

Abstract
Context:
Hurthle cell cancer (HCC) is an understudied cancer with poor prognosis.
Objective:
Our objective was to elucidate the genomic foundations of HCC.
Design and Setting:
We conducted a large-scale integrated analysis of mutations, gene expression profiles, and copy number alterations in HCC at a single tertiary-care cancer institution.
Methods:
Mass spectrometry-based genotyping was used to interrogate hot spot point mutations in the most common thyroid oncogenes: BRAF, RET, NRAS, HRAS, KRAS, PIK3CA, MAP2K1, and AKT1. In addition, common oncogenic fusions of RET and NTRK1 as well as PAX8/PPARγ and AKAP9-BRAF were also assessed by RT-PCR. Global copy number changes and gene expression profiles were determined in the same tumor set as the mutational analyses.
Results:
We report that the mutational, transcriptional, and copy number profiles of HCC were distinct from those of papillary thyroid cancer and follicular thyroid cancer, indicating HCC to be a unique type of thyroid malignancy. Unsupervised hierarchical clustering of gene expression showed the 3 groups of Hurthle tumors (Hurthle cell adenoma [HA], minimally invasive Hurthle cell carcinoma [HMIN], and widely invasive Hurthle cell carcinoma [HWIDE] clustered separately with a marked difference between HWIDE and HA. Global copy number analysis also indicated distinct subgroups of tumors that may arise as HWIDE and HMIN. Molecular pathways that differentiate HA from HWIDE included the PIK3CA-Akt-mTOR and Wnt/β-catenin pathways, potentially providing a rationale for new targets for this type of malignancy.
Conclusions:
Our data provide evidence that HCC may be a unique thyroid cancer distinct from papillary and follicular thyroid cancer.
Hurthle cell cancer (HCC) is comparatively understudied and accounts for 3% to 4% of all thyroid cancers (1). This cancer develops from Hurthle cells, which are believed to represent a common metaplastic change in thyroid follicular epithelium that has been damaged. Characteristically, they are large cells with hyperchromatic nuclei and an abundant granular cytoplasm containing large numbers of mitochondria (2). Hurthle cells can form adenomas and carcinomas. Malignancy cannot be diagnosed without the identification of capsular or vascular invasion. Vascular invasion is the hallmark finding of HCC, and it can be classified into a minimally invasive type and a widely invasive type according to the extent of vascular invasion. Clinically, the widely invasive form of HCC is the most important because they can be locally invasive, can metastasize into the lymphatics of the neck, and have a high incidence of distant metastasis to the lung, liver, and bone (3–5). Most importantly, they are often refractory to radioactive iodine (6, 7) and also have poor chemosensitivity. As a result, patients with the widely invasive form of HCC have a poor prognosis compared with papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC) with a recurrence rate of 31% and disease-specific mortality rate of 25% (8, 9). If patients present with distant metastases, the mortality rate is as high as 80% (9).
The World Health Organization classification on thyroid malignancies categorizes HCC as an oncocytic variant of follicular carcinoma (10). This would suggest HCC has similar genetic abnormalities to those of follicular carcinoma. However, a comparative genomic analysis has never been carried out in a comprehensive fashion. Despite some progress, particularly on mutational alterations in mitochondrial DNA, the molecular foundations of HCC are still relatively ill-defined (11–14). In addition, HCC is not being studied by The Cancer Genome Atlas project. Because the widely invasive form of HCC has a poor prognosis, a detailed genetic analysis of HCC is important because it will allow the identification of the molecular pathways altered in this cancer and hence identify targets for new treatment. The objective of our study was therefore to carry out a genome-wide analysis of Hurthle cell neoplasms by using mutation genotyping, gene expression profiling, and global copy number analysis to characterize the cytogenetic, transcriptional, and mutational events in different stages of this disease.
Patients and Methods
Patient characteristics and tumor samples
Hurthle cell adenomas (HAs) and carcinomas (HCCs) were obtained from the Memorial Sloan-Kettering Cancer Center tumor bank. Use of all tumors and patient data was approved by the Institutional Review Board. Tumor tissues consisted of high quality fresh-frozen samples. Hematoxylin and eosin slides were made, and a pathologist, specialized in thyroid pathology (R.G.), then confirmed the diagnosis of each tumor. Hurthle cell tumors were defined as tumors composed of >75% oncocytic cells lacking the nuclear features of PTC. Hurthle cell tumors without capsular or vascular invasion were categorized as adenomas (HAs). Tumors classified as HCC were divided as follows: 1) minimally invasive HCC (HMIN) defined as encapsulated tumor harboring <4 foci of angio-invasion (foci of vascular invasion that were closely adjacent to one another were counted as separate foci) and lacking both gross invasion as well as vascular invasion of extrathyroid vessels or 2) widely invasive HCC (HWIDE) if the tumor was grossly invasive, had extrathyroid angio-invasion and/or was encapsulated with 4 or more foci of vascular invasion. This categorization is specific to this study and consists of a modification of the HCC stratification used in the current National Cancer Center Network guidelines (15). Fresh-frozen tumor samples from 8 patients with HA, 9 patients with HMIN, and 10 patients with HWIDE were identified. Regions with cancer were marked and then microdissected from 10-μm frozen section slides to ensure a consistent tumor cell content of greater than 70%. DNA and RNA were then extracted using the AllPrep DNA/RNA Mini Kit (QIAGEN, Valencia, California), and DNA/RNA quality was verified by spectrophotometry (Nanodrop ND-1000, Wilmington, Delaware). Patient, treatment, and outcome characteristics were recorded for each tumor sample by a retrospective analysis of patient charts (Table 1).
Table 1.
