

Abstract
Expression of the Helicobacter pylori blood group antigen binding adhesin A (BabA) is more common in strains isolated from patients with peptic ulcer disease or gastric cancer, rather than asymptomatic colonization. Here we used mouse models to examine host determinants that affect H. pylori BabA expression. BabA expression was lost by phase variation as frequently in WT mice as in RAG2−/− mice that do not have functional B or T cells, and in MyD88−/−, TLR2−/− and TLR4−/− mice that are defective in toll like receptor signaling. The presence of other bacteria had no effect on BabA expression as shown by infection of germ free mice. Moreover, loss of BabA expression was not dependent on Leb expression or the capacity of BabA to bind Leb. Surprisingly, gender was the host determinant most associated with loss of BabA expression, which was maintained to a greater extent in male mice and was associated with greater bacterial load. These results suggest the possibility that loss of BabA expression is not driven by adaptive immunity or toll-like receptor signaling, and that BabA may have other, unrecognized functions in addition to serving as an adhesin that binds Leb.
Helicobacter pylori infects the gastric mucosa of about 50% of the world’s population1. The majority of those infected have only asymptomatic gastritis, but about 10% develop peptic ulcer and 1–3% develop gastric cancer1,2,3, which is the third most common cause of cancer death worldwide (~1 million cases per year). Given the large number of infected individuals, increasing development of antibiotic resistance4,5, and accumulating evidence that in some people H. pylori may be beneficial6,7,8,9, treatment of all infected individuals may not be warranted. Therefore, it is important to determine the elements of the host-H. pylori interaction that influence whether an individual will develop clinical disease or asymptomatic infection. One risk factor associated with more severe disease outcomes is the virulence factor, blood group antigen binding adhesin (BabA), which belongs to a family of H. pylori outer membrane proteins10 that also includes LabA11, SabA12, and the recently characterized HopQ13,14. BabA is a well-characterized adhesin15,16,17,18 that binds to ABO blood group antigens, fucosylated carbohydrates expressed on the gastric epithelium and the protective mucus layer. BabA exhibits highest affinity for Lewis b (Leb)19, owing to a polymorphic, three-pronged carbohydrate binding domain identified recently by X-ray structural analysis20,21. Epidemiologic studies of an association of BabA with disease22,23 are supported by in vitro evidence that BabA-mediated attachment to host gastric epithelium facilitates translocation of the CagA oncoprotein into host cells24. Translocation occurs via the type IV secretion system encoded on the cytotoxin associated gene pathogenicity island (cagPAI), itself a well-recognized risk factor for disease25,26,27,28,29.
BabA mediated attachment and development of disease are influenced by host expression of Lewis antigens, which is determined by a number of factors, including ABO blood type and secretor status30,31,32,33. The risk of ulcer is increased in individuals with blood group O, and in non-secretor individuals who do not express Leb and ABO antigens on gastric epithelial cells or on mucins30,31,32,33,34. Thus, disease outcome is related to both bacterial expression of the BabA adhesin and to ABO glycosylation on gastric epithelial cells and gastric mucins.
Previous studies have shown that H. pylori expression of BabA is lost during the first 2–12 weeks of infection using animal models such as rhesus macaques, gerbils, and mice35,36,37. Loss of BabA expression has been observed to occur by two mechanisms. In rhesus macaques, recombination between the babA gene and its babB paralog can result in duplication of all or part of the babB gene into the babA locus, resulting in loss of BabA expression and reciprocal overexpression of BabB. Alternatively, in mice and in macaques, slipped strand mispairing of a CT repeat region in the 5′ portion of the babA open reading frame (ORF) can lead to a frame shift, resulting in an early stop codon within the babA ORF and loss of BabA protein expression. That this phenomenon occurs across several animal models, and that loss of BabA expression can also be seen in human clinical isolates38, suggests that modulation of BabA expression is an important component of the H. pylori-host relationship.
Loss of BabA expression might occur as a bacterial mechanism of persistence to adapt to changing levels of inflammation, glycosylation patterns, or a combination of both. Here we used knockout and transgenic mouse models to determine the host factors that affect BabA expression. Surprisingly, the results suggest that adaptive immune responses and toll-like receptor signaling play little if any role in loss of BabA expression, and that the capacity to bind Leb is not required, but that gender- specific physiological differences may be important.
Methods
Bacterial strains and growth conditions
A complete list of bacterial strains used in this study is provided in Table 1. H. pylori cultures were maintained on Brucella agar (Becton, Dickinson and Company, Sparks, MD) supplemented with 5% heat inactivated newborn calf serum (Gibco, Grand Island, NY) and antibiotics (Sigma-Aldrich, Inc., St. Louis, MO), either TVPA (5 μg/mL trimethoprim, 10 μg/mL vancomycin, 2.5 units/mL polymyxin B and 2.5 μg/mL amphotericin B) for laboratory adapted cultures or ABPNV (100 μg/mL vancomycin, 3.3 μg/mL polymixin B, 200 μg/mL bacitracin, 10.7 μg/mL nalidixic acid and 10 μg/mL amphotericin B) for primary cultures from infected mice. All H. pylori cultures were maintained at 37 °C in a CO2 incubator or in an AnoxomatTM jar (Advanced Instruments, Inc., Norwood, MA) adjusted to contain 5% oxygen, 7.6% carbon dioxide, and 7.6% hydrogen39.
Table 1. Bacterial strains.
| Strain | Description | Antibiotic Resistancea | Source (Reference) |
|---|---|---|---|
| J166 | Wild Type | 35 | |
| J99 | Wild Type | 78 | |
| 26695 | Wild Type | 36,78 | |
| J166ΔbabAupstream | J166 with the upstream portion of babA replaced by CAT_rpsL (transformed with pBabA) | Cm | This study |
| J166sc | Single colony isolate of J166 | Str | This study |
| J1667CT | J166 engineered with 7 CT repeats in the 5′ end of babA | Str | This study |
| J1668CT | J166 engineered with 8 CT repeats in the 5′ end of babA | Str | This study |
| J166ΔbabA | J166 with babA replaced by CAT_rpsL | Cm | 20 |
| J166 BabACL2 | J166 with Cys 189 and 197 in BabA replaced with Ala | Str | 20 |
| J166 BabACL28CT | J166BabACL2 engineered to have 8 CT repeats in the 5′ end of babA, non-binding BabA is expressed | Str | This study |
| J166 BabACL29CT | J166BabACL2 engineered to have 9 CT repeats in the 5′ end of babA, BabA is not expressed | Str | This study |
aCm, chloramphenicol; Str, streptomycin.
