolved by SDS-PAGE (10%) and autoradiography as well as by Western blotting using an anti-GRIP1 antibody. The reticulocyte lysate inputs (2%) are shown. B and C, Coimmunoprecipitation assays were performed on lysates of COS-1 cells transfected with expression plasmids encoding HA-GRIP1 (7.0 μg) and CREB (2.0 μg) using anti-CREB antibody (α-CREB, B) or untransfected COS-1 cells using anti-nuclear receptor coactivator 2 antibody (α-GRIP1, C). A corresponding normal serum (normal IgG) was used as a negative control. Proteins from the immunoprecipitates (IP) were resolved by SDS-PAGE and Western blotting using anti-HA (α-HA, B) or anti-TIF2 (α-GRIP1, C) antibody to detect GRIP1 and anti-CREB (α-CREB) antibody to detect CREB, as indicated, and 10% or 15% of the input cell lysates are shown. *Bands for a protein that was nonspecifically pulled along in immunoprecipitation complexes by both the normal serum and anti-CREB antibody. D, Mammalian hybrid assays were carried out in COS-1 cells grown in 24-well plates. Cells were transfected with the expression plasmids encoding GAL4 (0.7 μg), GAL4-CREB (0.7 μg), VP16 (0.7 μg), and VP16-GRIP1 (0.7 μg) as indicated, together with the GAL4-luc reporter construct (0.6 μg). Cells were harvested after 48 hours of transfection and subjected to luciferase assays. The results are the mean ± SD of triplicate transfections and are representative of 3 independent experiments.
The interaction between CREB and GRIP1 was further examined by coimmunoprecipitation of both overexpressed as well as endogenous proteins (Figure 7, B and C). The results from both experiments were consistent with an interaction between CREB and GRIP1. Finally, to provide additional evidence of an interaction between the 2 proteins, we performed mammalian 2-hybrid assays (Figure 7D). GAL4-CREB activated the expression of the luciferase reporter gene, consistent with the constitutive properties of CREB in activating both basal and phosphorylation-dependent transcription (51, 52). Moreover, the GAL4-CREB transcriptional activity was significantly stimulated by coexpression of VP16-GRIP1 and not by VP16, suggesting that GRIP1 binds to CREB. Taken together, our results indicate that CREB interacts directly with GRIP1.
Transcriptional activity is dispensable, whereas the bZIP domain of CREB is required for GRIP1 interaction and degradation
CREB function is attributed to several of its functional and structural domains including the glutamine-rich domains (Q1 and Q2), the KID, and the C-terminal bZIP domain (Figure 8A) (53). The transcription factor is known to mediate the effect of cAMP/PKA through its phosphorylation at the Ser-133 residue located in the KID, followed by its dimerization and binding to cAMP-responsive elements on the promoter of target genes (30). First, we examined whether the transcriptional activity of CREB was associated with its ability to downregulate GRIP1 through using CREB dominant-negative mutants in which either Ser-133 residue is mutated to Ala (CREB S133A) or the basic DNA-binding region is mutated through a single aa replacement and insertion (KCREB). CREB S133A blocks Ser-133 phosphorylation while KCREB forms an inactive dimer with endogenous CREB and is unable to bind DNA. Similar to wt CREB, coexpression of the CREB dominant-negative mutants led to a dose-dependent inhibition of GRIP1-mediated transcription from GAL4-luc (Figure 8B). Moreover, our in vitro interaction data showed that KCREB as well as CREB S133A physically associated with GRIP1 (Figure 8C). The results suggest that CREB mediates GRIP1 degradation independently of its PKA-mediated Ser-133 phosphorylation, DNA-binding, or transcriptional activity.
Figure 8.