Patient and Treatment Characteristics of Samples Used
| Patient ID | Age at Diagnosis, y | Gender | Surgery Type | RAI, Yes/No | Recurrence | Site of Recurrence | Disease Status | Follow-up Time, mo |
|---|---|---|---|---|---|---|---|---|
| HA1 | 47 | Female | Thyroid lobectomy | No | No | NED | 0 | |
| HA2 | 54 | Male | Total thyroidectomy | No | No | NED | 61 | |
| HA3 | 53 | Female | Thyroid lobectomy | No | No | NED | 0 | |
| HA4 | 75 | Male | Thyroid lobectomy | No | No | NED | 6 | |
| HA5 | 44 | Female | Thyroid lobectomy | No | No | NED | 6 | |
| HA6 | 68 | Female | Thyroid lobectomy | No | No | NED | 17 | |
| HA7 | 52 | Female | Total thyroidectomy | No | No | NED | 26 | |
| HA8 | 56 | Male | Total thyroidectomy | No | No | NED | 0 | |
| HMIN_1 | 34 | Female | Thyroid lobectomy | No | No | NED | 27 | |
| HMIN_2 | 64 | Female | Thyroid lobectomy | No | No | DOC | 132 | |
| HMIN_3 | 52 | Female | Thyroid lobectomy | No | No | NED | 24 | |
| HMIN_4 | 61 | Male | Total thyroidectomy | Yes | No | NED | 77 | |
| HMIN_5 | 63 | Male | Total thyroidectomy | Yes | No | NED | 34 | |
| HMIN_6 | 67 | Female | Total thyroidectomy | No | No | NED | 61 | |
| HMIN_7 | 68 | Male | Total thyroidectomy | No | No | NED | 54 | |
| HMIN_8 | 61 | Female | Thyroid lobectomy | No | No | NED | 42 | |
| HMIN_9 | 57 | Male | Total thyroidectomy | No | No | NED | 24 | |
| HWIDE_1 | 81 | Female | Total thyroidectomy | No | No | DOC | 95 | |
| HWIDE_2 | 65 | Male | Total thyroidectomy | Yes | No | NED | 90 | |
| HWIDE_3 | 67 | Female | Total thyroidectomy | Yes | No | DOC | 47 | |
| HWIDE_4 | 81 | Female | Total thyroidectomy | Yes | No | NED | 36 | |
| HWIDE_5 | 46 | Male | Total thyroidectomy | Yes | No | NED | 47 | |
| HWIDE_6 | 73 | Male | Total thyroidectomy | Yes | No | NED | 48 | |
| HWIDE_7 | 71 | Male | Total thyroidectomy | Yes | Yes | Neck | AWD | 46 |
| HWIDE_8 | 57 | Male | Total thyroidectomy | Yes | Yes | Lung | AWD | 38 |
| HWIDE_9 | 40 | Female | Total thyroidectomy | Yes | No | NED | 43 | |
| HWIDE_10 | 64 | Female | Thyroid lobectomy | Yes | Yes | Bone | AWD | 43 |
Abbreviations: RAI, radioactive iodine; NED, no evidence of disease; AWD, alive with disease; DOC, dead of other cause.
MassARRAY genotyping of mutations in HA, HMIN, and HWIDE tumors
Characterization of mutations in Hurthle cell tumors
Mass spectrometry-based genotyping assays (Sequenom MassARRAY; Sequenom, San Diego, California) were used to interrogate hot spot point mutations in the most common thyroid oncogenes: BRAF, RET, NRAS, HRAS, KRAS, PIK3CA, MAP2K1, and AKT1 (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org). HRAS exon 1 mutations were analyzed by Sanger sequencing. These assays are as described by Ricarte Filho et al (16).
Screening for oncogenic rearrangements
Common oncogenic fusions of RET and NTRK1 as well as PAX8/PPARγ and AKAP9-BRAF were also assessed by RT-PCR (Supplemental Table 2). We used tumor cDNA as template for quantitative PCR to analyze for unbalanced expression of exons 10 to 11 relative to 12 to 13 of RET, which flank the rearrangement site in intron 11. Samples with RET 10–11 < 12–13 expression were then screened for specific RET recombination events using primers bracketing the respective fusion points of RET/PTC1, RET/PTC2, and RET/PTC3. Positive controls were cDNAs from TPC1 cells (that express RET/PTC1), PCCL3 cells expressing RET/PTC2, and a PTC sample expressing RET/PTC3. We screened for PAX8/PPARγ, AKAP9-BRAF, and NTRK1 fusions by RT-PCR using specific primers as previously described (17, 18). The primers used for this analysis are shown in Supplemental Table 2. cDNAs from tumor samples harboring each one of these rearrangements were used as positive controls. GAPDH was used as an internal control.
Comparative genomic hybridization analysis
We used the Agilent 1M array comparative genomic hybridization (CGH) platform for global copy number analysis (www.agilent.com/genomics) according to the manufacturer's instructions. The data were normalized, and then the Cy5 to Cy3 ratio for each probe was expressed as log2(Cy5/Cy3). Normalized data were then analyzed using RAE, a computational segmentation framework that adapts parameters to individual tumors, and then identifies statistically significant regions of interest across tumors (19). Candidate copy number alterations are based on a false discovery rate (FDR) Q-value. Regions with FDR ≤10% were plotted. Analysis and scores were calculated as described (19). These regions were then compared with known copy number alterations in PTC and FTC from published array CGH data. Clustering analysis of the copy number data was done using GenePattern.
Gene expression profiling
All gene expression analyses were performed with the HG-U133A Plus version 2.0 (Affymetrix, Santa Clara, California) array as described by the manufacturer. The microarray data were quantile-normalized (20), and the gene expression values were estimated using the robust multi-array average method (21). Moderated t statistics (22) were used to test whether genes were differentially expressed between the groups of interest. P value < .001 was considered statistically significant. Genes with P values < .001 were used for hierarchical clustering analysis. The expression values for each gene were centered and scaled before analysis. Complete-link algorithm was used for clustering, and Pearson correlation coefficient was used as the similarity distance.