Site directed mutagenesis
Isogenic mutants of H. pylori J166 were engineered with 7 (no ORF) or 8 (ORF) CT repeats in the 5′ coding region of babA using a contraselection approach modified from that previously described36. Briefly, 244 base pairs upstream of the babA translational start site and 39 base pairs of 5′ babA gene sequence were replaced with a CAT_rpsL cassette in a streptomycin resistant strain of H. pylori J166, using the plasmid pBabA as previously described39. The CAT_rpsL cassette was amplified using primers RpsLF and CamR (Table 2)36. The pBabA plasmid was constructed by ligating CAT_rpsL sequence between two PCR amplicons. The first was a 1211 bp amplicon containing hypD and intergenic sequence 244 bp upstream of babA in H. pylori J166 (primers HypDF and BabApromR, Table 2), and the second amplicon contained 996 bp of babA starting 39 bp downstream of the babA start site (primers BabAF and BabAR, Table 2). The resulting babA knockout strain was then transformed using genomic DNA from primary mouse output strains containing either 7 or 8 CT repeats in the 5′ coding region of babA. Transformants containing the desired number of CT repeats were selected by plating on streptomycin followed by replica plating to confirm loss of chloramphenicol resistance. The same approach was used to generate 8 (ORF) or 9 (no ORF) CT isogenic variants of J166 BabACL2, a site directed mutant of J166 in which Cys to Ala replacements at residues 189 and 197 result in a BabA protein that is expressed but cannot bind Leb 20. All mutants were sequenced using the method of Sanger to confirm the correct CT repeat structure.
Table 2. Primer sequences.
| Primer | Restriction Site | Sequence (5′-3′)a |
|---|---|---|
| Construction of pBabA | ||
| RpsLF | SacI | AAC GAGCTC GAT GCT TTA TAA CTA TGG ATT AAA CAC |
| CamR | BamHI | AAC GGATCC TTA TCA GTG CGA CAA ACT GGG AT |
| HypDF | NotI | AAC GCGGCCGC AGC CAC AAA ACC TCT AAA GA |
| BabApromR | SacI | AAC GAGCTC GGG GTA TTT TGA AAT AAC TCT C |
| BabAF | BamHI | AAC GGATCC TTG CTC CAC GCT GAA GAC |
| BabAR | XhoI | AAC CTCGAG GAC GCT CGT TTG ATT GAC CA |
| Leb genotyping | ||
| hGh-F | AGC TGG CCT TTG ACA CCT ACC AGG | |
| hGh-R | TCT GTT GTG TTT CCT CCC TGT TGG | |
| CT repeat length determination | ||
| BabAF14 | GCA TCA AGC AAG CGA TAA CTT TAC TAA | |
| BabARJC2 | TTT GCC GTC TAT GGT TTG G | |
aRestriction sites are underlined.
Mouse strains, housing and breeding
The local Institutional Animal Care and Use Committee approved all mouse experiments performed at Should read, University of California, Davis and the University of Michigan. Experiments performed at Umeå University, Umeå, Sweden were approved by Umeå Ethical Committee on Animal Research. All methods involving mice were performed according to the locally approved guidelines. Mice (Supplementary Table S1) were housed in sterilized, ventilated microisolator cages and given autoclaved water and irradiated food ad libitum as previously described36. FVB/N mice heterozygous for human α-1,3/4-fucosyltransferase gene (Leb transgenic mice)40 were bred to FVB/N WT mice and genotyped using tail snips obtained at weaning. DNA was extracted from tail snips by digestion in lysis buffer (50 mM KCl, 10 mM Tris, pH 8.5; 2 mM EDTA, Sigma-Aldrich, St. Louis, MO; 0.45% NP-40, Roche Diagnostics, Indianapolis, IN; 0.45% Tween-20, Bio-Rad, USA; and 1 mg/mL proteinase K, Roche Diagnostics, Indianapolis, IN) for 2 hr or overnight. Undigested fragments were removed by centrifugation (8,500 g for 10 min) and proteinase K was heat inactivated at 98 °C for 10 min. The resulting digest was used as template for PCR detection of human α-1,3/4-fucosyltransferase gene using primers hGh-F and hGh-R (Table 2), which were previously described41. Germ free C57BL/6 mice were raised and housed in soft-sided bubble isolators at the germ free mouse facility at the University of Michigan. Germ-free status was verified by aerobic and anaerobic cultures at least weekly, and all mice remained free of bacteria (other than the inoculated H. pylori strain) throughout the experiment.
Experimental H. pylori challenge and sample collection
H. pylori harvested from agar plates grown overnight (18–24 hr) was used to inoculate liquid cultures to an optical density A600 (OD) of 0.05 to 0.1. Liquid cultures were grown in Brucella broth (Becton, Dickinson and Company, Sparks, MD) supplemented with 5% NCS and TVPA shaking at 60–100 rpm overnight (18–24 hr) to an OD of 0.3 to 0.7. Bacteria from overnight liquid cultures were centrifuged (7,000 g for 10 min) and suspended in Brucella broth at approximately 1010 CFU/mL. For competition experiments where the inoculum contained equal parts of BabA expressing and deficient H. pylori, liquid cultures were grown to matching OD values prior to centrifugation and resuspension. In a subset of experiments, dilutions of the inoculum mixture were plated and 16 colonies were selected and sequenced at the 5′end of the babA locus to empirically determine the ratio of BabA expressing bacteria in the inoculum. On average the inoculum contained 45% BabA expressing bacteria with a standard deviation of 11%. This variability reflects both variation in the growth and survival of the cultures and technical variation in the detection of BabA expression in the output colonies. Mice were challenged at 10 to 14 weeks of age with 250 μL of suspension by oral gavage with a 20 gauge, 38mm animal feeding needle (Fisher Scientific)36. Mice were euthanized by intraperitoneal injection of 5 mg pentobarbitol-Na and 0.6 mg phenytoin-Na (Beuthanasia-D) and stomachs were removed and dissected into 2 to 4 longitudinal sections36,42. One quarter of the stomach tissue was placed in 10% phosphate buffered formalin (Fisher Scientific) for histological analysis and one half to one quarter was weighed and homogenized in Brucella broth for H. pylori culture and calculation of H. pylori colony forming units per gram of stomach tissue (CFU/g). The comparison between male and female mice was performed twice, as indicated in the figure legends and supplementary files. The remainder of the experiments was conducted exclusively in female mice.
Detection of BabA expression
In H. pylori J166, 8 CT repeats at the 5′ end of babA yield an ORF, with expression of BabA and attachment to Leb 36; 7 or 9 CT repeats does not because it produces a stop codon at position 49 or 79, respectively. Therefore, the proportion of BabA-expressing H. pylori in each mouse was determined by sequencing the CT repeat region of babA, which was selectively confirmed by RIA analysis using methods previously described36,43. Briefly, for CT analysis, a DNA fragment was sequenced from multiple individual colonies from each mouse after PCR amplification with primers BabAF14 and BabARJC2 (Table 2), which yields a 1046 bp product containing the CT repeats. On average 8 colonies were collected per mouse and analyzed in this way (minimum of 3 and maximum of 20). The percentage of the total colonies collected per mouse with a babA ORF is displayed as a single point in each figure describing BabA expression in mice. For RIA analysis, total cultured dilutions from mouse stomach homogenate (sweeps) were collected and 1 mL of an OD600 = 0.1 suspension of the mixture was incubated with a cocktail of 125I radiolabeled Leb conjugated to human serum albumin (Leb-HSA). The ratio of bound Leb-HSA (radioactivity measured in the bacterial pellet) to free Leb-HSA (radioactivity measured in the supernatant) was used to estimate BabA expression of the H. pylori community43.