The bZIP domain of CREB is essential for interaction with and degradation of GRIP1. A, Schematic illustrations of the full-length CREB (CREB wt) with functional domains, and the studied truncated/deletion mutants: CREB ΔQ1 (deletion of Q1), CREB ΔQ2 (deletion of Q2), CREB ΔKID (deletion of KID), CREB ΔbZIP (deletion of bZIP), and CREB bZIP (only bZIP). The approximate aa positions for each functional domain are indicated (uppermost). Q1 and Q2 indicate glutamine-rich domains. B, COS-1 cells were transiently transfected with a GAL4-responsive luciferase reporter construct (GAL4-luc) (1.1 μg) together with expression plasmids encoding GAL4-GRIP1 (1.0 μg) and KCREB or CREB S133A (0.01, 0.02, 0.05, 0.1, 0.5, and 1.0 μg) for 48 hours. Luciferase assays were performed, and the results are presented as mean ± SD of triplicate transfections from a representative experiment. C, In vitro protein interaction assays. [35S]Methionine-labeled CREB proteins (wt, CREB S133A, KCREB, ΔQ1, ΔQ2, ΔKID, ΔbZIP, and bZIP) were incubated with recombinant 6xHis-GRIP1 or 6xHis-peptide as described in Materials and Methods. Interaction complexes were pulled down using Ni-Sepharose beads and resolved by SDS-PAGE followed by autoradiography. Two percent of input was also included. Ratios between densitometries of input and interaction band are indicated below each panel. D, COS-1 cells were transfected with expression plasmids encoding HA-GRIP1 (2.0 μg) and Flag-tagged CREB wt, ΔbZIP, or bZIP (0.5 μg). Cell lysates were analyzed by Western blotting using anti-HA (GRIP1), anti-Flag (Flag-CREB), and anti-GAPDH antibodies.
We next attempted to identify CREB domains involved in the regulation of GRIP1 through examining GRIP1 interaction with CREB deletion mutants lacking one of the structural and functional domains (ΔQ1, ΔQ2, ΔKID, and ΔbZIP) as well as with a CREB fragment containing only the bZIP domain as shown in Figure 8A. We observed specific binding signals suggesting that GRIP1 interacted with CREB ΔQ1, ΔQ2, ΔKID, and CREB bZIP (Figure 8C). In contrast, we did not detect any interaction between GRIP1 and CREB ΔbZIP, suggesting that the bZIP domain is required for the interaction between the 2 proteins. The role of the bZIP domain was further examined by coexpression of the CREB mutants and GRIP1 in COS-1 cells. In line with the interaction data, GRIP1 protein level was reduced by overexpression of CREB mutants containing the bZIP (CREB wt and CREB bZIP), whereas it was not affected by overexpression of the mutant lacking this domain (Figure 8D). The results suggest that the bZIP domain is essential for CREB to interact with and mediate GRIP1 degradation.
Discussion
Regulation of the SRCs by intracellular signaling is an important mechanism in modulation of NR functions and the expression of their target genes (6). We have previously shown that activation of PKA initially enhances the recruitment of GRIP1 to an estrogen-responsive gene promoter, followed by an increased turnover and proteasomal degradation of GRIP1 (26, 27). In the present study, we demonstrate that CREB downregulates GRIP1, leading to changes in the expression of a subset of ERα target genes in MCF-7 breast cancer cells. It is apparent that CREB mediates GRIP1 degradation in response to prolonged PKA activation and that this downregulation modulates GRIP1 availability affecting ERα-mediated transcriptional activities. Studies have reported that PKA activation suppresses estradiol-dependent proliferation of MCF-7 cells through modulating transcription of ERα target genes (28) and that the peptide hormone glucagon-like peptide-1 activates cAMP and inhibits breast cancer cell growth (54). It is possible that the inhibitory effect of cAMP/PKA on breast cancer cell proliferation is mediated through downregulation of GRIP1. CREB is overexpressed in breast tumors of patients with poor prognosis, metastatic disease, and nodal involvement (55). CREB also stimulates proliferation, migration, and invasion of metastatic MDA-MB-231 breast cancer cells (56). In contrast, a recent study showed that knockdown of GRIP1 resulted in acceleration of liver tumorigenesis, suggesting that GRIP1 is a tumor suppressor of liver cancer (57). Whether CREB-mediated downregulation of GRIP1 is associated with metastasis and progression of breast tumors remains to be elucidated.