Comparisons of expression profiles for HA vs HMIN, HA vs HWIDE, and HMIN vs HWIDE were performed (Supplemental Tables 3–5) and a hierarchical clustering analysis for genes with P values < .001 and a fold change differential of ×1.8 performed. For the HA vs HWIDE comparison, select genes of significance were internally validated using real-time PCR with the iCycler System (Bio-Rad Instruments, Hercules, California) using SYBR green detection. Primers for each gene were designed using Primer-Blast (www.ncbi.nlm.nih.gov/BLAST/) and are shown in Supplemental Table 6 with optimum annealing temperature. Quantitative RT-PCR analysis was performed using the ΔΔCt method (23). As a reference gene, the housekeeping gene GADPH was amplified using the primers forward 5′-AAGGTGAAGGTCGGAGTCAA-3′ and reverse 5′-AATGAAGGGGTCATTGATGG-3′ and an annealing temperature of 60°C.
Concept module mapping with Oncomine, Gene Set Enrichment Analysis (GSEA), and Ingenuity Pathway Analysis
Concept module mapping was performed as follows. The differential gene expression signature identified from our analysis for HA vs HWIDE, HA vs HMIN, and HMIN vs HWIDE (P value < .001 and differential fold change of ×1.8) was imported into Oncomine (http://www.oncomine.org) to search for associations with molecular concept signatures derived from independent cancer profiling studies. We report statistically significant overlaps of our gene expression signature with the top-ranking gene expression signatures of clinical outcome using percentile cutoffs (10%). Q-value is calculated as previously described (24). GSEA was performed with GSEA software version 2.0.7 (www.broadinstitute.org/cancer/software/gsea). We assessed the significance of the gene sets with the following parameters: number of permutations = 1000, and permutation type = phenotype with an FDR Q-value cutoff of 25%. The most differentially expressed genes from statistically significant gene sets were identified with the leading edge subset that consists of genes with the most contribution to the enrichment score of a particular gene set. For Ingenuity Pathway Analysis, the same differential gene expression signature for HA vs HWIDE was imported into IPA (http://ingenuity.com/) and then correlated with genes associated with vascular invasion to search for interacting genes.
Comparison of transcriptomes of HWIDE, PTC, and FTC tumors
Using GEO (gene expression omnibus: http://www.ncbi.nlm.nih.gov/geo/), expression array profiles for PTC (25) and FTC (26) were obtained and then compared with the transcriptomes for the HWIDE profile. Datasets were identified using the same Affymetrix platform (HG-U133A and 2.0) and are shown in Supplemental Table 7.
Using published gene expression profiles for normal thyroid tissue, the differential gene expression profiles for PTC, FTC, and HWIDE were compared with normal thyroid. To reduce any potential batch effect, the expression data were first normalized, and then the differential gene expressions identified with Significance Analysis of Microarray (SAM) (27) using MultiExperiment Viewer (MeV) with 100 permutations. The complete lists of genes upregulated and downregulated for PTC, FTC, and HWIDE compared with normal are shown in Supplemental Tables 8, 9, and 10. Differentially expressed genes were used to identify the key pathways.
Immunohistochemistry
The 5-μm sections from each tissue block were deparaffinized and subjected to the following procedures. For phospho-AKT, the sections were subjected to a heat-induced epitope retrieval for 30 minutes with citrate buffer (pH 6) and then incubated overnight at 4οC with the phospho-AKT (Ser473)(736E11) monoclonal antibody (catalog item 3787; Cell Signaling Technology, Danvers, Massachusetts) at 1:25 dilution. The procedure was then carried out manually using a rabbit secondary antibody (Vector Laboratories, Burlingame, California) diluted at 1:500 (60 minutes at room temperature) followed by the avidin-biotin complex (ABC) method as a detection system (30 minutes at room temperature) and 3,3′-diaminobenzidine (DAB) as a chromogen. For β-catenin, β-catenin (clone 14) monoclonal antibody (catalog item 760-4242; Cell Marque, Rocklin, California) prediluted was used. The procedure was carried out on the Ventana Discovery XT semiautomated staining system using with CC1-standard pretreatment time (Ventana Medical Systems, Oro Valley, Arizona). The Chromo Map DAB detection system was used (catalog item 760-159; Ventana Medical Systems).
Results
Somatic mutations in Hurthle tumors identified by Sequenom-based genotyping and gene rearrangement analysis
Figure 1C shows the genetic alterations identified in the Hurthle cell tumors (n = 27) compared with reported genetic alterations in other types of thyroid cancer. Sequenom analysis showed no Hurthle cell tumor had a BRAF mutation and there were no RET/PTC rearrangements. Because HAs and HCCs are thought to be oncocytic variants of follicular cancer, we expected to detect a high proportion of RAS mutations or Pax8-PPARγ rearrangements (18, 28–30). However, only 3 of 27 tumors (11%) had a RAS mutation, 1 HMIN (NRASQ61R) and 2 HWIDE tumors (NRASQ61R and NRASQ61K) (Figure 1, A and B), and there were no Pax8-PPARγ rearrangements. Mutations in PI3KCA and PTEN, which occur in 10% to 30% and 10% of FTCs, respectively, were also not detected.
Figure 1.

A, Summary of mutations detected by Sequenom genotyping in HA, HMIN, and HWIDE tumors. Among the mutations known to exist in PTC and FTC, only NRAS mutations were identified in Hurthle cell tumors. Codon changes are noted. B, Example of NRAS mutation detected by Sequenom genotyping in Hurthle cell tumor HWIDE2. Peaks represent MassARRAY intensity at the site of mutation. C, Summary of genetic alterations in Hurthle cell tumors compared with other types of thyroid cancers (PTC, FTC, poorly differentiated thyroid cancer, and anaplastic thyroid cancer).