In vitro analysis of BabA attachment to Leb
H. pylori expression of functional BabA protein was assayed by in vitro attachment to Leb using an ELISA as previously described22,35,43. Briefly, digoxigenin (Roche Applied Biosciences) labeled H. pylori cells were applied to Leb-HSA coated wells in a 96 well polystyrene plate. Unbound bacteria were removed by washing and bound bacteria were detected with anti-digoxigenin Fab fragments conjugated to horseradish peroxidase (POD) (Roche Applied Biosciences) followed by incubation with 2,2′-azino-di(3-ethyl-benzthiazoline-6-sulfonate) (ABTS). Color change was measured by subtraction of absorbance at 490 nm from 405 nm. Attachment ratio values were reported as an average of readings from two Leb positive wells divided by two Leb negative wells.
Statistical Analysis
Unless otherwise indicated, Mann-Whitney U, Fisher’s exact test or analysis of variance was performed using Graphpad Prism Software. Bacterial colonization (CFU/gram) was log transformed prior to analysis. The proportion of BabA expressing colonies was logit transformed using R software44 and 0 values were remapped to a proportion of 0.025 prior to analysis of variance. If no bacteria were isolated from a given mouse, the colonization level was shown at the limit of detection. P < 0.05 was considered statistically significant.
Results
Adaptive immunity does not affect H. pylori BabA expression
Loss of H. pylori BabA expression by phase variation might result from adaptive immune pressure directed against BabA. When loss of BabA expression occurs by a gene conversion event in which babB is duplicated into the babA locus35,36, this might represent a form of antigenic variation. Both occur commonly in bacteria and other pathogens to avoid adaptive immunity45,46,47,48. To test this hypothesis, we measured BabA expression in output strains after inoculation of H. pylori J166 into wild type (WT) and RAG2−/− mice, which do not develop mature B or T lymphocytes49. H. pylori colonized RAG2−/− mice at a significantly higher level than WT (Fig. 1a), supporting previous evidence that adaptive immunity, including mature T lymphocytes50,51, is important for control of H. pylori infection. To examine BabA expression, 3 to 6 H. pylori colonies were isolated from the stomachs of RAG2−/− and WT mice (N = 4–7) at 2 and 8 weeks post infection (PI). Since we previously showed that loss of BabA expression in mice occurs only by phase variation and not by gene conversion36, we determined BabA expression by DNA sequence analysis of the 5′ region that contains the CT dinucleotide repeats, where 8 repeats corresponds to a babA open reading frame (ORF) and attachment to Leb 36. Although there was considerable variability among individual mice 2 weeks PI, by 8 weeks PI BabA expression was lost in 8 of 8 mice (Fig. 1b). No differences were found between RAG2−/− and WT mice. The experiment was repeated with similar results (Fig. 1c,d). These data indicate that although the adaptive immune response controls H. pylori infection, it does not play a role in selection against BabA expressing bacteria in mice.
Figure 1. Loss of BabA expression in mice is not affected by B or T cell response.

H. pylori colonization was significantly greater in female RAG2−/− compared to WT mice at 2 and 8 weeks PI when the results were pooled over time, P < 0.0001 (a), but loss of the babA ORF was unaffected (b). Each point represents the percentage of colonies (average of 8 colonies total) from a single mouse determined by sequencing to express a full length babA ORF. Similar results were obtained when the experiment was repeated (c,d), although the difference in colonization level was less significant, P < 0.05 (c). Data were analyzed by two-way ANOVA of log (a,c) or logit (b,d) transformed values. *P < 0.05, **P < 0.01.
H. pylori BabA expression is lost in both conventional and germ free mice
It is now recognized that H. pylori infection occurs in the context of a gastric microbial community, which is less complex than that in the gut, but is probably autochthonous52,53,54,55 and may affect the outcome of infection. For example, in INS-GAS mice, which overexpress gastrin under the insulin promoter56, H. pylori infection induces more severe gastrointestinal intraepithelial neoplasia when animals have conventional microbiota compared to germ free counterparts57. Additionally, differences in intestinal microbiota composition in mice from different vendors have been shown to impact the immune response to pathogens58. Since adaptive immunity did not select for loss of BabA expression, and there was substantial variability among individual mice during the first two weeks of infection, we considered the possibility that the gastric microbial community could affect BabA expression. To determine whether microbiota could influence H. pylori expression of BabA, we infected germ free and conventionally raised C57BL/6 mice with H. pylori J166, and examined BabA expression in output colonies from mice sacrificed 3 weeks PI. Similar to the results 2 weeks PI (Fig. 1), there was marked variability, but no statistical differences in loss of BabA expression between conventional and germ free C57BL/6 mice (Fig. 2). These results indicate that the microbiota does not affect H. pylori expression of BabA, nor influence the variability among individual mice.
Figure 2. H. pylori loss of BabA expression occurs equally in germ free and conventionally raised mice.
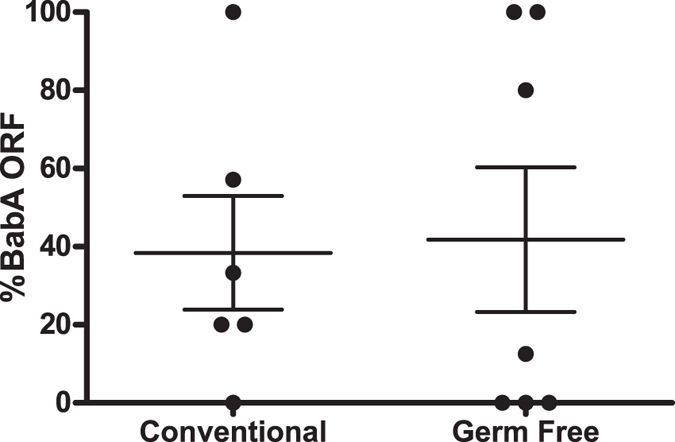
Percent BabA expressing H. pylori is shown for conventional and germ free female C57BL/6 mice inoculated with H. pylori J166 and sacrificed 3 weeks PI. No significant differences were found between the experimental groups.
The H. pylori J166 inoculum is heterogeneous at the BabA locus
The marked variability in BabA expression among output colonies (Figs 1 and 2) might result from stochastic or bottleneck effects if the inoculum contains a significant population of phase variants with no babA ORF. This possibility was supported by the finding that sequence of 5–6 individual colonies from 3 instances of liquid culture grown from the same H. pylori J166 stock contained variable percentages of BabA expressing clones, ranging from 67 to 100%. We therefore re-isolated a J166 single colony (designated J166SC) with a babA ORF (8 CT repeats) and compared the outcome of infection with J166sc to that with the original J166 inoculum containing a mixture of phase variants. After infection for between 2 and 8 weeks, loss of BabA expression was observed in 6 of 11 (55%) mice infected with the original stock of J166, but only 1 of 12 (8%) mice infected with J166sc (P < 0.05, Fisher’s exact test). Since J166SC might differ from J166 in other ways, in addition to the number of CT repeats in babA, we used contraselection to generate isogenic clones of J166SC that had either 8 CT repeats (J1668CT), encoding a babA ORF, or 7 CT repeats (J1667CT), which was out of frame and led to loss of BabA expression. J1668CT and J1667CT showed similar growth rates in liquid culture (Supplementary Figure S1), but, as expected, only J1668CT attached to Leb (Supplementary Figure S2). For all subsequent experiments, H. pylori inoculation was performed as a competition experiment in which mice were infected with an equal mixture of J1668CT and J1667CT (designated J1668CT:7CT).