The PKA/CREB-dependent downregulation of GRIP1 is mediated through 2 domains localized in the N-terminal (aa 347–758) and the C-terminal (aa 1121–1462) regions of GRIP1. The aa 347 to 758 region is rich in phosphorylation-prone Ser and Thr residues and shows a low level of sequence homology, with the exception of the LXXLL motifs, compared with corresponding regions of SRC-1 and SRC-3. Although the LXXLL motifs, which are essentially involved in the interaction with NRs (58), seem to be bypassed in PKA-regulated degradation of GRIP1 (26) (present study), little is known about the importance of the aa 347 to 758 sequence element for GRIP1 function and regulation. A recent study identified 6 glucocorticoid-induced and 1 constitutive phosphorylation site within this protein region, which may dictate GRIP1 function in glucocorticoid receptor-dependent transcriptional control (23). It remains to be investigated whether these phosphorylation sites are associated with PKA/CREB-stimulated degradation of GRIP1. The aa 1121 to 1462 region, which contains the conserved autonomous AD2 with high degree of homology with SRC-1 and SRC-3, is well documented to be pivotal for interaction between GRIP1 and other coactivator proteins and for enhancement of NR transcriptional activity (34, 59–62). Our previous study showed that PKA downregulated the coactivator function of GRIP1 but not SRC-1 and SRC-3 (25). Furthermore, it has been reported that the stability of SRC-3 is specifically regulated by other protein kinases including p38 MAPK (63), glycogen synthase kinase 3 (64), and protein kinase C (65), through phosphorylation and ubiquitin-proteasomal degradation pathway, in an NR-associated context. Therefore, it seems likely that PKA/CREB-mediated proteasomal degradation is specific for GRIP1.
We demonstrated also a direct association between CREB and GRIP1 through CREB-bZIP domain, which is important for GRIP1 degradation. Previous studies have reported that CREB interacts with and modulates the transcriptional activity of NRs such as the AR (66) and steroidogenic factor-1 (67) as well as several coactivator proteins including CBP/p300 and coactivator-associated arginine methyltransferase 1 (68–70). CBP/p300 associates with both CREB and GRIP1 (37, 38, 68) and possesses intrinsic ubiquitin ligase activity and is involved in p53 polyubiquitination and proteasome degradation (71, 72). Thus, CBP could potentially be involved in the degradation of GRIP1 through association with CREB. However, we observed that a CREB mutant lacking the CBP-binding KID (51) was still able to interact with GRIP1 and that GRIP1 mutants lacking the CBP-binding AD1 (39) were subjected to degradation by CREB. These findings suggest that CBP is not involved in CREB-mediated degradation of GRIP1. We propose that CREB mediates GRIP1 degradation through protein-protein interaction with GRIP1 at the bZIP domain of CREB. The bZIP domain is composed of a cluster of basic residues important for DNA binding and heptad leucine repeats that form a zipper region responsible for CREB homodimerization or heterodimerization (53, 73). It has been shown that the bZIP domain is involved in interactions with several other proteins including the CRTCs (74). It is well known that activation of PKA leads to CRTC3 dephosphorylation and translocation to the nucleus where it associates with CREB and stimulates gene expression. Likewise, prolonged PKA activation triggers degradation of CRTC3 (75, 76). However, it remains to be elucidated whether any of the CRTCs are associated with the regulation of GRIP1 by PKA and CREB. Other members of the CREB/cAMP responsive element modulator/activating transcription factor family have been shown to interact with ubiquitin ligases (77–79). It is therefore possible that CREB may stimulate the effect of ubiquitin-protein ligases. So far, no interaction of CREB with a ubiquitin-protein ligase has been reported. Moreover, it has been reported that other proteins including bZIP transcription factors induce proteasomal degradation of their interacting partners through recruitment of ubiquitin- and/or the 26S proteasome components (80–83). CREB might also act through such a mechanism being a docking signal to target GRIP1 to the proteasome, but this remains to be studied in more detail.