Global copy number diversity in Hurthle tumors
Regions of chromosomal gain and loss are shown in Supplemental Table 11 (a more detailed list is shown in Supplemental Table 12 and Supplemental Gene Tables 1–6). This table also lists genes and microRNAs for each significant chromosomal region. Genomic regions showing amplification were chromosomes 4p16, 5p15-5q35,6p25, 7p15-7q36, 8p21-23, 10p13, 12p13-q24, 16q23, and 17p13-q25. Chromosomal regions of loss included 4q24, 6p23, 7p15, 9q33, 13q14, and 16q23. In particular, there were very large regions of amplification on chromosome 5 (174 gene region and 650 gene region), chromosome 7 (128 gene region and 776 gene region), chromosome 12 (995 genes), and chromosome 17 (471 gene region, 322 gene region, 59 gene region, 252 gene region). The RAE plot showing amplified and deleted genes of interest is shown in Supplemental Figure 1. A summary of chromosomal regions of gain and loss in HCC compared with PTC and FTC is shown in Figure 2. Regions of gain are shown in red, and regions of loss are shown in green.
Figure 2.

Summary of chromosomal regions of gain and loss in HCC compared with PTC and FTC. Regions of gain are shown in red, and regions of loss are shown in green.
A number of chromosomes demonstrated increases in number, including chromosomes 5, 7, 10, 12, 17, and 20, suggesting the presence of either chromosomal instability or selection for increased copies of these chromosomes. Clustering analysis of the copy number data revealed 3 main groups of Hurthle tumors (Figure 3). These were tumors where large regions of amplification were predominant (subtype 3), tumors where deletions were predominant (subtype 1), and tumors with a mix of focal chromosomal alterations (subtype 2). Four widely invasive tumors were subtype 3, 5 were subtype 2, and 1 was subtype 1. The 3 tumors that were clinically more aggressive with distant metastases were of subtype 2. We analyzed all 22 chromosomes to determine whether individual chromosomal gains/losses could differentiate HA, HMIN, and HWIDE. The differential pattern for all chromosomes closely resembled the clustering pattern observed for all 22 autosomes. We were unable to identify any pattern of chromosomal gains/losses that could specifically differentiate HA from HMIN and from HWIDE.
Figure 3.
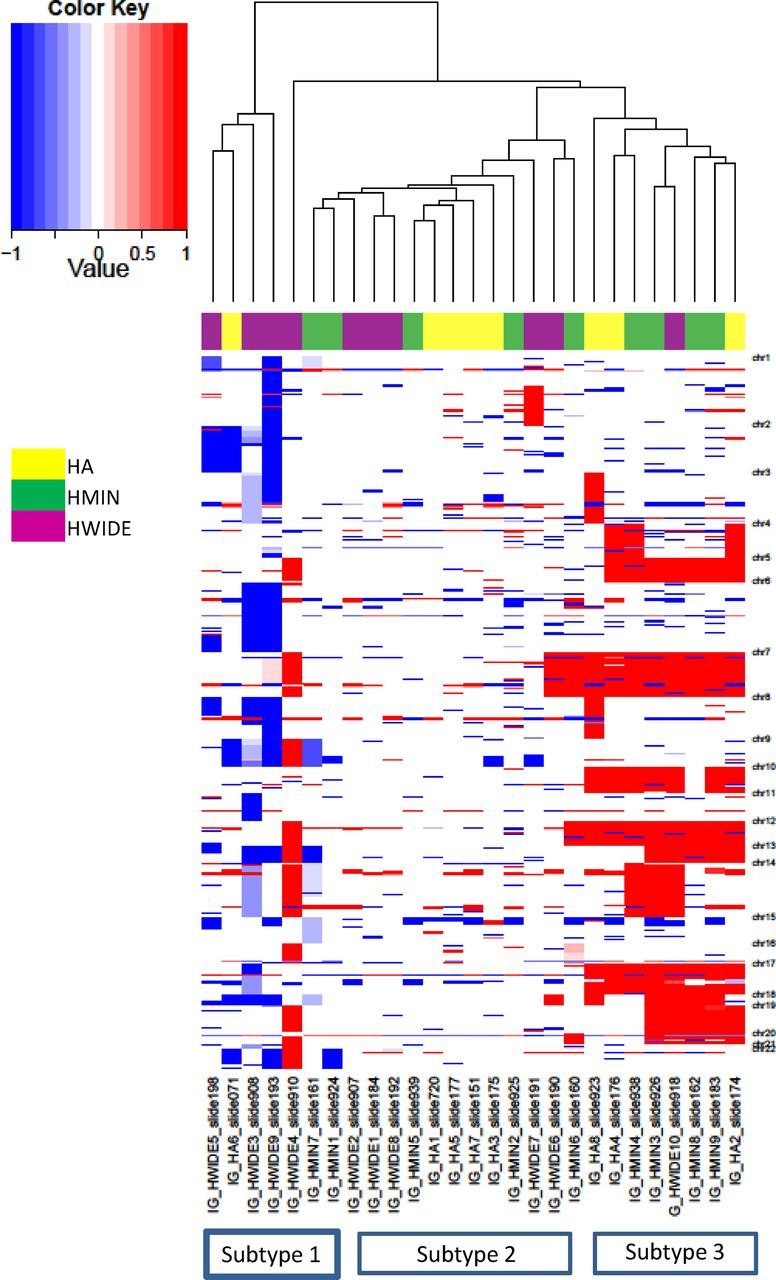
Diversity of the Hurthle cell tumor copy number landscape. Clustering of copy number alterations for each tumor. Amplifications (red) and deletions (blue) are indicated across the 22 autosomes. Three main groups of Hurthle tumors are identified: tumors where large regions of amplification are predominant (subtype 3 shown on the right), tumors where deletions are predominant (subtype 1 shown on the left), and tumors with more focal chromosomal alterations (subtype 2).
Transcriptomes of Hurthle cell tumors reveal differentially active pathways
Principal component analysis using the Partek Genomics Suite (http://www.partek.com/software) was carried out to determine the similarity of HA, HMIN, and HWIDE tumors (Figure 4A). The HMIN tumors as a group were very similar to the HA tumors. In contrast, the HWIDE tumors clustered away from the HA and HMIN groups, although there were 3 tumors similar to the HMIN group. This indicated that the HWIDE tumors were quite different in their transcriptional profiles from each other and from both HA and HMIN.
Figure 4.