BabA expression decreases within 24 hours during acute infection
Previous experiments indicated that loss of BabA expression can occur within the first two weeks of H. pylori infection (Fig. 1). We next examined this more precisely by inoculating WT C57BL/6 mice with J1668CT:7CT and examining BabA expression in output colonies recovered from mice sacrificed between 6 hrs and 14 days PI. Loss of BabA expression was detected as early as 6 hrs PI, was dominant by 3 days PI, and nearly uniform at 2 weeks PI (Fig. 3a). To confirm that this was not specific to a particular mouse strain, we performed the same experiment in WT FVB/N mice. Loss of BabA expression was similar in FVB/N mice (Fig. 3b) to what we previously observed in C57BL/6 mice (Fig. 3a), with colonies from 10 of 11 mice (91%) showing no BabA expression by 2 weeks PI. To confirm that sequencing the babA CT repeat region in a limited number of output colonies is representative, we performed RIA analysis, which detects Leb binding in the total H. pylori population cultured from the stomach. Leb binding analyzed by RIA correlated closely with CT analysis (Pearson R2 = 0.57, P < 0.0001), and demonstrated a similar loss in BabA expression over time (Fig. 3c). Together, these data suggest that there is a strong selection against BabA expression in mice that occurs within hours to a few days after inoculation.
Figure 3. H. pylori loss of BabA expression occurs early during infection.
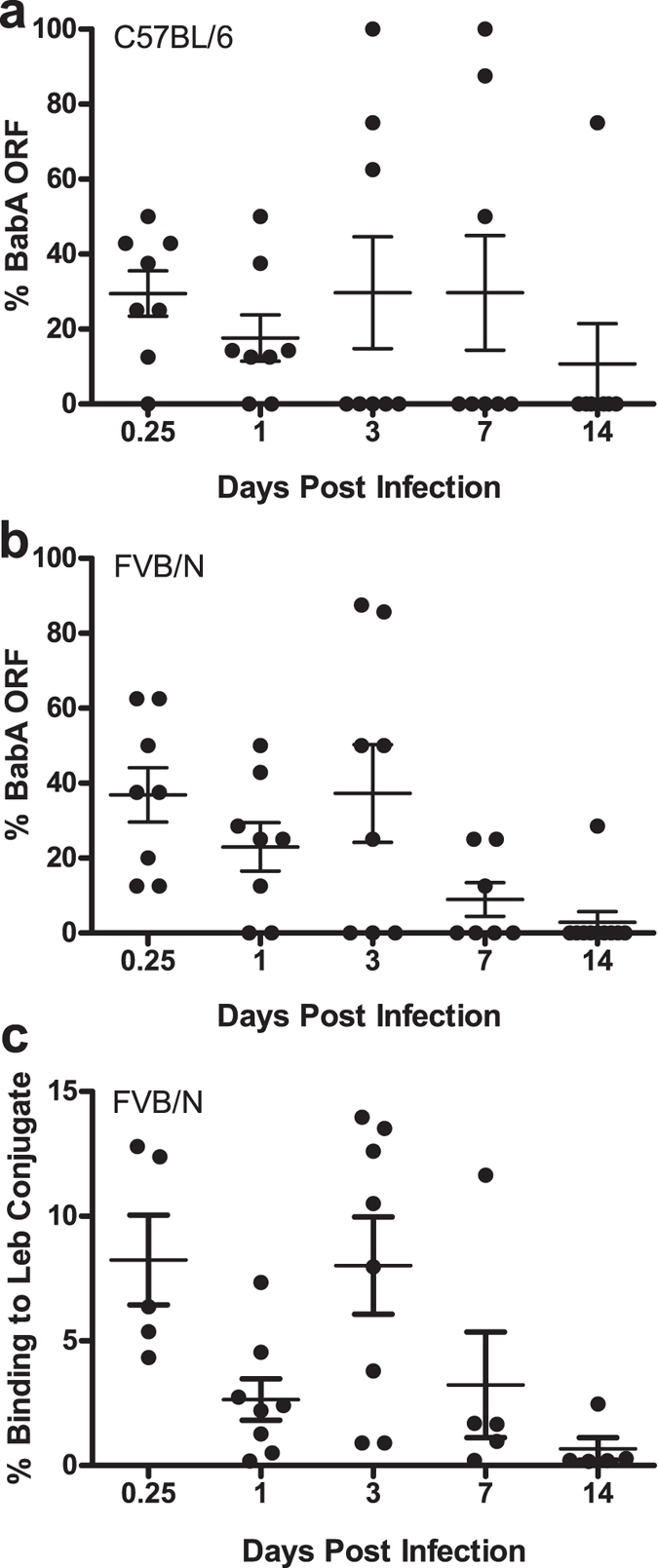
Percentage of BabA expressing H pylori detected by CT repeat analysis in female C57BL/6 (a) and female FVB/N (b) mice inoculated with J1668CT:7CT and sacrificed from 6 hrs to 14 days PI. (c) H. pylori populations cultured from the stomachs of FVB/N mice shown in B, analyzed for binding to Leb-HSA conjugate by RIA analysis.
Role of toll like receptor (TLR) signaling in BabA expression
Early selection, hours to days post infection, against BabA expression is consistent with the absence of a role for adaptive immunity in selecting against BabA expression (Fig. 1), and suggests that innate immunity might play a role. Attachment to host epithelial cells has been shown to induce expression of IL-824, suggesting that attachment increases host innate inflammatory responses and perhaps selects against strains expressing BabA. Moreover, we previously found that BabA expression was retained to a greater extent in C3H/HeJ mice36, which express a defective toll like receptor 4 (TLR4)59. Together, these data suggest the possibility that TLR signaling could be involved in driving loss of BabA expression. To examine this, BabA expression was analyzed 2 weeks PI in C57BL/6 WT mice and compared to mice with a homozygous deletion of MyD88 (MyD88−/−), toll like receptor 2 (TLR2−/−), or TLR4−/−. By 2 weeks PI, no BabA expressing bacteria were detected in the large majority of mice from each experimental group (Fig. 4), indicating that loss of BabA expression is under strong negative selection even in the absence of TLR signaling.
Figure 4. TLR signaling does not influence H. pylori BabA expression.
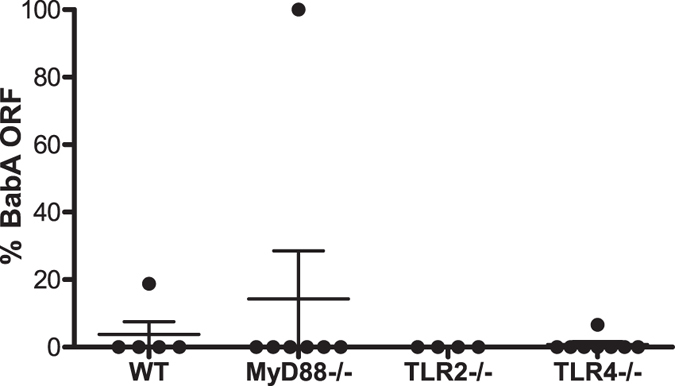
Percentage of BabA expressing H. pylori detected in female C57BL/6 WT, MyD88−/−, TLR2−/− and TLR4−/− mice inoculated with J1668CT:7CT and sacrificed 2 weeks PI. No significant differences were found between any of the experimental groups.
