

ABSTRACT
The transcription factor Pdx1 is required for multiple aspects of pancreatic organogenesis. It remains unclear to what extent Pdx1 expression and function depend upon trans-activation through 5′ conserved cis-regulatory regions and, in particular, whether the mammal-specific Area II (−2139 to −1958 bp) affects minor or major aspects of organogenesis. We show that Area II is a primary effector of endocrine-selective transcription in epithelial multipotent cells, nascent endocrine progenitors, and differentiating and mature β cells in vivo. Pdx1ΔAREAII/− mice exhibit a massive reduction in endocrine progenitor cells and progeny hormone-producing cells, indicating that Area II activity is fundamental to mounting an effective endocrine lineage-specification program within the multipotent cell population. Creating an Area II-deleted state within already specified Neurog3-expressing endocrine progenitor cells increased the proportion of glucagon+ α relative to insulin+ β cells, associated with the transcriptional and epigenetic derepression of the α-cell-determining Arx gene in endocrine progenitors. There were also glucagon and insulin co-expressing cells, and β cells that were incapable of maturation. Creating the Pdx1ΔAREAII state after cells entered an insulin-expressing stage led to immature and dysfunctional islet β cells carrying abnormal chromatin marking in vital β-cell-associated genes. Therefore, trans-regulatory integration through Area II mediates a surprisingly extensive range of progenitor and β-cell-specific Pdx1 functions.
KEY WORDS: Cis-regulatory function, Lineage diversification, Pancreatic endocrine progenitors, Pdx1 enhancer Area II, Mouse
Summary: A crucial enhancer element in the Pdx1 promoter, termed Area II, is required for endocrine progenitor specification, α/β-cell lineage diversification and β-cell maturation in mice.
INTRODUCTION
Various pancreas-enriched transcription factors have been linked to the programs that direct the differentiation of early and later pancreas progenitors into functional islet β cells (e.g. Sox9, Nkx6.1, Neurog3, Pdx1) (reviewed by Pan and Wright, 2011). Although the integrated, likely highly cross-regulatory gene-regulatory networks are not well defined, the dynamic expression pattern of Pdx1, and profound defects incurred with global or cell type-specific inactivation (e.g. Fujitani et al., 2006; Gannon et al., 2001; Hale et al., 2005; Kodama et al., 2016; Offield et al., 1996; Swift et al., 1998), clearly point to its pervasive and orchestrating role during organogenesis. Thus, a Pdx1 null mutation in human (Stoffers et al., 1997a,b) or mouse (Jonsson et al., 1994; Offield et al., 1996) results in pancreatic agenesis, and a heterozygous mutation leads to human early-onset diabetes (Stoffers et al., 1997a,b). Moreover, conditional deletion of Pdx1 has revealed the requirement for this transcription factor in several of the later stages of pancreatic endocrine cell development and in adult islet β-cell function (reviewed by Pan and Wright, 2011).
Much of Pdx1 transcriptional regulation appears to be exerted by trans-acting factors acting within four conserved upstream cis-regulatory regions (termed Areas I-IV), located within 6.5 kb of the transcriptional start site (Gannon et al., 2001; Gerrish et al., 2000; Van Velkinburgh et al., 2005). Whereas Areas I, III and IV are present in widely differing vertebrate species, Area II is restricted, somewhat surprisingly, to mammals (Gerrish et al., 2000). In mouse, combined deletion of Areas I-II-III (Pdx1ΔI-II-III) in vivo produces severely deficient Pdx1 expression and impairs formation of the early pancreatic buds (Fujitani et al., 2006), an effect similar to the pancreatic agenesis in Pdx1 germline nulls (Offield et al., 1996). Complementary experiments showed that Pdx1 expression driven by Areas I-II-III, with only a small portion of Area IV, restored full pancreatic development to Pdx1 null mice (Boyer et al., 2006; Gannon et al., 2001). These results imply that the embryonic Pdx1 expression required for complete production of a differentiated pancreatic organ is principally, if not exclusively, regulated by Areas I-II-III.
Enhancer-like activities for Areas I, II and III have been documented in reporter assays in β-cell lines and a limited number of transgenic mouse assays. Such studies assigned β-cell-specific enhancer-like activities to Area II. For example, while Area I or Area II imparted β-cell-specific activation in cell lines (Gerrish et al., 2000), only Area II independently directed expression to islet β cells in vivo, although expression was variegated. When placed together, Areas I and II seemed to show functional interactions that were now able to induce high Pdx1 expression throughout the entire β-cell population from around embryonic day (E) 13.5, which represents the start of the major phase of insulin+ cell production (Van Velkinburgh et al., 2005). Whereas the region representing Areas I-II-III is bivalently marked in early endodermal progenitors, it is subsequently derepressed in nascent pancreatic progenitors leading to a relative deficit of repressive chromatin markings (van Arensbergen et al., 2010; Xie et al., 2013; Xu et al., 2011). Together with Area I-II-III transgene analysis (Wiebe et al., 2007), these findings supported the idea that Areas I-II-III are involved in driving Pdx1 expression in pancreatic endocrine as well as exocrine progenitors.
Although these combined findings support a central role for Area II in driving Pdx1 transcription, the effect of removing just Area II from the endogenous gene remained untested. It was therefore uncertain whether this mammal-specific cis-regulatory region operates broadly over multiple steps of the entire organogenesis process, or perhaps plays a more nuanced or ʻtuning role' directed at producing the correct number of islet β cells or keeping them functioning normally. We addressed this issue in vivo with a newly derived targeted allele carrying a precise Area II deletion, termed Pdx1ΔAREAII (Pdx1ΔII). By combining Pdx1ΔII with null and conditionally inactivated floxed Pdx1 alleles, we established that the mammal-restricted Area II is essential to Pdx1 transcription during several distinct phases of pancreatic organogenesis and islet endocrine cell ontogeny. Although previous findings pointed to a β-cell-selective role for Area II, a germline global deletion massively affected all pancreatic endocrine progenitors and progeny. Endocrine-selective reduction of gene activity by removing Area II affected endocrine cell-type allocation, and severely debilitated maturation of β cells. We report effects on chromatin marking status of Pdx1 and key genes directly or indirectly targeted by Pdx1 caused by reducing the level of Pdx1. These studies establish that Area II is a potent contributor to all endocrine-specific functions of Pdx1, including endocrine progenitor specification, β-cell versus α-cell lineage allocation, and β-cell maturation. Our findings are discussed with respect to the possible unique regulatory significance of this compact cis-regulatory region in pancreas formation in mammals as compared with other vertebrates.