In conclusion, we have identified CREB as a novel factor in the proteasomal regulation of GRIP1. We have mapped the domains of GRIP1 that are involved in the ubiquitin-proteasomal degradation and shown that CREB interacts with GRIP1 and mediates its proteasomal degradation in a bZIP domain-dependent manner. Our study ascribes a novel function for CREB as a protein that promotes proteasomal degradation of an interaction partner.
Acknowledgments
We thank Dr J. Lund, Dr F. Schaufele, Dr G. S. McKnight, Dr E. Treuter, Dr R. H. Goodman, and Dr R. A. Maurer for the supplied plasmids. Technical assistance from Thomas Helland, Carol Cook, Bente Skjellstad, Anne M. Sellevold, Alicia Folloso, and Anita Ivarsflaten is highly appreciated.
This work was funded by the Research Council of Norway, the Norwegian Cancer Society, Samarbeidsorganet Helse Vest HF, Meltzerfondet, and KG Jebsen Center for Diabetes Research.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- aa
- amino acids
- AD
- activation domain
- AR
- androgen receptor
- bHLH-PAS
- N-terminal basic helix-loop-helix Per-Arnt-Sim
- bZIP
- basic leucine zipper
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- CRTC
- CREB-regulated transcription coactivator
- DMEM
- Dulbecco's modified Eagle's medium
- DMSO
- dimethylsulfoxide
- ERα
- estrogen receptor α
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GR
- glucocorticoid receptor
- GRIP1
- glucocorticoid receptor-interacting protein 1
- HA
- hemagglutinin
- His-Ub
- His-tagged ubiquitin
- KID
- kinase-inducible domain
- NID
- NR-interaction domain
- NR
- nuclear receptor
- PKA
- cAMP dependent protein kinase
- qRT-PCR
- quantitative RT-PCR
- SDS
- sodium dodecyl sulfate
- shRNA
- short hairpin RNA
- siRNA
- small interference RNA
- SRC
- steroid receptor coactivator
- TIF2
- transcriptional intermediary factor 2
- wt
- wild type.
References
- 1. McKenna NJ , O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. [DOI] [PubMed] [Google Scholar]
- 2. Hong H , Kohli K , Trivedi A , Johnson DL , Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci U S A. 1996;93:4948–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Voegel JJ , Heine MJ , Zechel C , Chambon P , Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- 4. Norris JD , Fan D , Stallcup MR , McDonnell DP. Enhancement of estrogen receptor transcriptional activity by the coactivator GRIP-1 highlights the role of activation function 2 in determining estrogen receptor pharmacology. J Biol Chem. 1998;273:6679–6688. [DOI] [PubMed] [Google Scholar]
- 5. Xu J , O'Malley BW. Molecular mechanisms and cellular biology of the steroid receptor coactivator (SRC) family in steroid receptor function. Rev Endocr Metab Disord. 2002;3:185–192. [DOI] [PubMed] [Google Scholar]
- 6. York B , O'Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem. 2010;285:38743–38750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gehin M , Mark M , Dennefeld C , Dierich A , Gronemeyer H , Chambon P. The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol Cell Biol. 2002;22:5923–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeong JW , Lee KY , Han SJ , et al. . The p160 steroid receptor coactivator 2, SRC-2, regulates murine endometrial function and regulates progesterone-independent and -dependent gene expression. Endocrinology. 2007;148:4238–4250. [DOI] [PubMed] [Google Scholar]
- 9. Picard F , Gehin M , Annicotte J , et al. . SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell. 2002;111:931–941. [DOI] [PubMed] [Google Scholar]
- 10. Jeong JW , Kwak I , Lee KY , et al. . The genomic analysis of the impact of steroid receptor coactivators (SRCs) ablation on hepatic metabolism. Mol Endocrinol. 2006;20:1138–1152. [DOI] [PubMed] [Google Scholar]