H. pylori BabA expression is greater in male mice
Glycosylation levels and patterns are altered early during H. pylori infection, with loss of fucosylation and increase in sialylation due to inflammation-induced changes in glycosyltransferases12,36,60,61,62. Specific glycans can affect not only adherence, but also H. pylori growth and gene expression63. We therefore compared BabA expression in WT mice, which do not express the Leb antigen, to transgenic mice that express Leb in the gastric mucosa40. Male and female Leb transgenic mice and their wild type littermates were infected with J1668CT:7CT and sacrificed 2 weeks PI. H. pylori colonization levels were greater in males (Fig. 5a, P < 0.001), but there was no difference between WT and Leb mice. BabA expression was also similar in WT and Leb mice, but again there was a significant effect of gender (Fig. 5b, P < 0.0001). Female mice were predominantly colonized by H. pylori lacking BabA expression, similar to what we observed previously (Figs 3 and 4), while BabA expression was greater in male mice (Fig. 5b). Expression of BabA in H. pylori isolated from male Leb mice was significantly greater than that from either female Leb mice or female WT mice (Tukey’s post test, P = 0.002 and P = 0.001, respectively). Increased colonization and a small increase in Leb binding were also observed in male Leb relative to female Leb mice 4 weeks PI using H. pylori strain J99 (Supplementary Figure S3), indicating that this phenomenon is robust across H. pylori strains that express functional BabA.
Figure 5. H. pylori colonization and BabA expression are greater in male than female FVB/N mice.
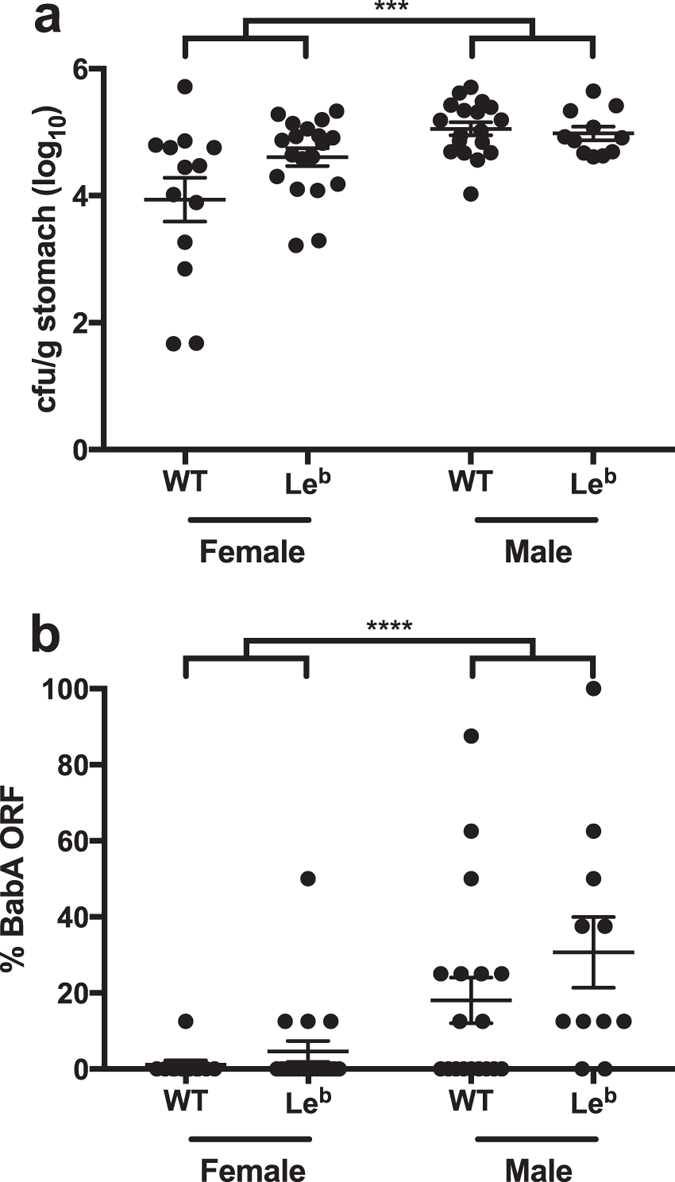
Male and female Leb transgenic FVB/N mice and their WT littermates were inoculated with J1668CT:7CT and sacrificed 2 weeks PI. H. pylori colonization density (a) and expression of BabA (b) were greater in male than in female mice. ***P < 0.001, ****P < 0.0001 (Two-way ANOVA of logit (a) or log (b) transformed values).
Loss of H. pylori BabA expression is not dependent on BabA attachment to Leb
Since loss of BabA expression in mice is not significantly affected by expression of Leb, we hypothesized that it might also be unaffected by the capacity of BabA to bind Leb. We recently demonstrated that BabA residues Cys189 and Cys197 form a redox-sensitive disulfide-clasped loop designated CL2, which is essential to bind the Leb α-1-2-linked fucose residue20. Cys to Ala replacement at BabA residues 189 and 197 in H. pylori J166 (designated BabACL2) was sufficient to eliminate all Leb binding activity, though the protein was expressed on the cell surface at levels similar to WT20. To examine the effect of Leb binding on BabA expression in mice, we used contraselection to generate isogenic strains of BabACL2 with either 8 (J166 BabACL28CT) or 9 (BabACL29CT) CT repeats. The BabA expression status predicted by the CT repeat number for each strain was confirmed by western blot (Supplementary Figure S4). The elimination of Leb binding activity in all strains was confirmed by ELISA (Supplementary Figure S4). These strains were combined in equal proportions (BabACL28CT:9CT) and used to infect female Leb transgenic mice and their WT littermates, which were sacrificed 2 weeks PI. For comparison, WT and Leb mice were also infected with J1668CT:7CT. There were no differences in colonization levels between BabACL28CT:9CT compared to J1668CT:7CT (Fig. 6a) in WT or in Leb mice. Strikingly, there was strong selection for loss of BabA expression, even in the absence of the capacity to bind Leb (Fig. 6b).
Figure 6. Loss of BabA expression in mice occurs even in the absence of capacity to bind Leb.