RESULTS
Loss of Area II does not affect Pdx1 regulation of overall pancreas size
An Area II-specific deletion was generated within the endogenous locus (Pdx1ΔII) to determine the requirement for this transcriptional control region in Pdx1 expression and function (Fig. S1). Mice of several genotypes were derived (Fig. 1A-C).
Fig. 1.

Glucose levels of different Pdx1 mutant classes. (A-C) Schematic of Pdx1+/−, Pdx1ΔII/− and Pdx1ΔII/ ΔII genotypes. (D-E′) Glucose level (mg/100 ml) of different ad libitum-fed Pdx1 mutant classes at early postnatal stages (D,D′), 4 weeks (E) and 5-6 months (E′) of age. *P<0.05.
Homozygous Pdx1ΔII/ΔII mice showed no gross changes during development nor postnatal physiological abnormalities in younger adults, with pancreas size and islet cluster size/number being normal, although the various hormone-secreting islet cell types were arranged differently from control islets, exhibiting the well-known abnormal ʻmixed-islet' phenotype (Fig. S2A,B). Pdx1ΔII/ΔII β cells were immature at 4 weeks of age, suggested by the lack of the key maturation markers MafA and Glut2 (Slc2a2), along with abnormal maintenance of the adult α-cell marker MafB (Fig. S2C-E). However, older, 5- to 6-month-old male Pdx1ΔII/ΔII mice became distinctly hyperglycemic (Fig. 1E,E′), indicating an age-dependent requirement for this control domain in islet β-cell function.
Crossing existing Pdx1+/− mice with Pdx1ΔII/+ mice led to the production of Pdx1ΔII/− animals, in which Pdx1 protein is only derived from one Pdx1ΔII allele, thereby creating a more sensitized condition compared with Pdx1+/−. Pdx1ΔII/− neonates were also indistinguishable from control Pdx1+/− littermates by outward appearance, and their gastrointestinal tract anatomy was indistinguishable from control littermates, including a pancreas of normal size (Fig. 2A,B). They did not, however, survive beyond postnatal day (P) 3 and were hyperglycemic at all time points measured before death (Fig. 1D,D′). Pdx1 protein production in Pdx1ΔII/− pancreatic tissue was markedly diminished compared with the Pdx1+/− state (Fig. 2C-D′), and there was a large decrease in the total number of all endocrine cells, and a greater representation of glucagon (Gcg)-expressing cells (Fig. 2E,F,O).
Fig. 2.

Endocrine-specific defects in the Pdx1ΔII/− pancreas. (A,B) Overall morphology of the Pdx1+/− and Pdx1ΔII/− pancreatic/duodenal region at P1. (C-D′) Gcg and Pdx1 expression (single Pdx1 channel and merge) in P1 endocrine clusters. (E,F) Ins and Gcg expression at P1. Arrows indicate Ins+ Gcg+ co-expressing cells. Note that the large cluster of hormonal cells shown in F was not commonly found in Pdx1ΔII/− and is shown for comparison. (G,H) Pdx12E3-lacZ reporter for Pdx1 transcriptional activities in E13.5 Pdx1+/− and Pdx1ΔII/− pancreata. (I-J′) Sox9 and Pdx1 expression at E13.5. (K-L′) Decreased Neurog3+ cell numbers are observed in E14.5 Pdx1ΔII/− pancreas (L,L′). (M,N) Morphometric analysis of Sox9+ cells and Neurog3+ cells at E14.5. *P<0.05. (O) Ins+:Gcg+ cell ratios in Pdx1+/− and Pdx1ΔII/− at P1.
Because Pdx1 autoregulates its own expression through the neighboring Area I control region (Gerrish et al., 2000), we used a Pdx1 exon 2 lacZ knock-in null allele (Pdx12E3-lacZ; Offield et al., 1996) and whole-mount embryo analysis to examine whether this reduced Pdx1 condition affected the spatiotemporal pattern of Pdx1 expression in Pdx1ΔII/2E3-lacZ embryos. E13.5 control embryos (Pdx12E3-lacZ/+) showed β-galactosidase expression throughout the normal Pdx1 expression domain, spanning from caudal stomach to the rostral duodenum and including the pancreas and bile duct (Fig. 2G). The spatial pattern in Pdx1ΔII/2E3-lacZ embryos was equivalent, but of lower intensity (Fig. 2H), suggesting decreased β-galactosidase production related to the known interactions between Areas I and II and between Areas I, II and III (Gerrish et al., 2001).