- 11. Chopra AR , Louet JF , Saha P , et al. . Absence of the SRC-2 coactivator results in a glycogenopathy resembling Von Gierke's disease. Science. 2008;322:1395–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moedder UI , Monroe DG , Fraser DG , et al. . Skeletal consequences of deletion of steroid receptor coactivator-2/transcription intermediary factor-2. J Biol Chem. 2009;284:18767–18777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Osborne CK , Bardou V , Hopp TA , et al. . Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–361. [DOI] [PubMed] [Google Scholar]
- 14. Fleming FJ , Myers E , Kelly G , et al. . Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J Clin Pathol. 2004;57:1069–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haugan Moi LL , Hauglid Flageng M , Gandini S , et al. . Effect of low-dose tamoxifen on steroid receptor coactivator 3/amplified in breast cancer 1 in normal and malignant human breast tissue. Clin Cancer Res. 2010;16:2176–2186. [DOI] [PubMed] [Google Scholar]
- 16. Karmakar S , Foster EA , Smith CL. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-α transcriptional activity. Endocrinology. 2009;150:1588–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karmakar S , Foster EA , Blackmore JK , Smith CL. Distinctive functions of p160 steroid receptor coactivators in proliferation of an estrogen-independent, tamoxifen-resistant breast cancer cell line. Endocr Relat Cancer. 2011;18:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Girault I , Lerebours F , Amarir S , et al. . Expression analysis of estrogen receptor α coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin Cancer Res. 2003;9:1259–1266. [PubMed] [Google Scholar]
- 19. Lopez GN , Turck CW , Schaufele F , Stallcup MR , Kushner PJ. Growth factors signal to steroid receptors through mitogen-activated protein kinase regulation of p160 coactivator activity. J Biol Chem. 2001;276:22177–22182. [DOI] [PubMed] [Google Scholar]
- 20. Gregory CW , Fei X , Ponguta LA , et al. . Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279:7119–7130. [DOI] [PubMed] [Google Scholar]
- 21. Frigo DE , Basu A , Nierth-Simpson EN , et al. . p38 mitogen-activated protein kinase stimulates estrogen-mediated transcription and proliferation through the phosphorylation and potentiation of the p160 coactivator glucocorticoid receptor-interacting protein 1. Mol Endocrinol. 2006;20:971–983. [DOI] [PubMed] [Google Scholar]
- 22. Chopra AR , Kommagani R , Saha P , et al. . Cellular energy depletion resets whole-body energy by promoting coactivator-mediated dietary fuel absorption. Cell Metab. 2011;13:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobrovolna J , Chinenov Y , Kennedy MA , Liu B , Rogatsky I. Glucocorticoid-dependent phosphorylation of the transcriptional coregulator GRIP1. Mol Cell Biol. 2012;32:730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kotaja N , Karvonen U , Janne OA , Palvimo JJ. The nuclear receptor interaction domain of GRIP1 is modulated by covalent attachment of SUMO-1. J Biol Chem. 2002;277:30283–30288. [DOI] [PubMed] [Google Scholar]
- 25. Børud B , Hoang T , Bakke M , Jacob AL , Lund J , Mellgren G. The nuclear receptor coactivators p300/CBP/cointegrator-associated protein (p/CIP) and transcription intermediary factor 2 (TIF2) differentially regulate PKA-stimulated transcriptional activity of steroidogenic factor 1. Mol Endocrinol. 2002;16:757–773. [DOI] [PubMed] [Google Scholar]
- 26. Hoang T , Fenne IS , Cook C , et al. . cAMP-dependent protein kinase regulates ubiquitin-proteasome-mediated degradation and subcellular localization of the nuclear receptor coactivator GRIP1. J Biol Chem. 2004;279:49120–49130. [DOI] [PubMed] [Google Scholar]