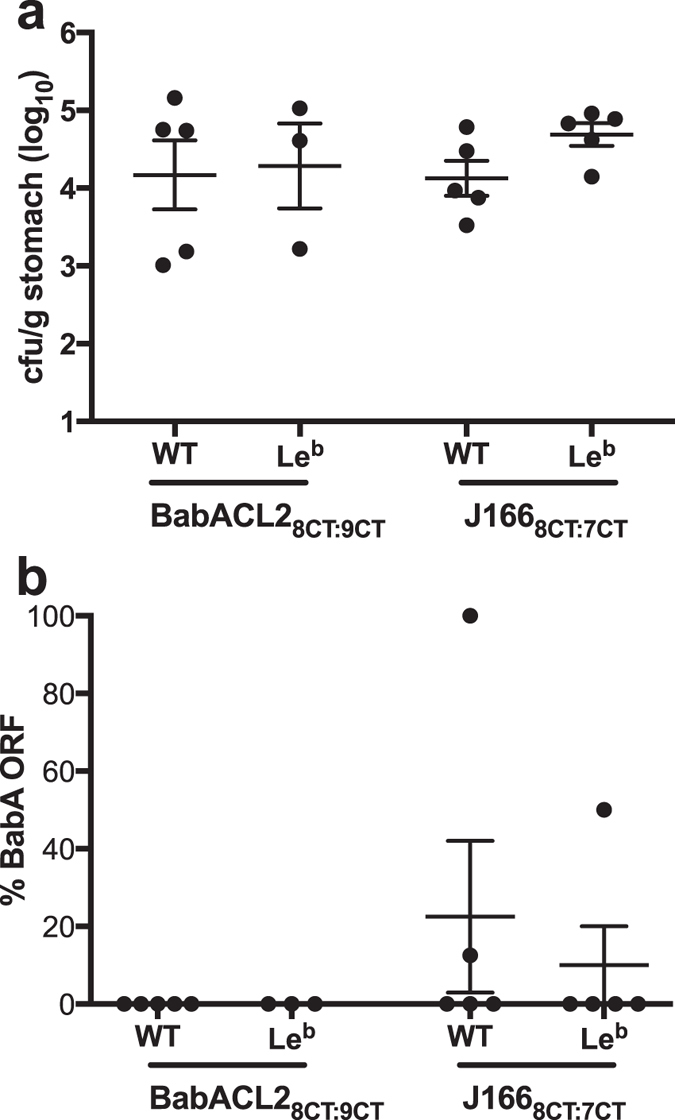
Female Leb transgenic FVB/N mice and their WT littermates were inoculated with BabACL28CT:9CT or J1668CT:7CT and sacrificed 2 weeks PI. Colonization density (a) and selection for loss of BabA expression (b) were similar in WT and Leb mice inoculated with J166 and BabACL2, which expresses BabA but does not attach to Leb. No significant differences were found between any of the experimental groups.
Discussion
We previously found that expression of BabA was lost either by phase variation or by gene conversion within the first 2–12 weeks during experimental H. pylori infection of non-human primates, mice, and gerbils35,36. Similar observations have been made by others37,64. Clinical isolates of H. pylori also show remarkable diversity at the babA locus, which may encode a “specialist” adhesin that binds only blood group O/Leb, or a “generalist” that also binds blood group A/ALeb and B/BLeb 19,20. In other cases, BabA may be expressed but not bind any known blood group antigen, may be present as a pseudogene, or even be absent from the genome altogether36. We recently suggested that in order to persist in the human stomach, H. pylori must face what we called an “attachment dilemma”, in which the benefits of adherence to the gastric epithelium such as escape from luminal acid and nutrient acquisition must be balanced with the costs, particularly interaction with the host immune response65. This is likely a dynamic process where BabA expression can be lost but also regained, either by phase variation, or perhaps by reintroduction of a copy of babA that has been archived in a small proportion in the population66. The diversity and dynamic nature of host glycosylation further adds to the complexity. With this perspective, we hypothesized that modulation of BabA expression and attachment to Leb would be driven by the host immune response and glycan expression. Here we set out to test this hypothesis.
Surprisingly, our results suggest that neither adaptive immunity, toll like receptor mediated host immune responses, Leb expression, nor even the capacity of BabA to bind Leb affected loss of BabA expression in mouse models. BabA expression was lost by phase variation equally in WT mice as in RAG2−/− mice that do not have functional B or T cells, and in MyD88−/− mice that cannot signal via all TLRs except TLR 3. Initial experiments suggested that loss of BabA expression required up to 8 weeks of colonization (Fig. 1), which might suggest involvement of host immunity, but competition experiments demonstrated selection for loss of BabA expression as early as one day PI (Fig. 3). Although we have not specifically examined the role of innate immune cells such as polymorphonuclear leukocytes (PMNs) and macrophages, our results suggest that adaptive immunity and TLR mediated immune responses do not select for loss of BabA expression. Moreover, loss of BabA expression is not dependent on Leb expression or the capacity of BabA to bind Leb, which suggests the possibility that BabA may have other, unrecognized functions.
Instead, we found that gender is the host determinant most associated with loss of BabA expression, which was maintained to a greater extent in male mice than in females, and was also associated with greater bacterial load (Fig. 5 and Supplementary Figure S3). These data highlight the importance of conducting host-pathogen interaction studies in both genders when using a mouse model, which is a common experimental design problem that has gained increased interest in recent years67. Male gender is a well-known risk factor for H. pylori-associated disease, including gastric adenocarcinoma68 and peptic ulcer, though the male predominance in ulcers appears to be declining as its prevalence decreases69. Animal models also provide support for male predominance of gastric cancer70. Although less studied, there is also epidemiologic evidence that male gender is a risk factor for H. pylori infection71, and that, as we observed, H. pylori bacterial load in mouse models is higher in males than in females72,73.
The mechanistic link between host gender and H. pylori expression of BabA remains to be determined. There are numerous differences in gastric physiology between males and females74, including pH, transit time, and enzyme expression, though it is unclear how these or other differences might affect relative fitness of H. pylori BabA expression. Similarly, gender dependent differences in glycosylation patterns have been described in humans75,76,77, suggesting the possibility that differences in mucosal glycan expression between male and female mice might participate in selection against BabA expression. However, since capacity to bind Leb is not required for loss of BabA expression, the effect of gender differences in glycosylation may involve as yet uncharacterized lectin functions of BabA.
Additional Information
How to cite this article: Kable, M. E. et al. Host Determinants of Expression of the Helicobacter pylori BabA Adhesin. Sci. Rep. 7, 46499; doi: 10.1038/srep46499 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by grants R01 AI081037 and R01 AI070803 from the National Institutes of Health (NIH) to JVS. TB is supported by grants from Vetenskapsrådet (VR/M) and Cancerfonden. We thank Matthew Nguyen for performing DNA extractions.
Footnotes
TB is founder of Helicure and a member of its scientific advisory board.
Author Contributions M.E.K., J.V.S. and T.B. wrote the main text of the manuscript. L.M.H. edited text, designed primers and contributed ideas. C.M.S. contributed Figure 1. S.L.D. collected data and performed a preliminary analysis for Figure 5. O.R. contributed Supplementary Figure S3. AS performed the RIA analysis throughout. K.A.E. contributed Figure 2. M.E.M. bred and characterized the transgenic Lewis B mouse colony for Figure 5. P.G. developed strain J166 BabACL2 and contributed ideas.