The total size of the Pdx1ΔII/− pancreatic epithelium was not significantly changed compared with Pdx1+/− (Fig. S3C,E,H) and was essentially equivalent to the wild type (Fujitani et al., 2006; Offield et al., 1996). In addition, the pancreatic epithelium in E12.5 Pdx1ΔII/− and Pdx1+/− embryos was indistinguishable in the formation and developmental advancement of the epithelial plexus (Fig. 2J, Fig. S3B, Fig. S4A-D). The production of several progenitor stage transcription factors required for key aspects of pancreas growth and differentiation was also normal [e.g. Sox9, HNF6 (Onecut1), Gata4; Fig. S4A-D]. Moreover, similar numbers of lineage-labeled pancreatic epithelial cells were found at E12.5 in Pdx1ΔII/− and Pdx1+/− pancreatic tissues (using embryos carrying the Pdx1TgCre-R26REYFP system for Pdx1 lineage tracing; Fig. S3C) (Gu et al., 2002). Pdx1 lineage-labeled cells from both Pdx1ΔII/− and Pdx1+/− genotypes at E13.5 were flow-sorted and Pdx1 expression was determined by qRT-PCR using allele-specific primers that do not detect transcript from the Pdx1 null allele (Table S2). Whereas mRNA from the Pdx1ΔII allele was ∼32% of the control level (Fig. S3D), mRNAs of several other lineage-selective factors found in multipotent progenitor cells were unchanged (e.g. Sox9, HNF1β, HNF6; Fig. S3D) (Haumaitre et al., 2005; Lynn et al., 2007; Zhang et al., 2009), which was corroborated by immunoanalysis (Fig. S4). Interestingly, only the α-cell-determining factor Arx (Collombat et al., 2007, 2003) was significantly upregulated in qRT-PCR analysis (Fig. S3D). Further, normal expression of ductal and acinar markers was found in P1 Pdx1ΔII/− pancreas (Fig. S5). These results indicate that the greatly reduced Pdx1 mRNA of the Pdx1ΔII/− mutant was still sufficient for the formation of the exocrine acinar and ductal cell lineages, which constitute ∼98% of the organ mass.
Endocrine islet cell formation is severely compromised in the Pdx1ΔII/− state
The severe hyperglycemia in Pdx1ΔII/− newborns (Fig. 1), suggestive of β-cell dysfunction and/or reduced β-cell mass, prompted an examination of endocrine cell formation and differentiation. The E14.5-E15.5 Pdx1ΔII/− pancreata showed a massively decreased number of Neurog3+ endocrine progenitor cells (20-40% of control; Fig. 2N, Fig. S3F). Neurog3+ cells give rise to postmitotic endocrine precursors that express Pax6 (Sander et al., 1997; St-Onge et al., 1997), and both Neurog3 and Pax6 mRNA levels were specifically reduced in Pdx1ΔII/− epithelium (Fig. S3D). This large decrease in endocrine progenitors is likely to have resulted in the 70% decline in total endocrine cell number at P1 (Fig. S3G).
Any one of several scenarios could result in a reduction in endocrine progenitor numbers. First, because Neurog3+ cells arise from a Sox9+ bipotent progenitor pool (Kopp et al., 2011), reduced Sox9+ cell production would lead to fewer endocrine progenitors. However, the number of Sox9+ cells in E13.5 pancreata was similar between genotypes (Fig. 2M) and the development of the epithelial plexus was relatively normal, as described above. In addition, the 16% decrease in the mitotic index of Sox9+ cells (as assessed by phospho-histone H3 staining; Fig. S4F) would not account for the profound loss of endocrine progenitors observed in the Pdx1ΔII/− mutant. Second, general epithelial or endocrine progenitor stage apoptosis is unlikely to contribute to the failure of cells to enter the endocrine lineage in Pdx1ΔII/− pancreata, as only rare TUNEL+ cells were detected (Fig. S4C,D). Third, and most likely, the threshold of Pdx1 protein production in the Pdx1ΔII/− pancreatic epithelium was insufficient to activate Neurog3 (Oliver-Krasinski et al., 2009). Pertinent here is the previous finding that Pdx1 augments the expression of other essential early epithelial regulatory factors, such as Sox9 and HNF1β, which together are required for normal Neurog3 transcription and endocrine specification (Oliver-Krasinski et al., 2009), yet the Pdx1LOW condition in the Pdx1ΔII/− pancreata did not affect Sox9 or HNF1β production (Fig. 2M, Fig. S3D), only Neurog3+ cell formation (Fig. 2N,L′). Since the birth of endocrine cells affects islet size significantly (Jo et al., 2012), the formation of smaller islets in the Pdx1ΔII/− pancreas is likely to have resulted from the massive reduction of endocrine progenitors.
Area II deletion reduces Pdx1 levels and disturbs α versus β lineage allocation
Although the greatly reduced numbers of endocrine cells produced in Pdx1ΔII/− mutants were nonetheless assembled into islet-like clusters by P1, there was impairment in the normal core versus mantle distribution of the various hormone cell types in the rarely found larger clusters (Fig. 2F), again yielding the mixed-islet phenotype. In addition, the majority of hormone-producing cells were found in small clusters of ∼5-7 cells, broadly dispersed throughout the pancreas (Fig. 2D). Many insulin (Ins) and Gcg co-expressing cells were also detected in P1 Pdx1ΔII/− mutants (arrows in Fig. 2F), representing 5.3% of total Ins+ or Gcg+ cells, as compared with only 0.5% in Pdx1+/−. This phenotype suggests that the lower level of Pdx1 produced in mutant β cells fails to fully repress the α-cell program (Gao et al., 2014).
The Pdx1LOW condition associated with the Pdx1ΔII/− state caused a significant alteration in the proportion of α (Gcg+) and β (Ins+) cell types. At P1, whereas islets in Pdx1+/− control pancreas contained β and α cells in a 69:31% ratio, in Pdx1ΔII/− it was 58:42% (Fig. 2O). Unfortunately, Pdx1ΔII/− animals died too soon after birth to assess the quality of these β cells by expression of various β-cell maturation markers. However, the overall hyperglycemia and increased number of Ins and Gcg co-expressing cells strongly suggested far-reaching defects in β-cell lineage progression and, consequently, in the postnatal physiological activity of the islet β-cell population.