A, Principal component analysis of expression data derived from Hurthle cell tumors. The subclasses HA, HMIN, and HWIDE are noted in the color legend. B, Heatmap showing differential gene expression between HA, HMIN (Min Inv Ca), and HWIDE (Widely Inv Ca). HMIN shows a similar profile to HA, whereas HWIDE shows a more divergent expression profile. Histogram from clustering analysis is shown at the top. Relative expression is noted in the legend.
Unsupervised hierarchical clustering (Figure 4B) also showed that the 3 groups of tumors clustered separately from each other with some differences between the HA and HMIN tumors but a more marked difference between HWIDE and the other 2 groups. Supplemental Table 13 shows the number of differentially expressed genes between HA, HMIN, and HWIDE tumors. The complete list of differentially expressed genes with a P value < .001 for HA vs HMIN, HA vs HWIDE, and HMIN vs HWIDE are shown in Supplemental Tables 3–5. Because the greatest number of gene expression differences were in the HA vs HWIDE comparison (henceforth called the HWIDE signature), we focused further analysis on this dataset. This comparison showed 403 gene expression differences; 324 genes were underexpressed in the HWIDE tumors and 79 genes overexpressed compared with the HA tumors. Select genes were validated by RT-PCR. Genes selected for validation were PFKFB2, CTNNB1, PRO1073, SULT131, MALAT1, ITCH, RHOU, TXN, ZNF567, SHB, AYTL1, and GATA6. Primers and PCR conditions used for RT-PCR validation are shown in Supplemental Table 6 and Supplemental Figures 2 and 3.
Genetic programs involving vascular invasion and β-catenin in Hurthle cell tumor subsets
Concept module mapping of the HWIDE signature using Oncomine showed that there were several strongly significant gene sets that were enriched (Supplemental Table 14). The top program was a CTNNB1 (β-catenin)-driven signature resulting from expression of β-catenin (31) (Q-value = 1.5 × 10−23) (Figure 5 and Supplemental Table 15). This implicates β-catenin in HCC oncogenesis.
Figure 5.

Correlation of HWIDE signature with select gene sets in Oncomine database. The significant gene overlap is represented by −log of the P value. CTNNB1 transfection, outlined in red, has the most significant P value with a −log P value of 27.7. The complete concept datasets are shown in Supplemental Table 13.
To determine the molecular context of β-catenin pathway enrichment, we made use of Ingenuity Pathway Analysis. Analysis of genes involved in vascular invasion with the HWIDE signature showed that β-catenin is intimately involved in processes regulating vascular invasion. Supplemental Figure 4, A and B, shows the results from this analysis. Another concept highlighted from Oncomine analysis was the temsirolimus-sensitive signature (31) (genes relating to temsirolimus sensitivity), which had 60 overlapping genes with a significant Q-value of 1.2 × 10−8. Temsirolimus is a mammalian target of rapamycin (mTOR) inhibitor and recent phase I studies at our institute have shown patients with distant metastases from widely invasive HCC responded to this drug (unpublished data). The gene set was also analyzed using GSEA and the top datasets with significant overlapping genes are shown in Supplemental Table 16. There is also significant overlap with genes upregulated in HepaRG cells (liver cancer) expressing active forms of mTOR (23-gene overlap; P = 5.08 × 10−6). This again was evidence that mTOR may be important in HCC pathogenesis. Significant overlap with genes downregulated in poorly differentiated thyroid cancer (31-gene overlap; P = 1.6 × 10−7) and genes downregulated in anaplastic thyroid cancer (18-gene overlap; P = 3.4 × 10−4) were also found. Concept analysis of the HMIN gene signature (HMIN vs HA) using Oncomine (Supplemental Table 17a) and GSEA (Supplemental Table 17b) showed different concepts compared with the HWIDE signature. This provides some evidence that the progression of HCC from HA to HMIN and HWIDE may not be a stepwise progression model. In contrast, concept analysis of HWIDE vs HMIN showed similar concepts to the HWIDE vs HA differential expression set (Supplemental Table 18, a and b). Additional concepts of interest identified included sorafenib, torcetrapib, pazopanib, and foratinib sensitivity signatures.
Comparison of transcriptomes of HWIDE, PTC, and FTC tumors
Principal component analysis (Figure 6A) and hierarchical clustering (Figure 6, B and C) showed the transcriptome for PTC and FTC was very different from that of HWIDE. Distinct clusters were observed consisting of HCC, PTC, or FCC, but not groups with mixed histology. This provides further evidence that HWIDE tumors have a very different molecular profile from that of PTC and FTC.
Figure 6.

A, Principal component analysis of expression data of HWIDE tumors compared with PTC and FTC from GEO (gene expression omnibus) using Partek Genomics Suite. B, Hierarchical clustering dendrogram of HWIDE expression profile compared with FTC and PTC samples. C, Heatmap showing differential gene expression between HWIDE expression profile compared with FTC and PTC samples from GEO. Tumors are arranged along the top and genes along the left.
Differentially expressed genes for PTC, FTC, and HWIDE compared with normal thyroid were used to identify the key pathways in each tumor. The principal pathways enriched in PTC, FTC, and HWIDE are shown in Supplemental Tables 19, 20, and 21 and Supplemental Figure 5. Comparison of these tumor groups showed activity of fundamentally different biological processes. Interestingly, HCCs possess strong phosphoinositide 3-kinase (PI3K)/Akt signaling and Wnt/β-catenin signatures.
Discussion
The objective of our study was to dissect the mutational, cytogenetic, and gene expression changes that are present in both benign and malignant Hurthle cell tumors and compare these alterations to the other more common thyroid malignancies such as PTC and FTC.
In PTC, the predominant genetic alterations are activating mutations of the BRAF gene encoding the B type Raf kinase (32) occurring in 40% to 49% of patients with classical PTC (33). In our analysis of 27 Hurthle cell tumors, we identified no BRAF mutations. The second most common genetic alteration in PTC are chromosomal RET rearrangements occurring in 20% to 30% of PTCs. At least 15 different RET hybrid oncogene variants have been described (RET/PTC 1–12, 1L, 3r2, and 3r3). The variants RET/PTC1 and RET/PTC3 frequently occur in conventional PTC and postradiation PTC, the others are more rare. Of note is the fact that BRAF mutations and RET/PTC rearrangements are mutually exclusive (32, 34). In our study, we found no RET/PTC rearrangements.