References
- Kusters J. G., van Vliet A. H. & Kuipers E. J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol Rev. 19, 449–490, doi: 10.1128/CMR.00054-05 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipponen P. et al. Cumulative 10-year risk of symptomatic duodenal and gastric ulcer in patients with or without chronic gastritis. A clinical follow-up study of 454 outpatients. Scand. J. Gastroenterol. 25, 966–973, doi: 10.3109/0036552900897621 (1990). [DOI] [PubMed] [Google Scholar]
- Basso D., Plebani M. & Kusters J. G. Pathogenesis of Helicobacter pylori infection. Helicobacter 15 Suppl 1, 14–20, doi: 10.1111/j.1523-5378.2010.00781.x (2010). [DOI] [PubMed] [Google Scholar]
- Hsiang J. et al. Increasing primary antibiotic resistance and ethnic differences in eradication rates of Helicobacter pylori infection in New Zealand–a new look at an old enemy. N. Z. Med. J. 126, 64–76 (2013). [PubMed] [Google Scholar]
- Vakil N. & Vaira D. Treatment for H. pylori infection: new challenges with antimicrobial resistance. J. Clin. Gastroenterol. 47, 383–388, doi: 10.1097/MCG.0b013e318277577b (2013). [DOI] [PubMed] [Google Scholar]
- Arnold I. C. et al. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Invest. 121, 3088–3093, doi: 10.1172/JCI45041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amedei A., Codolo G., Del Prete G., de Bernard M. & D’Elios M. M. The effect of Helicobacter pylori on asthma and allergy. J. Asthma Allergy 3, 139–147, doi: 10.2147/JAA.S8971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther J., Dave M., Higgins P. D. & Kao J. Y. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm. Bowel Dis. 16, 1077–1084, doi: 10.1002/ibd.21116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins P. D. et al. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm. Bowel. Dis. 17, 1398–1408, doi: 10.1002/ibd.21489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm R. A. et al. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68, 4155–4168 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossez Y. et al. The lacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J. Infect. Dis. 210, 1286–1295, doi: 10.1093/infdis/jiu239 (2014). [DOI] [PubMed] [Google Scholar]
- Mahdavi J. et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297, 573–578, doi: 10.1126/science.1069076 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koniger V. et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2, 16188, doi: 10.1038/nmicrobiol.2016.188 (2016). [DOI] [PubMed] [Google Scholar]
- Javaheri A. et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2, 16189, doi: 10.1038/nmicrobiol.2016.189 (2016). [DOI] [PubMed] [Google Scholar]
- Boren T., Falk P., Roth K. A., Larson G. & Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 262, 1892–1895, doi: 10.1126/science.8018146 (1993). [DOI] [PubMed] [Google Scholar]
- Ilver D. et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279, 373–377 (1998). [DOI] [PubMed] [Google Scholar]
- Bjornham O., Bugaytsova J., Boren T. & Schedin S. Dynamic force spectroscopy of the Helicobacter pylori BabA-Lewis b binding. Biophys. Chem. 143, 102–105, doi: 10.1016/j.bpc.2009.03.007 (2009). [DOI] [PubMed] [Google Scholar]
- Subedi S. et al. Expression, purification and X-ray crystallographic analysis of the Helicobacter pylori blood group antigen-binding adhesin BabA. Acta crystallographica. Section F, Structural biology communications 70, 1631–1635, doi: 10.1107/S2053230X14023188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm-Hurtig M. et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 305, 519–522, doi: 10.1126/science.1098801 (2004). [DOI] [PubMed] [Google Scholar]
- Moonens K. et al. Structural Insights into Polymorphic ABO Glycan Binding by Helicobacter pylori. Cell Host Microbe 19, 55–66, doi: 10.1016/j.chom.2015.12.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage N. et al. Structural basis of Lewis(b) antigen binding by the Helicobacter pylori adhesin BabA. Sci. Adv. 1, e1500315, doi: 10.1126/sciadv.1500315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard M. et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. USA 96, 12778–12783, doi: 10.1073/pnas.96.22.12778 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz C. et al. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 61, 1903–1909 (2001). [PubMed] [Google Scholar]
- Ishijima N. et al. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J. Biol. Chem. 286, 25256–25264, doi: 10.1074/jbc.M111.233601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviles-Jimenez F. et al. In vivo expression of Helicobacter pylori virulence genes in patients with gastritis, ulcer, and gastric cancer. Infect. Immun. 80, 594–601, doi: 10.1128/IAI.05845-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S. et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2, 155–164, doi: 10.1126/science.287.5457.1497 (2000). [DOI] [PubMed] [Google Scholar]
- Blaser M. J. et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55, 2111–2115 (1995). [PubMed] [Google Scholar]
- Odenbreit S. et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500, doi: 10.1126/science.287.5457.1497 (2000). [DOI] [PubMed] [Google Scholar]
- Segal E. D., Cha J., Lo J., Falkow S. & Tompkins L. S. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA 96, 14559–14564, doi: 10.1073/pnas.96.25.14559 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden S. et al. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 4, e2, doi: 10.1371/journal.ppat.0040002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo M. et al. Infection by Helicobacter pylori expressing the BabA adhesin is influenced by the secretor phenotype. J. Pathol. 215, 308–316, doi: 10.1002/path.2363 (2008). [DOI] [PubMed] [Google Scholar]
- Magalhaes A. et al. Fut2-null mice display an altered glycosylation profile and impaired BabA-mediated Helicobacter pylori adhesion to gastric mucosa. Glycobiology 19, 1525–1536, doi: 10.1093/glycob/cwp131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruge J. L. et al. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 95, 3925–3930, doi: 10.1073/pnas.95.7.3925 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball P. A. Influence of the secretor and Lewis genes on susceptibility to duodenal ulcer. Br. Med. J. 2, 948–950, doi: 10.1136/bmj.2.5310.948 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnick J. V., Hansen L. M., Salama N. R., Boonjakuakul J. K. & Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc. Natl. Acad. Sci. USA 101, 2106–2111, doi: 10.1073/pnas.0308573100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styer C. M. et al. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect. Immun. 78, 1593–1600, doi: 10.1128/IAI.01297-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno T. et al. Effects of blood group antigen-binding adhesin expression during Helicobacter pylori infection of Mongolian gerbils. J. Infect. Dis. 203, 726–735, doi: 10.1093/infdis/jiq090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbeck J. C., Hansen L. M., Fong J. M. & Solnick J. V. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect. Immun. 74, 4375–4378, doi: 10.1128/IAI.00485-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. E., Lam A., Bhatnagar S. & Solnick J. V. Environmental determinants of transformation efficiency in Helicobacter pylori. J. Bacteriol. 196, 337–344, doi: 10.1128/JB.00633-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk P. G., Bry L., Holgersson J. & Gordon J. I. Expression of a human alpha-1,3/4-fucosyltransferase in the pit cell lineage of FVB/N mouse stomach results in production of Leb-containing glycoconjugates: a potential transgenic mouse model for studying Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 92, 1515–1519, doi: 10.1073/pnas.92.5.1515 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl M. A. et al. Host-dependent Lewis (Le) antigen expression in Helicobacter pylori cells recovered from Leb-transgenic mice. J. Exp. Med. 206, 3061–3072, doi: 10.1084/jem.20090683 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal M. et al. Helicobacter pylori Activates and Expands Lgr5(+) Stem Cells Through Direct Colonization of the Gastric Glands. Gastroenterology 148, 1392–1404 e1321, doi: 10.1053/j.gastro.2015.02.049 (2015). [DOI] [PubMed] [Google Scholar]
- Aspholm M. et al. Helicobacter pylori adhesion to carbohydrates. Method Enzymol 417, 293–339, doi: 10.1016/S0076-6879(06)17020-2 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, available at http://www.R-project.org/ (2015).