We next analyzed pancreas with the genotype Neurog3Cre;Pdx1FloxE2/ΔII (referred to as Pdx1endo-ΔII) in which inactivation of the conditional null Pdx1FloxE2 allele (Gannon et al., 2008) leads to Pdx1 protein being derived only from the Pdx1ΔII allele in the endocrine progenitors and their descendants (Fig. 3B). These mice lived much longer than Pdx1ΔII/− mice, indicating a less profound deficit when restricting the loss of Area II to the endocrine lineage. The total endocrine cell number at P1 in Pdx1endo-ΔII pups was 72% of that in controls (Fig. 3F). Nonetheless, there was a noticeable restoration of islet size distribution in favor of a size increase and now with an essential absence of the many small clusters found in Pdx1ΔII/− pancreata (compare Fig. 3D with Fig. 2D). There was an alteration in the relative proportion of β versus α cells that was strikingly similar to that seen in P1 Pdx1ΔII/− animals: a 60:40% β to α ratio in Pdx1endo-ΔII mutant tissue as compared with 73:27% for controls (Fig. 3E). In addition, ∼5.5% of these endocrine cells showed Ins and Gcg co-expression, versus 0.5% in controls. The effect on lineage allocations seemed to be restricted to α and β cells, as we found no quantifiably significant difference in the numbers of somatostatin or pancreatic polypeptide cell types (Fig. S6). Pdx1endo-ΔII mice were hyperglycemic from P1 until at least 4 weeks of age (Fig. 4A,B), with islet dysfunction probably reflecting the inability of the Pdx1LOW condition to facilitate β-cell maturation. For example, although expression of the β-cell factor Nkx6.1 was relatively normal (Fig. 4D′), mutant β cells at 4 weeks of age lacked an important transcriptional regulator of this process, MafA, while a factor that is normally enriched in mouse α but not β cells, MafB, was still apparent (Fig. 4E,F). Moreover, the size spectrum of Pdx1endo-ΔII islets, although relatively similar to controls at P1 (Fig. 3), appeared at 4 weeks to have shifted noticeably in favor of being much smaller (e.g. Fig. 4C′), suggesting postnatal defects in islet expansion.
Fig. 3.
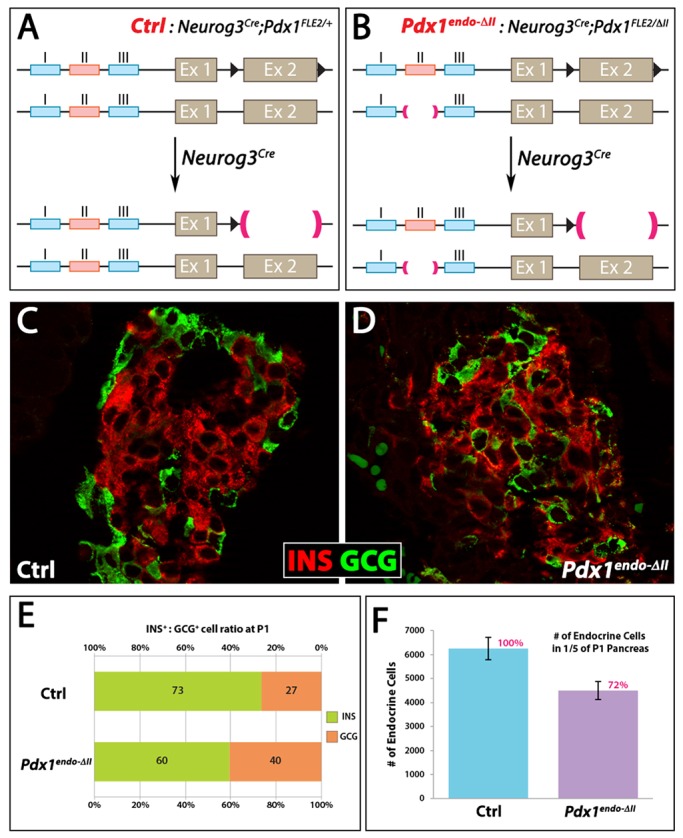
Pdx1endo-ΔII mice show defects in islet organization and in the β:α cell ratio. (A,B) Schematic presentation of Neurog3Cre;Pdx1FloxE2/ΔII (Pdx1endo-ΔII) and Neurog3Cre;Pdx1FloxE2/+ control. In Pdx1endo-ΔII, the endocrine progenitors and their descendant endocrine cells were Pdx1ΔII/− upon Neurog3-mediated recombination. (C,D) Ins and Gcg expression in P1 islets. The mantle and core structure (C) was not obvious in Pdx1endo-ΔII islets (D). (E) Quantitative estimation of β:α cell ratio at P1 (by Ins and Gcg expression, respectively). (F) Total numbers of endocrine cells in P1 pancreata.
Fig. 4.

Immature β cells in Pdx1endo-ΔII mice. (A,B) Glucose level at P1-3 (A) and 4 weeks of age (B) for the indicated genotypes. *P<0.05. (C-F′) Four-week-old Pdx1endo-ΔII β cells showed low to no Pdx1 (C′) and MafA (E′), abnormally sustained MafB (F′), and no changes in Nkx6.1 (D′).
Derepression of Arx in Pdx1endo-ΔII endocrine progenitors
We further probed the function of Pdx1 in controlling β-cell versus α-cell fate choice by introducing into Pdx1endo-ΔII mice a Neurog3GFP knock-in allele (Lee et al., 2002) and combining this with immunodetection of CD133 (Prom1), a lumenal apical surface marker (Benitez et al., 2014; Sugiyama et al., 2007), to flow-sort Neurog3-expressing endocrine progenitors. This process enriched for endocrine progenitor cells by guarding against the inclusion of cells that had passed well beyond the endocrine progenitor state but still contained a long-lived GFP signal from the Neurog3GFP knock-in reporter (Fig. S7).