- 27. Fenne IS , Hoang T , Hauglid M , et al. . Recruitment of coactivator glucocorticoid receptor interacting protein 1 to an estrogen receptor transcription complex is regulated by the 3′,5′-cyclic adenosine 5′-monophosphate-dependent protein kinase. Endocrinology. 2008;149:4336–4345. [DOI] [PubMed] [Google Scholar]
- 28. Al-Dhaheri MH , Rowan BG. Protein kinase A exhibits selective modulation of estradiol-dependent transcription in breast cancer cells that is associated with decreased ligand binding, altered estrogen receptor alpha promoter interaction, and changes in receptor phosphorylation. Mol Endocrinol. 2007;21:439–456. [DOI] [PubMed] [Google Scholar]
- 29. Fontana JA , Miksis G , Miranda DM , Durham JP. Inhibition of human mammary carcinoma cell proliferation by retinoids and intracellular cAMP-elevating compounds. J Natl Cancer Inst. 1987;78:1107–1112. [PubMed] [Google Scholar]
- 30. Mayr B , Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. [DOI] [PubMed] [Google Scholar]
- 31. Sands WA , Palmer TM. Regulating gene transcription in response to cyclic AMP elevation. Cell Signal. 2008;20:460–466. [DOI] [PubMed] [Google Scholar]
- 32. Screaton RA , Conkright MD , Katoh Y , et al. . The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. [DOI] [PubMed] [Google Scholar]
- 33. Altarejos JY , Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen D , Ma H , Hong H , et al. . Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. [DOI] [PubMed] [Google Scholar]
- 35. Lee YH , Campbell HD , Stallcup MR. Developmentally essential protein flightless I is a nuclear receptor coactivator with actin binding activity. Mol Cell Biol. 2004;24:2103–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johannessen M , Delghandi MP , Seternes OM , Johansen B , Moens U. Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain. Cell Signal. 2004;16:1187–1199. [DOI] [PubMed] [Google Scholar]
- 37. Voegel JJ , Heine MJ , Tini M , Vivat V , Chambon P , Gronemeyer H. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J. 1998;17:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma H , Hong H , Huang SM , et al. . Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol. 1999;19:6164–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang SM , Cheng YS. Analysis of two CBP (cAMP-response-element-binding protein-binding protein) interacting sites in GRIP1 (glucocorticoid-receptor-interacting protein), and their importance for the function of GRIP1. Biochem J. 2004;382:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baumann CT , Ma H , Wolford R , et al. . The glucocorticoid receptor interacting protein 1 (GRIP1) localizes in discrete nuclear foci that associate with ND10 bodies and are enriched in components of the 26S proteasome. Mol Endocrinol. 2001;15:485–500. [DOI] [PubMed] [Google Scholar]
- 41. Yan F , Gao X , Lonard DM , Nawaz Z. Specific ubiquitin-conjugating enzymes promote degradation of specific nuclear receptor coactivators. Mol Endocrinol. 2003;17:1315–1331. [DOI] [PubMed] [Google Scholar]
- 42. Perissi V , Aggarwal A , Glass CK , Rose DW , Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. [DOI] [PubMed] [Google Scholar]
- 43. Deroo BJ , Archer TK. Proteasome inhibitors reduce luciferase and beta-galactosidase activity in tissue culture cells. J Biol Chem. 2002;277:20120–20123. [DOI] [PubMed] [Google Scholar]
- 44. Tsai HW , Katzenellenbogen JA , Katzenellenbogen BS , Shupnik MA. Protein kinase A activation of estrogen receptor α transcription does not require proteasome activity and protects the receptor from ligand-mediated degradation. Endocrinology. 2004;145:2730–2738. [DOI] [PubMed] [Google Scholar]