- van Belkum A., van Leeuwen W., Scherer S. & Verbrugh H. Occurrence and structure-function relationship of pentameric short sequence repeats in microbial genomes. Res Microbiol 150, 617–626, doi: 10.1016/s0923-2508(99)00129-1 (1999). [DOI] [PubMed] [Google Scholar]
- Stern A. & Meyer T. F. Common mechanism controlling phase and antigenic variation in pathogenic neisseriae. Mol Microbiol 1, 5–12, doi: 10.1111/j.1365-2958.1987.tb00520.x (1987). [DOI] [PubMed] [Google Scholar]
- Telford J. L. Bacterial genome variability and its impact on vaccine design. Cell Host Microbe 3, 408–416, doi: 10.1016/j.chom.2008.05.004 (2008). [DOI] [PubMed] [Google Scholar]
- Vink C., Rudenko G. & Seifert H. S. Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol Rev 36, 917–948, doi: 10.1111/j.1574-6976.2011.00321.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y. et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867, doi: 10.1016/0092-8674(92)90029-c (1992). [DOI] [PubMed] [Google Scholar]
- Muller A. & Solnick J. V. Inflammation, immunity, and vaccine development for Helicobacter pylori. Helicobacter 16 Suppl 1, 26–32, doi: 10.1111/j.1523-5378.2011.00877.x (2011). [DOI] [PubMed] [Google Scholar]
- Taylor J. M., Ziman M. E., Canfield D. R., Vajdy M. & Solnick J. V. Effects of a Th1- versus a Th2-biased immune response in protection against Helicobacter pylori challenge in mice. Microb Pathog 44, 20–27, doi: 10.1016/j.micpath.2007.06.006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. E. & Solnick J. V. The gastric microbial community, Helicobacter pylori colonization, and disease. Gut microbes 5, 345–350, doi: 10.4161/gmic.28573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S. et al. Bacterial community mapping of the mouse gastrointestinal tract. PloS one 8, e74957, doi: 10.1371/journal.pone.0074957 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. E. et al. The impact of Helicobacter pylori infection on the gastric microbiota of the rhesus macaque. PloS one 8, e76375, doi: 10.1371/journal.pone.0076375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. E., Dieter J. A., Luo Z., Baumgarth N. & Solnick J. V. Predicting the outcome of infectious diseases: variability among inbred mice as a new and powerful tool for biomarker discovery. mBio 3, e00199–00112, doi: 10.1128/mBio.00199-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. C. et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 118, 36–47, doi: 10.1016/s0016-5085(00)70412-4 (2000). [DOI] [PubMed] [Google Scholar]
- Lofgren J. L. et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 140, 210–220, doi: 10.1053/j.gastro.2010.09.048 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498, doi: 10.1016/j.cell.2009.09.033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088, doi: 10.1126/science.282.5396.2085 (1998). [DOI] [PubMed] [Google Scholar]
- Cooke C. L. et al. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterology 137, 1061–1071, 1071 e1061-1068, doi: 10.1053/j.gastro.2009.04.014 (2009). [DOI] [PubMed] [Google Scholar]
- Navabi N., Johansson M. E., Raghavan S. & Linden S. K. Helicobacter pylori infection impairs the mucin production rate and turnover in the murine gastric mucosa. Infect Immun 81, 829–837, doi: 10.1128/IAI.01000-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos N. T. et al. Helicobacter pylori induces beta3GnT5 in human gastric cell lines, modulating expression of the SabA ligand sialyl-Lewis x. J Clin Invest 118, 2325–2336, doi: 10.1172/JCI34324 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog E. C. et al. Human gastric mucins differently regulate Helicobacter pylori proliferation, gene expression and interactions with host cells. PloS one 7, e36378, doi: 10.1371/journal.pone.0036378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. Analysis of a single Helicobacter pylori strain over a 10-year period in a primate model. Int J Med Microbiol 305, 392–403, doi: 10.1016/j.ijmm.2015.03.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. E., Boren T. & Solnick J. V. Life at the margins: modulation of attachment proteins in Helicobacter pylori. Gut microbes 2, 42–46, doi: 10.4161/gmic.2.1.14626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom A. et al. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci USA 101, 16923–16928, doi: 10.1073/pnas.0404817101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton J. A. & Collins F. S. Policy: NIH to balance sex in cell and animal studies. Nature 509, 282–283, doi: 10.1038/509282a (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson W. F. et al. Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA 303, 1723–1728, doi: 10.1001/jama.2010.496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata J. H. & Nogawa A. N. Meta-analysis of risk factors for peptic ulcer. Nonsteroidal antiinflammatory drugs, Helicobacter pylori, and smoking. J Clin Gastroenterol 24, 2–17 (1997). [DOI] [PubMed] [Google Scholar]
- Fox J. G. et al. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res 63, 942–950 (2003). [PubMed] [Google Scholar]
- de Martel C. & Parsonnet J. Helicobacter pylori infection and gender: a meta-analysis of population-based prevalence surveys. Dig Dis Sci 51, 2292–2301, doi: 10.1007/s10620-006-9210-5 (2006). [DOI] [PubMed] [Google Scholar]
- Aebischer T., Laforsch S., Hurwitz R., Brombacher F. & Meyer T. F. Immunity against Helicobacter pylori: significance of interleukin-4 receptor alpha chain status and gender of infected mice. Infect Immun 69, 556–558, doi: 10.1128/IAI.69.1.556-558.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Every A. L. et al. Localized suppression of inflammation at sites of Helicobacter pylori colonization. Infect Immun 79, 4186–4192, doi: 10.1128/IAI.05602-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire A. C., Basit A. W., Choudhary R., Piong C. W. & Merchant H. A. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int J Pharm 415, 15–28, doi: 10.1016/j.ijpharm.2011.04.069 (2011). [DOI] [PubMed] [Google Scholar]
- Pucic M. et al. Changes in plasma and IgG N-glycome during childhood and adolescence. Glycobiology 22, 975–982, doi: 10.1093/glycob/cws062 (2012). [DOI] [PubMed] [Google Scholar]
- Knezevic A. et al. Effects of aging, body mass index, plasma lipid profiles, and smoking on human plasma N-glycans. Glycobiology 20, 959–969, doi: 10.1093/glycob/cwq051 (2010). [DOI] [PubMed] [Google Scholar]
- Brinkman-Van der Linden C. M. et al. Oral estrogen treatment induces a decrease in expression of sialyl Lewis x on alpha 1-acid glycoprotein in females and male-to-female transsexuals. Glycobiology 6, 407–412, doi: 10.1093/glycob/6.4.407 (1996). [DOI] [PubMed] [Google Scholar]
- Alm R. A. et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180, doi: 10.1038/16495 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.