Certain islet-enriched transcription factors have been linked to hormone-selective endocrine lineage commitment, acting within or shortly after the Neurog3-expressing state (Desgraz and Herrera, 2009; Nelson et al., 2007). Arx has been linked to α-cell fate (Collombat et al., 2007, 2003), whereas Pdx1, Pax4 and Nkx6.1 are potent instructors of the β-cell fate (Collombat et al., 2009; Nelson et al., 2007; Schaffer et al., 2013; Yang et al., 2011). Consequently, we flow-sorted Neurog3GFP and CD133 co-positive progenitors from E14.5 Pdx1endo-ΔII pancreata to assess abnormalities in the early-stage lineage bias toward the α-cell pathway. Pdx1 mRNA was substantially decreased (∼50%) in this progenitor population, as were Neurog3, Nkx6.1 and Hnf6 (Fig. 5A). Although in a slightly different cellular context, Pdx1 has been shown to be a direct effector of Nkx6.1 and Hnf6 expression (Schaffer et al., 2013; Teo et al., 2015), and as early-stage endocrine progenitors these cells are expected to express both Nkx6.1 and Hnf6. It is likely that the reduction in expression of these other factors was at least partly caused by a direct transcriptional effect because, for example, both Pdx1 and HNF6 are reported to positively control Neurog3 expression (Jacquemin et al., 2000; Oliver-Krasinski et al., 2009). Arx mRNA was notably elevated over controls. We cannot explain why Pax4 was unaltered (Fig. 5A), but the degree of alteration in Arx might not be translated into the known negative transcriptional effect on Pax4 (Collombat et al., 2003). At the level of chromatin marking, we always found a significant decrease in three marks examined within Area II in Pdx1endo-ΔII endocrine progenitors, an expected outcome because of the deletion of Area II (that is, there is one copy of Area II in Pdx1endo-ΔII versus two copies in control). There was a significant increase in the histone activating marks H3K4me1 and H3K27ac, and a decrease in the H3K27me3 repressive mark, within the Arx UR2 enhancer (Fig. 4B,C, Fig. S8), which is a control region important in maintaining the mature β-cell state (Dhawan et al., 2011). Consistent with the unaltered Pax4 mRNA levels (Fig. 5A), no significant alterations in the levels of activating or repressive histone marks were detected within its respective enhancer regions in Neurog3GFP and CD133 co-positive progenitors at E14.5 (Fig. 5B,C, Fig. S8).
Fig. 5.
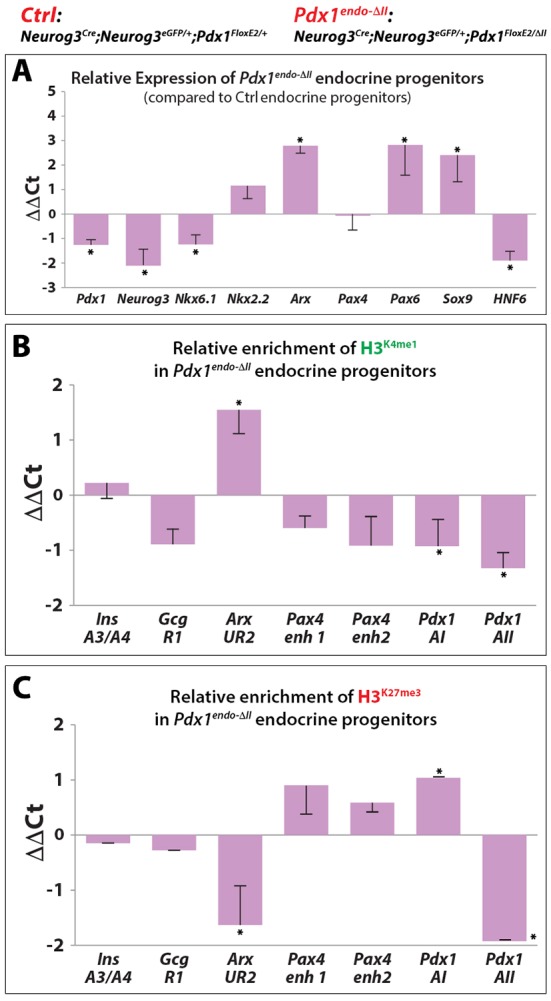
Loss of Pdx1 Area II leads to derepression of Arx in Pdx1endo-ΔII endocrine progenitors. (A) qRT-PCR assessment of relative expression of key epithelial factors comparing sorted Pdx1endo-ΔII endocrine progenitor cells with control. (B,C) ChIP assays showing relative enrichment of H3K4me1 and H3K27me3 (comparing mutant endocrine progenitor cells with the control) at enhancer regions of key transcription factors. *P<0.05.
Area II mediates the higher Pdx1 expression essential for β-cell maturation
As a more selective test of the requirement for Area II within β cells that have progressed relatively further into their lineage, we generated a β-cell-specific Pdx1ΔII/− condition (Pdx1beta-ΔII) by placing the RIP2Cre transgene (in which Cre is driven by the rat Ins2 promoter) in combination with Pdx1FloxE2/ΔII, and examining the effect on islet composition and function (Fig. 6A,A′). These mice showed normal pancreatic organogenesis prior to β-cell specification, as expected. Postnatal Pdx1beta-ΔII animals were hyperglycemic, with very rare Ins+ Gcg+ cells observed (Fig. 6B). Not all β cells showed the marked decrease in Pdx1 protein because of variegated Cre production (Fig. 6D′, Fig. S9C) (Pan et al., 2015). However, in β cells that were YFP+ flow-sorted from Pdx1beta-ΔII;R26REYFP pancreas at P1, we recorded an 88% reduction in Pdx1 mRNA compared with controls (Fig. 6F).
Fig. 6.

Pdx1beta-ΔII β cells are immature and show transcriptional and epigenetic alterations. (A,A′) Schematic presentation of RIP2Cre;Pdx1FloxE2/ΔII (Pdx1beta-ΔII) and RIP2Cre;Pdx1FloxE2/+ (Ctrl). In Pdx1beta-ΔII, the β cells were Pdx1ΔII/− upon recombination. (B,B′) MafA expression was lost in E17.5 Pdx1beta-ΔII β cells (B′). (C-E′) MafB, Ins and Pdx1 expression at P1 (C,C′) and P7 (D-E′). Arrows (D′) indicate abnormal Ins+ MafB+ Pdx1LOW cells. Glut2 was largely diminished in Pdx1beta-DII β cells (E′) compared with the control (E). (F) qRT-PCR analysis of key factors involved in α/β identity and β-cell maturation of Pdx1beta-ΔII β cells isolated by FACS. (G-I) ChIP-qPCR analysis of the enrichment of histone marks H3K27me3, H3K4me1 and H3K27ac at the enhancer region of key factors involved in α/β identity and β-cell maturation from isolated Pdx1ΔII/− β cells. *P<0.05.