- 45. Drabsch Y , Hugo H , Zhang R , et al. . Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc Natl Acad Sci U S A. 2007;104:13762–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Collins C , Rommens JM , Kowbel D , et al. . Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci U S A. 1998;95:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beardsley DI , Kowbel D , Lataxes TA , et al. . Characterization of the novel amplified in breast cancer-1 (NABC1) gene product. Exp Cell Res. 2003;290:402–413. [DOI] [PubMed] [Google Scholar]
- 48. Boulay A , Breuleux M , Stephan C , et al. . The Ret receptor tyrosine kinase pathway functionally interacts with the ERα pathway in breast cancer. Cancer Res. 2008;68:3743–3751. [DOI] [PubMed] [Google Scholar]
- 49. de Jager T , Pelzer T , Muller-Botz S , Imam A , Muck J , Neyses L. Mechanisms of estrogen receptor action in the myocardium. Rapid gene activation via the ERK1/2 pathway and serum response elements. J Biol Chem. 2001;276:27873–27880. [DOI] [PubMed] [Google Scholar]
- 50. Baron V , Adamson ED , Calogero A , Ragona G , Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Quinn PG. Distinct activation domains within cAMP response element-binding protein (CREB) mediate basal and cAMP-stimulated transcription. J Biol Chem. 1993;268:16999–17009. [PubMed] [Google Scholar]
- 52. Roesler WJ , Graham JG , Kolen R , Klemm DJ , McFie PJ. The cAMP response element binding protein synergizes with other transcription factors to mediate cAMP responsiveness. J Biol Chem. 1995;270:8225–8232. [DOI] [PubMed] [Google Scholar]
- 53. Johannessen M , Delghandi MP , Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. [DOI] [PubMed] [Google Scholar]
- 54. Ligumsky H , Wolf I , Israeli S , et al. . The peptide-hormone glucagon-like peptide-1 activates cAMP and inhibits growth of breast cancer cells. Breast Cancer Res Treat. 2012;132:449–461. [DOI] [PubMed] [Google Scholar]
- 55. Chhabra A , Fernando H , Watkins G , Mansel RE , Jiang WG. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007;18:953–958. [PubMed] [Google Scholar]
- 56. Son J , Lee JH , Kim HN , Ha H , Lee ZH. cAMP-response-element-binding protein positively regulates breast cancer metastasis and subsequent bone destruction. Biochem Biophys Res Commun. 2010;398:309–314. [DOI] [PubMed] [Google Scholar]
- 57. O'Donnell KA , Keng VW , York B , et al. . A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109:E1377–E1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ding XF , Anderson CM , Ma H , et al. . Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): multiple motifs with different binding specificities. Mol Endocrinol. 1998;12:302–313. [DOI] [PubMed] [Google Scholar]
- 59. Koh SS , Chen D , Lee YH , Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–1098. [DOI] [PubMed] [Google Scholar]
- 60. Li H , Kim JH , Koh SS , Stallcup MR. Synergistic effects of coactivators GRIP1 and β-catenin on gene activation: cross-talk between androgen receptor and Wnt signaling pathways. J Biol Chem. 2004;279:4212–4220. [DOI] [PubMed] [Google Scholar]
- 61. Huang SM , Huang CJ , Wang WM , Kang JC , Hsu WC. The enhancement of nuclear receptor transcriptional activation by a mouse actin-binding protein, alpha actinin 2. J Mol Endocrinol. 2004;32:481–496. [DOI] [PubMed] [Google Scholar]
- 62. Kotaja N , Vihinen M , Palvimo JJ , Janne OA. Androgen receptor-interacting protein 3 and other PIAS proteins cooperate with glucocorticoid receptor-interacting protein 1 in steroid receptor-dependent signaling. J Biol Chem. 2002;277:17781–17788. [DOI] [PubMed] [Google Scholar]
- 63. Gianni M , Parrella E , Raska I , et al. . P38MAPK-dependent phosphorylation and degradation of SRC-3/AIB1 and RARα-mediated transcription. EMBO J. 2006;25:739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu RC , Feng Q , Lonard DM , O'Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. [DOI] [PubMed] [Google Scholar]