In follicular adenoma and carcinoma, one of the most frequent genetic mutations is RAS mutation, which activates the RAS-RAF-MEK pathway. This has been reported to occur in 45% of follicular tumors, whereas it is relatively rare (10%) in PTC. The World Health Organization classification of thyroid malignancy currently classifies Hurthle cell tumors as an oncocytic variant of follicular adenoma and carcinoma (10), and therefore we may expect RAS mutations to occur in Hurthle cell tumors with a similar frequency. However, in our study, only 3 of 27 tumors had a RAS mutation. Talini et al (35) have also reported that H-, K-, and NRAS mutations were uncommon in Hurthle cell tumors. Other mutations that have been reported to occur in follicular carcinoma are PIK3CA (10%–30%); these were also not detected in our Hurthle tumors. PPARγ rearrangements, reported to occur in 25% to 60% of follicular cancers, were also not detected in our Hurthle cell tumors. Therefore, our targeted mutational analysis, focusing on the most important mutations occurring in thyroid carcinogenesis, suggests Hurthle cell tumors are a different class of thyroid malignancy at the genetic level. However, our mutational analysis using the Sequenom platform may have some limitations because we focused only on the most important mutations reported to occur in thyroid carcinogenesis. It is therefore possible other mutations may have been missed using this technique.
We then investigated the global copy number alterations that occur in Hurthle cell tumors. Figure 2 shows a summary of common regions of gain and loss in PTC (36, 37), FTC (38, 39), and HCC. The patterns of chromosomal aberrations in HCC were quite different from that observed in PTC and FTC. From our data, the most striking feature was large regions of gain on chromosomes 5, 7, 12, and 17. Chromosomal instability could explain the CGH findings in our tumors. Clustering analysis of the copy number data revealed 3 main groups of Hurthle tumors; these were tumors where large regions of amplification were predominant (subtype 3), tumors where deletions were predominant (subtype 1), and tumors with a mix of focal chromosomal alterations (subtype 2). This suggests that these groups have distinct molecular etiologies and could be consistent with a model in which Hurthle tumors develop either into HMIN or HWIDE in a de novo fashion rather than through a stepwise progression model where all HWIDEs develop from preexisting HMINs. This finding may have particular clinical significance because it suggests that a progression from minimal to widely invasive phenotype is not a primary manifestation of this malignancy. However, due to the small number of tumors involved in our study, we cannot completely discount that a progression model may still exist.
To determine whether there were any similarities between HWIDE and PTC and FTC at the transcriptome level, principal component analysis and hierarchical clustering was carried out using expression data for PTC and FTC from GEO. This showed no correlation between HWIDE and PTC or FTC. However, these data should be interpreted with caution because the invasiveness of the PTC and FTC tumors used in the comparison are not known (T stage, vascular invasion, or extrathyroid extension). Therefore, the 3 tumor types may not be matched and thus the differences we observed may not be due to differences in histology. Using normal thyroid tissue as a control, the differential gene expression profiles and resultant pathway analysis showed that HWIDE tumors were quite different from PTC and FTC. This again was further confirmatory evidence that could suggest that HCC is a unique thyroid malignancy. We have therefore shown by targeted analysis of mutations, global copy number analysis, and expression profiling that HCC is different from the other common types of thyroid cancer.
Our data provide us with some ideas on possible mechanisms for HCC development. In thyroid carcinogenesis, the 2 main signaling pathways are the RAS-RAF-MEK and the PI3K-AkT-mTOR pathways. The large amplification on chromosomes 7 and 12 could potentially activate the RAS-RAF-MEK pathway by amplification of the BRAF gene located on chromosome 7 or amplification of KRAS located on chromosome 12. In our dataset, amplification of BRAF and KRAS was observed in 12 and 10 tumors, respectively. Amplification of BRAF, rather than activating mutations, has been reported as a potential mechanism of activation by Ciampo et al (40). Amplification of BRAF has also recently been reported as a mechanism of resistance to melanomas sensitive to BRAF inhibitor therapy (41). Amplification of chromosome 10, which harbors the RET gene, was amplified in 10 tumors, and this could also be a mechanism for activation of the RAS-RAF-MEK pathway. The large amplifications seen on chromosomes 5, 7, 12, and 17 could also potentially activate the PI3K-AkT-mTOR pathway by amplification of PIK3CG (chromosome 7-amplified in 12 tumors), AGAP2 (chromosome 12-amplified in 10 tumors), RAPTOR (chromosome 17-amplified in 8 tumors), RICTOR (chromosome 5-amplified in 8 tumors), RHEB (chromosome 7-amplified in 11 tumors), GOLPH3 (chromosome 5-amplified in 8 tumors), and MDM2 (chromosome 12-amplified in 10 tumors). Other genes amplified included EGFR (chromosome 12-amplified in 12 tumors), CDK4 (chromosome 12-amplified in 10 tumors), and ERBB2 (chromosome 17-amplified in 8 tumors). Indeed, of the 10 most significant amplifications reported in human cancers by Beroukhim et al (42), Hurthle tumors exhibited amplification of 5 of these genes (namely ERBB2, CDK4, MDM2, EGFR, and KRAS). Clearly, additional studies are needed to examine these possibilities. We have cross-matched the genes amplified on CGH with the expression profiling data. Five of 12 tumors with BRAF amplification and 4 of 10 tumors with KRAS amplification had high mRNA expression (Supplemental Figure 6). In the PI3K-AkT-mTOR pathway, 5 of 8 tumors with amplification of RICTOR, 4 of 8 tumors with amplification of GOLPH3, and 4 of 10 tumors with amplification of MDM2 had high mRNA expression. Not all tumors with copy number gain on the affected chromosomes had increases in expression, suggesting that copy number is one of many qualities that can affect gene expression. This relationship between overexpression and broad areas of amplification has been well documented in the literature. Alternatively, some of the increases in expression may be small and not particularly well represented via microarray.