Although there were no obvious defects in endocrine cell formation or β/α proportional representation (data not shown), Pdx1beta-ΔII β cells displayed classical characteristics of immaturity, including low levels of MafA (Fig. 6B′,F) and of the β-cell-specific glucose transporter Glut2 (Fig. 6E′,F), and sustained MafB expression (Fig. 6C′,F). The effect on MafA and MafB is likely to be direct because Pdx1 binds to control regions to either positively or negatively influence transcription (Gao et al., 2014; Raum et al., 2006). In addition, Gcg, Brn4 (Pou3f4) and Arx expression was increased in YFP-sorted Pdx1beta-ΔII β cells (Fig. 6F). The Pdx1 Area I enhancer showed an enrichment of the activating marks H3K4me1 and H3K27ac, whereas Pdx1 transcription was significantly decreased (Fig. 6G-I). This suggests that Area I alone, in the absence of Area II, is insufficient to drive adequate Pdx1 transcription. There was a reduction in the activating marks H3K4me1 and H3K27ac and an increase in repressive H3K27me3 levels within the Mafa and Ins enhancers (Fig. 6G-I). Reciprocally, increased H3K4me1/H3K27ac and reduced H3K27me3 were observed at enhancers of the pro-α-cell loci Arx, Mafb and Gcg (Fig. 6G-I). Therefore, the action of the mammal-specific Area II is apparently essential to produce enough Pdx1 to repress a cohort of α-cell-associated genes, with a central influence being the Pdx1-MafA positive-feedback loop.
DISCUSSION
Overall, our data demonstrate that the Area II enhancer-like module within the Pdx1 gene functions to augment the role of Pdx1 at three key stages of mammalian pancreatic endocrine and β-cell differentiation. These include: (1) the creation of an epithelial state to allow the emergence of the correct number of Neurog3-expressing endocrine lineage cells during the secondary transition period of organogenesis; (2) Pdx1-mediated repression of Arx for directing lineage selection as cells move into the β-cell lineage; and (3) Pdx1HIGH expression within β cells that drives switching of Mafb to Mafa expression, and affects other genes acting as principal components of the developmental maturation and physiological maintenance of the β-cell state. These conclusions were supported by the observation that reducing Pdx1 production disturbs the epigenetic configuration and expression of fate-determining genes that have been strongly connected to endocrine cell differentiation and/or islet cell function.
Because Area II is only found in mammals, the degree of conservation of the mechanisms that ensure appropriate spatiotemporal Pdx1 expression in lower vertebrates, such as chickens, for producing the correct number and type of islets of Langerhans, or directing entry into and long-term pursuance of β-cell versus α-cell programs, emerges as an issue of evolutionary interest. We speculate that Area II imparts unique properties to Pdx1 expression, not only affecting the gene itself but also by influencing the expression of other key regulators through distinct transcriptional interactions (Papantonis et al., 2012; Schoenfelder et al., 2010). The Area II control region could have been evolutionarily acquired in mammals to increase the absolute number of β cells or islets, thereby affecting the spectrum of islet sizes, relative intra-islet organization of the various hormone-secreting cell types, or functional β-cell heterogeneity. It is possible that such effects could then indirectly lead to altered cellular communication with the neural and vascular systems. Presumably, non-mammals use non-Area II-dependent processes to produce sufficient appropriately structured islets to function efficiently under the normal physiological parameters and rigors of their own ecological niche. An anomaly within mammals is the desert sand rat Psammomys obesus, which is proposed not to have a Pdx1 gene (Leibowitz et al., 2001). We speculate that genome sequencing might detect a highly divergent version, in which case it will be interesting to determine the degree of conservation of its putative Areas I-II-III-IV cis-regulatory domains.
Pdx1 expression in pancreas organogenesis
Whereas complete removal of Pdx1 Areas I-II-III causes pancreatic agenesis (Fujitani et al., 2006; Offield et al., 1996), the remaining Areas I, III and IV working synthetically in the Pdx1ΔII state were sufficient to drive early pancreatic multipotent progenitor specification and apparently full-scale exocrine acinar and ductal differentiation. Thus, the organization of the Pdx1 transcriptional regulatory landscape in the Pdx1ΔII mutant does not prevent transcription factor and co-factor interactions essential for the early specification and differentiation of non-endocrine cell types, presumably including genomic regulatory interactions that are mediated by chromatin looping (e.g. Pasquali et al., 2014). Previous studies indicate that the broad Pdx1 expression within embryonic pancreatic buds and in the developing acinar compartment involves trans-acting contributions from the Ptf1a transcription factor within Area III and likely also Area IV (Gannon et al., 2001; Wiebe et al., 2007).
Pdx1 and Area II function in endocrine fate selection and progression into maturity
The massive failure of endocrine progenitor formation in the ʻpancreas global' Pdx1ΔII/− germline heteroallelic animals is likely to be due to failures in Pdx1-mediated trans-activation of Neurog3 in the developing pancreatic epithelium. These results indicate that a relatively high Pdx1 level within the epithelium is needed to engage the Neurog3HIGH state, leading to subsequent commitment toward all endocrine cell lineages (Yang et al., 2011). Moreover, the data from the endocrine-specific deletion of Area II function suggest that increased Pdx1 works within endocrine progenitors to direct progeny cells away from the α-cell fate, prominently through the repression of Arx expression and a negative influence via chromatin marking. It is possible that the degree of reduction in the relative number of β cells (versus α cells) in the Pdx1endo-ΔII condition was constrained by our method of creating this state, which used Cre-based inactivation of a conditional protein null allele. Pdx1 protein could thus have persisted in lineage-committing cells at a high enough level, over sufficient time, for large numbers of cells to enter the β-cell lineage. Subsequently, at later stages of ontogeny, the lower Pdx1ΔII-derived protein level could be sufficient to prevent large-scale drift to another endocrine fate (note that substantial numbers of Ins and Gcg co-expressing cells were found) but be inadequate for driving proper maturation to the final β-cell state. These widespread effects under lowered Pdx1 production occurred in the absence of any apparent effect on the level of mRNA for the β-cell determinant Pax4 (Fig. 4A); this Pdx1-Pax4 discrepancy has been discussed previously (e.g. by Yang et al., 2011), but caveats here include possible heterogeneous effects across the cell population, and the current unavailability of reliable antibodies to assess Pax4 protein levels at cellular resolution. The different chromatin marking on the enhancer regions of Arx and Pax4 in Pdx1endo-ΔII endocrine progenitor cells is likely to reflect the potential recruitment of different co-regulator complexes at these loci. It has been reported that Pdx1-recruited Brg-containing versus Brm-containing SWI/SNF complexes play opposing regulatory roles in β cells in vitro (McKenna et al., 2015). Therefore, we speculate that Pdx1 recruits its co-regulator complexes in a cell type- and gene-specific manner, leading to a diversified outcome in the chromatin architecture and transcriptional status of different target genes.