- 65. Yi P , Feng Q , Amazit L , et al. . Atypical protein kinase C regulates dual pathways for degradation of the oncogenic coactivator SRC-3/AIB1. Mol Cell. 2008;29:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim J , Jia L , Stallcup MR , Coetzee GA. The role of protein kinase A pathway and cAMP responsive element-binding protein in androgen receptor-mediated transcription at the prostate-specific antigen locus. J Mol Endocrinol. 2005;34:107–118. [DOI] [PubMed] [Google Scholar]
- 67. Manna PR , Eubank DW , Lalli E , Sassone-Corsi P , Stocco DM. Transcriptional regulation of the mouse steroidogenic acute regulatory protein gene by the cAMP response-element binding protein and steroidogenic factor 1. J Mol Endocrinol. 2003;30:381–397. [DOI] [PubMed] [Google Scholar]
- 68. Mayr BM , Canettieri G , Montminy MR. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci U S A. 2001;98:10936–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Krones-Herzig A , Mesaros A , Metzger D , et al. . Signal-dependent control of gluconeogenic key enzyme genes through coactivator-associated arginine methyltransferase 1. J Biol Chem. 2006;281:3025–3029. [DOI] [PubMed] [Google Scholar]
- 70. Makkonen KM , Pasonen-Seppanen S , Torronen K , Tammi MI , Carlberg C. Regulation of the hyaluronan synthase 2 gene by convergence in cyclic AMP response element-binding protein and retinoid acid receptor signaling. J Biol Chem. 2009;284:18270–18281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grossman SR , Deato ME , Brignone C , et al. . Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. [DOI] [PubMed] [Google Scholar]
- 72. Shi D , Pop MS , Kulikov R , Love IM , Kung AL , Grossman SR. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A. 2009;106:16275–16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Manna PR , Dyson MT , Stocco DM. Role of basic leucine zipper proteins in transcriptional regulation of the steroidogenic acute regulatory protein gene. Mol Cell Endocrinol. 2009;302:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Conkright MD , Canettieri G , Screaton R , et al. . TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. [DOI] [PubMed] [Google Scholar]
- 75. Liu Y , Dentin R , Chen D , et al. . A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Song Y , Altarejos J , Goodarzi MO , et al. . CRTC3 links catecholamine signalling to energy balance. Nature. 2010;468:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Firestein R , Feuerstein N. Association of activating transcription factor 2 (ATF2) with the ubiquitin-conjugating enzyme hUBC9. Implication of the ubiquitin/proteasome pathway in regulation of ATF2 in T cells. J Biol Chem. 1998;273:5892–5902. [DOI] [PubMed] [Google Scholar]
- 78. Lassot I , Segeral E , Berlioz-Torrent C , et al. . ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(βTrCP) ubiquitin ligase. Mol Cell Biol. 2001;21:2192–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wei Y , Jiang J , Liu D , et al. . Cdc34-mediated degradation of ATF5 is blocked by cisplatin. J Biol Chem. 2008;283:18773–18781. [DOI] [PubMed] [Google Scholar]
- 80. Gottlicher M , Heck S , Doucas V , et al. . Interaction of the Ubc9 human homologue with c-Jun and with the glucocorticoid receptor. Steroids. 1996;61:257–262. [DOI] [PubMed] [Google Scholar]
- 81. Furukawa M , Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kobayashi A , Kang MI , Okawa H , et al. . Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Isono O , Ohshima T , Saeki Y , et al. . Human T-cell leukemia virus type 1 HBZ protein bypasses the targeting function of ubiquitination. J Biol Chem. 2008;283:34273–34282. [DOI] [PMC free article] [PubMed] [Google Scholar]