The expression data analysis also highlights the importance of the PI3K-AkT-mTOR pathway. Concept analysis of the HWIDE gene signature using Oncomine and GSEA showed a very significant overlap with the mTOR pathway as well as with a temsirolimus-sensitive signature (genes relating to temsirolimus sensitivity). Ingenuity Pathway Analysis of the differential gene expression profile for HWIDE tumors to normal thyroid tissue also showed that the PI3K-AkT-mTOR pathway was highly active. This would be consistent with the copy number alterations on CGH. We have further assessed the PI3K-AkT-mTOR pathway using immunohistochemistry to assess expression of phospho-AKT for tumors that had amplification of genes RICTOR, AKT2, or GOLPH3 (Supplemental Figure 7). Of 6 tumors assessed, all had overexpression of phospho-AKT. Thus, activation of the mTOR pathway would appear to be a potential mechanism in HCC development. Oncomine also showed significant overlap with a bortezomib-sensitive signature (Q = 6.9 × 10−11) and topetecan-sensitive signature (Q = 7.9 × 10−13). β-Catenin may also be important because we saw a significant overlap with the gene expression profile from β-catenin–transfected human epithelial mammary cell line (31). Mutation of the β-catenin gene in thyroid cancer has been reported in anaplastic thyroid cancer (60%) and also in poorly differentiated thyroid cancer (25%) (43). Further evidence for the potential importance of β-catenin came from Ingenuity Pathway Analysis comparing HWIDE gene signature with genes involved in vascular invasion, a hallmark of widely invasive cancer. β-Catenin is well known to play a role in vascular invasion (44). This suggests that β-catenin may play a central role in regulating the differences in vascular phenotype that is the hallmark of HWIDE tumors, providing a molecular rationale for a pathological hallmark of HCCs.
In conclusion, our data provide evidence that HCC may be a unique thyroid cancer distinct from PTC and FTC. Our study comprises one of the most comprehensive integrated analyses to date of this relatively rare type of thyroid cancer. Clearly, further detailed analysis of the PI3K-AkT-mTOR and Wnt/β-catenin pathways in HCC development are warranted to identify possible therapeutic targets.
Acknowledgments
We would like to thank Agnes Viale and the Memorial Sloan Kettering Genomics Core for excellent technical support.
This work was supported by Dr Peter Scardino (Department of Surgery, MSKCC) and by Mr Richard Pechter of the Pechter Foundation. Special thanks goes to Mr Pechter and Dr Scardino for providing encouragement and inspiration for this work. Funding was also provided by the Louis Gerstner Foundation (T.A.C.).
Disclosure Summary: All authors have no conflicts of interest.
Footnotes
- CGH
- comparative genomic hybridization
- FDR
- false discovery rate
- FTC
- follicular thyroid carcinoma
- GSEA
- Gene Set Enrichment Analysis
- HA
- Hurthle cell adenoma
- HCC
- Hurthle cell cancer
- HMIN
- minimally invasive HCC
- HWIDE
- widely invasive HCC
- mTOR
- mammalian target of rapamycin
- PI3K
- phosphoinositide 3-kinase
- PTC
- papillary thyroid carcinoma.
References
- 1. Hundahl SA , Fleming ID , Fremgen AM , Menck HR. A National Cancer Data Base report on 58,856 cases of thyroid carcinoma treated in U.S., 1985–1995. Cancer. 1998;83:2638–2648. [DOI] [PubMed] [Google Scholar]
- 2. Montone KT , Baloch ZW , LiVolsi VA. The thyroid Hürthle (oncocytic) cell and its associated pathologic conditions: a surgical pathology and cytopathology review. Arch Pathol Lab Med. 2008;132:1241–1250. [DOI] [PubMed] [Google Scholar]
- 3. Ghossein RA , Hiltzik DH , Carlson DL , et al. Prognostic factors of recurrence in encapsulated Hurthle cell carcinoma of the thyroid gland: a clinicopathologic study of 50 cases. Cancer. 2006;106(8):1669–1676. [DOI] [PubMed] [Google Scholar]
- 4. Shaha AR , Shah JP , Loree TR. Patterns of nodal and distant metastasis based on histologic varieties in differentiated carcinoma of the thyroid. Am J Surg. 1996;172:692–694. [DOI] [PubMed] [Google Scholar]
- 5. Grossman RF , Clark OH. Hurthle cell carcinoma. Cancer Control. 1997;4:13–17. [DOI] [PubMed] [Google Scholar]
- 6. Lopez-Penabad L , Chiu AC , Hoff AO , et al. Prognostic factors in patients with Hürthle cell neoplasms of the thyroid. Cancer. 2003;97:1186–1194. [DOI] [PubMed] [Google Scholar]
- 7. Besic N , Vidergar-Kralj B , Frkovic-Grazio S , et al. The role of radioactive iodine in the treatment of Hürthle cell carcinoma of the thyroid. Thyroid. 2003;13:577–584. [DOI] [PubMed] [Google Scholar]
- 8. Carcangiu ML , Bianchi S , Savino D , Voynick IM , Rosai J. Follicular Hurthle cell tumors of the thyroid gland. Cancer. 1991;68:1944–1953. [DOI] [PubMed] [Google Scholar]
- 9. Kushchayeva Y , Duh QY , Kebebew E , Clark OH. Prognostic indications for Hürthle cell cancer. World J Surg. 2004;28:1266–1270. [DOI] [PubMed] [Google Scholar]
- 10. DeLellis RA , Lloyd RV , Heitz PU. World Health Organization Classification of Tumors. Pathology and Genetics: Tumors of Endocrine Organs. Lyon, France: IARC Press; 2004:69–72. [Google Scholar]
- 11. Maximo V , Sobrinho-Simoes M. Mitochondrial DNA 'common' deletion in Hürthle cell lesions of the thyroid. J Pathol. 2000;192:561–562. [DOI] [PubMed] [Google Scholar]
- 12. Máximo V , Botelho T , Capela J , et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. Br J Cancer. 2005;92:1892–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Musholt PB , Musholt TJ , Morgenstern SC , Worm K , Sheu SY , Schmid KW. Follicular histotypes of oncocytic thyroid carcinomas do not carry mutations of the BRAF hot spot. World J Surg. 2008;32(5):722–728. [DOI] [PubMed] [Google Scholar]
- 14. Musholt PB , Imkamp F , von Wasielewski R , Schmid KW , Musholt TJ. RET rearrangements in archival oxyphilic thyroid tumors:new insights in tumorigenesis and classification of Hurthle cell carcinomas? Surgery. 2003;134:881–889. [DOI] [PubMed] [Google Scholar]
- 15. Thyroid carcinomas (version 2012). In: Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network Web site. www.nccn.org Accessed December 2012.