Finding that a reduced proportion of β versus α cells formed in the absence of Area II suggests that other transcription factors (such as Nkx6.1, Nkx2.2 and MafB) working together with the low level of Pdx1 are involved in keeping embryonic endocrine cells on their correct differentiation tracks. In fact, Pdx1 has been shown to directly repress MafB expression in β cells, preventing entry, although possibly incomplete, to the α-like program (Gao et al., 2014; Yang et al., 2011). In terms of deriving the various islet cell types in their correct proportions, the principal effect of removing Area II function seemed to be on the β and α populations, without significantly affecting the other hormone-expressing cell types (although we did score an overall ∼25% reduction in endocrine cell numbers in the Pdx1endo-ΔII condition). Even though the amount of Pdx1 from a Pdx1ΔII allele could, with other β-cell lineage-driving factors, function to preserve largely appropriate lineage allocation, β-cell maturation was nonetheless prevented. Consistent with this idea, we found widespread β-cell dysfunction, without the increased proportional representation of α versus β cells or Ins+ Gcg+ bihormonal cells, with the Pdx1ΔII/− condition generated in insulin-expressing β cells via RIP2Cre. This demonstrates that, once the β-cell lineage is ʻappropriately entered', reductions in Pdx1 protein level as observed in this mutant only affect β-cell maturation and functional activity and not cell identity per se. The β cells under this condition did not drift en masse toward an α-like condition, unlike those under the complete inactivation of Pdx1 described by Gao et al. (2014).
The mixed-islet phenotype, with normally peripheral non-β cells improperly located more centrally within the islet, was detected in all Area II mutant classes (Pdx1ΔII/−, Pdx1ΔII/ΔII, Pdx1endo-ΔII and Pdx1beta-ΔII). We found no evidence that the intermingled α cells came from trans-differentiation of β cells (Fig. S10), but it is unclear what caused this islet architecture disturbance. This islet phenotype is relatively common and often ill-defined regarding the degree of intermixing, and it arises under multiple genetic or physiological perturbations, including β-cell-specific deletion of Pdx1, Foxa2 or Nkx6.1 (Gannon et al., 2008; Schaffer et al., 2013; Sund et al., 2001), persistent Hnf6 expression (Gannon et al., 2000), or under chronic hyperglycemia caused by altering KATP channel function or diet (Brereton et al., 2014; Roat et al., 2014; Shiota et al., 2005). Although the large effects at the mRNA level for the fate-instructive gene Arx (Fig. 4) played out into a more modest effect on β- versus α-cell numbers, this increase in α cells could contribute to islet cell intermixing. In addition, a large number of β cells that are profoundly deficient in moving along the final steps of β-cell differentiation might display altered preferences for other islet cell types as neighbors, allowing extensive intermingling of normally peripheral endocrine cells.
Interestingly, although all Pdx1ΔII/ΔII animals showed abnormal expression of key β-cell genes (Fig. S2C-E), only males developed hyperglycemia at a later stage (Fig. 1E′). In the present study, the cause of this sexual dimorphism phenotype is unclear. This dimorphism is observed in multiple rodent models (Amrani et al., 1998; Bell et al., 1994; Östenson et al., 1989; Yoshioka et al., 1997), possibly because of the protective effect of estrogen (Efrat, 1991) and the tendency for females in general to show increased glucose tolerance (Bonnevienielsen, 1982; Geisler et al., 2002).
MafA/B and Area II interactions in driving acquisition of β-cell maturity
Although MafA and MafB share high protein domain similarity, expression in Ins+ cells during early organogenesis, and the same binding specificity within Pdx1 Area II (Vanhoose et al., 2008), they have different functional requirements in β-cell development. In mouse, MafB predominantly functions embryonically and during early postnatal stages, whereas MafA is only required in postnatal β cells (Artner et al., 2010; Hang et al., 2014). It is likely that MafB normally occupies Area II motifs before Mafa expression is initiated in pro-β cells. The lower level of Pdx1 protein produced under Area II deficiency had a strong negative effect on Mafa but not Mafb expression, suggesting a relatively selective and reciprocal positive feedback between Pdx1 and Mafa in the β-cell lineage. This situation represents an interaction that normally functions to stabilize progression along the maturation phase of the β-cell differentiation program, via the activation of additional target genes.
MATERIALS AND METHODS
Mice
Information on mouse strains, including generation of the Pdx1ΔII allele, is provided in the supplementary Materials and Methods. All animals and embryos were PCR genotyped. Animal handling was under protocols approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee.
Immunohistochemistry, β-galactosidase staining and morphometric analysis
Tissues were prepared and β-galactosidase staining staining was performed as described (Fujitani et al., 2006). Information on antibodies and morphometric methods are provided in the supplementary Materials and Methods and Table S1. For data shown in Figs 2–6, no gender-specific phenotypes were detected. Statistical analysis was performed using single-factor ANOVA tests and significance determined by P<0.05.