- 16. Ricarte-Filho JC , Ryder M , Chitale DA , et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69(11):4885–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beimfohr C , Klugbauer S , Demidchik EP , Lengfelder E , Rabes HM. NTRK1 re-arrangement in papillary thyroid carcinomas of children after the Chernobyl reactor accident. Int J Cancer. 1999;March 15;80(6):842–847. [DOI] [PubMed] [Google Scholar]
- 18. Nikiforova MN , Biddinger PW , Caudill CM , et al. PAX8-PPARγ rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol. 2002;26(8):1016–1023. [DOI] [PubMed] [Google Scholar]
- 19. Taylor BS , Barretina J , Socci ND , et al. Functional copy-number alterations in cancer. PLoS One. 2008;September 11;3(9):e3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolstad BM , Irizarry RA , Astrand M , Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics. 2003;19(2):185–193. [DOI] [PubMed] [Google Scholar]
- 21. Irizarry RA , Bolstad BM , Collin F , Cope LM , Hobbs B , Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article 3. [DOI] [PubMed] [Google Scholar]
- 23. Silver N , Best S , Jiang J , Lay S. Selection of housekeeping genes for gene expression studies inhuman reticulocytes using real-time PCR. BMC Mol Biol. 2006;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rhodes DR , Kalyana-Sundaram S , Tomlins SA , et al. Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia. 2007;9:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He H , Jazdzewski K , Li W , et al. The role of microRNA genes in papillary thyroid cancer. Proc Natl Acad Sci U S A. 2005;102(52):19075–19080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giordano TJ , Au AY , Kuick R , et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clin Cancer Res. 2006;12(7 Pt 1):1983–1993. [DOI] [PubMed] [Google Scholar]
- 27. Tusher VG , Tibshirani R , Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeGellis RA. Pathology and genetics of thyroid carcinoma. J Surg Oncol. 2006;94:662–669. [DOI] [PubMed] [Google Scholar]
- 29. Esapa CT , Johnson SJ , Kendall-Taylor P , Lennard TW , Harris PE. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol (Oxf). 1999;50(4):529–535. [DOI] [PubMed] [Google Scholar]
- 30. Kroll TG , Sarraf P , Pecciarini L , et al. PAX8-PPARγ1 fusion oncogene in human thyroid carcinoma. Science. 2000;289(5483):1357–1360. [DOI] [PubMed] [Google Scholar]
- 31. Bild AH , Yao G , Chang JT , et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. [DOI] [PubMed] [Google Scholar]
- 32. Kimura ET , Nikiforova MN , Zhu Z , Knauf JA , Nikiforov YE , Fagin JA. High prevalence of BRaf mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- 33. Lee JH , Lee ES , Kim YS. Clinicopathologic significance of BRAF V600E mutation in papillary carcinoma of the thyroid: a metanalysis. Cancer. 2007;110:38–46. [DOI] [PubMed] [Google Scholar]
- 34. Soares P , Trovisco V , Rocha AS , et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578–4580. [DOI] [PubMed] [Google Scholar]
- 35. Talini G , Hsueh A , Liu S , Garcia-Rostan G , Speicher MR , Ward DC. Frequent chromosomal imbalance in thyroid oncocytic (Hurthle cell) neoplasms detected by comparative genomic hybridisation. Lab Invest. 1999;79:547–555. [PubMed] [Google Scholar]
- 36. Singh B , Lim D , Cigudosa JC , et al. Screening for genetic aberrations in papillary thyroid cancer by using comparative genomic hybridization. Surgery. 2000;128:888–894. [DOI] [PubMed] [Google Scholar]
- 37. Wreesmann VB , Sleczka EM , Socci ND , et al. Genome wide profiling of papillary thyroid cancer identifies MUC1 as an independent prognostic marker. Cancer Res. 2004;64:3780–3789. [DOI] [PubMed] [Google Scholar]
- 38. Hemmer S , Wasenius VM , Knuutila S , Franssila K , Joensuu H. DNA copy number changes in thyroid cancer. Am J Pathol. 1999;154:1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frisk T , Kytola S , Wallin G , Zedenius J , Larsson C. Low frequency of numerical chromosomal aberrations in follicular thyroid tumors detected by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25:349–353. [PubMed] [Google Scholar]
- 40. Ciampo R , Zhu Z , Nikiforov YE. BRAF copy number gains in thyroid tumors detected by fluorescence in siti hybridisation. Endocr Pathol. 2005;16:99–105. [DOI] [PubMed] [Google Scholar]
- 41. Shi H , Moriceau G , Kong X , et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beroukhim R , Mermel CH , Porter D , et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Rostan G , Tallini G , Herrero A , D'Aquila TG , Carcangiu ML , Rimm DL. Frequent mutation and nuclear localization of β-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811–1815. [PubMed] [Google Scholar]
- 44. Suzuki H , Masuda N , Shimura T , et al. Nuclear β-catenin expression at the invasive front and in the vessels predicts liver metastasis in colorectal carcinoma. Anticancer Res. 2008;28(3B):1821–1830. [PubMed] [Google Scholar]