FACS analysis, cell sorting, and low cell number ChIP
To generate single cells for FACS, embryos were dissected and dissociated with Accumax (Sigma) for 20-40 min at 37°C, then stopped with an equal volume of L15 (Gibco). The cells were filtered through a nylon mesh, mixed with CD133-PE (0.05 µg per 100 µl; eBioscience 12-1331) for 30 min at 4°C, then washed and purified on a BD FACSDiva cell sorter. ChIP was performed using the Low Cell Number ChIP Kit (Diagenode) as described (Xu et al., 2011), with antibodies against H3K4me1 (Millipore 07-436), H3K27ac (Millipore 07-360) or H3K27me3 (Millipore 07-449; 1 µg per ChIP). Primers are listed in Table S3. All ChIP-qPCR data were normalized to IgG controls.
qRT-PCR
RNA was isolated from flow-sorted pancreatic cells using Trizol (Invitrogen), followed by DNase treatment (Ambion), cDNA synthesis (iScript, Bio-Rad) and qPCR (SsoFast, Bio-Rad) using the primers listed in Table S2. Three samples per genotype per stage were collected and qPCR was performed at least twice on each sample to determine ΔCT. Results are shown as ΔCT±s.e.m. and were subjected to Student's t-test to determine significance (P<0.05).
Acknowledgements
We thank A. Leiter for Neurog3TgBAC-Cre and K. Kaestner for Neurog3EGFP mice; G. Gu for Neurog3 antibodies; C. R. Xu and K. Zaret for suggestions on ChIP analysis; and M. Gannon and members of the C.V.E.W. laboratory for discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Y.-P.Y. and C.V.E.W. developed the concept, designed and/or performed experiments, interpreted data and wrote the manuscript. M.A.M. designed and generated mice. R.S. developed the concept, designed experiments and critically read the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (NIH) [R01 DK050203 to R.S. and C.V.E.W., U01 DK72473 to M.A.M., T32 DK007563 to Y.-P.Y.] and from JDRF to Y.-P.Y. We acknowledge the Vanderbilt University Cell Imaging and Transgenic Mouse/ES Cell shared resources, which are supported in part by either the Vanderbilt University Medical Center Digestive Disease Research Center, the Vanderbilt Diabetes Research Training Center, or the Vanderbilt-Ingram Cancer Center [supported by NIH grants CA68485, DK20593, DK58404 and DK59637]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.143123.supplemental
References
- Amrani A., Durant S., Throsby M., Coulaud J., Dardenne M. and Homo-Delarche F. (1998). Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology 139, 1115-1124. 10.1210/en.139.3.1115 [DOI] [PubMed] [Google Scholar]
- Artner I., Hang Y., Mazur M., Yamamoto T., Guo M., Lindner J., Magnuson M. A. and Stein R. (2010). MafA and MafB regulate genes critical to {beta} cells in a unique temporal manner. Diabetes 59, 2530-2539. 10.2337/db10-0190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell R. C., Khurana M., Ryan E. A. and Finegood D. T. (1994). Gender differences in the metabolic response to graded numbers of transplanted islets of langerhans. Endocrinology 135, 2681-2687. [DOI] [PubMed] [Google Scholar]
- Benitez C. M., Qu K., Sugiyama T., Pauerstein P. T., Liu Y., Tsai J., Gu X., Ghodasara A., Arda H. E., Zhang J. et al. (2014). An integrated cell purification and genomics strategy reveals multiple regulators of pancreas development. PLoS Genet. 10, e1004645 10.1371/journal.pgen.1004645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnevienielsen V. (1982). Different effects of high glucose and high-fat diet on pancreatic insulin and glucagon in female and male-mice. Diabetes Metab. 8, 271-277. [PubMed] [Google Scholar]
- Boyer D. F., Fujitani Y., Gannon M., Powers A. C., Stein R. W. and Wright C. V. E. (2006). Complementation rescue of Pdx1 null phenotype demonstrates distinct roles of proximal and distal cis-regulatory sequences in pancreatic and duodenal expression. Dev. Biol. 298, 616-631. 10.1016/j.ydbio.2006.07.020 [DOI] [PubMed] [Google Scholar]
- Brereton M. F., Iberl M., Shimomura K., Zhang Q., Adriaenssens A. E., Proks P., Spiliotis I. I., Dace W., Mattis K. K., Ramracheya R. et al. (2014). Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat. Commu. 5, 4639 10.1038/ncomms5639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Mansouri A., Hecksher-Sorensen J., Serup P., Gradwohl G. and Gruss P. (2003). Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 17, 2591-2603. 10.1101/gad.269003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Hecksher-Sørensen J., Krull J., Berger J., Riedel D., Herrera P. L., Serup P. and Mansouri A. (2007). Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J. Clin. Invest. 117, 961-970. 10.1172/JCI29115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P., Xu X., Ravassard P., Sosa-Pineda B., Dussaud S., Billestrup N., Madsen O. D., Serup P., Heimberg H. and Mansouri A. (2009). The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 138, 449-462. 10.1016/j.cell.2009.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgraz R. and Herrera P. L. (2009). Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 136, 3567-3574. 10.1242/dev.039214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S., Georgia S., Tschen S.-I., Fan G. and Bhushan A. (2011). Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 20, 419-429. 10.1016/j.devcel.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrat S. (1991). Sexual dimorphism of pancreatic beta-cell degeneration in transgenic mice expressing an insulin-ras hybrid gene. Endocrinology 128, 897-901. 10.1210/endo-128-2-897 [DOI] [PubMed] [Google Scholar]
- Fujitani Y., Fujitani S., Boyer D. F., Gannon M., Kawaguchi Y., Ray M., Shiota M., Stein R. W., Magnuson M. A. and Wright C. V. E. (2006). Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 20, 253-266. 10.1101/gad.1360106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon M., Ray M. K., Van Zee K., Rausa F., Costa R. H. and Wright C. V. (2000). Persistent expression of HNF6 in islet endocrine cells causes disrupted islet architecture and loss of beta cell function. Development 127, 2883-2895. [DOI] [PubMed] [Google Scholar]
- Gannon M., Gamer L. W. and Wright C. V. E. (2001). Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev. Biol. 238, 185-201. 10.1006/dbio.2001.0359 [DOI] [PubMed] [Google Scholar]
- Gannon M., Ables E. T., Crawford L., Lowe D., Offield M. F., Magnuson M. A. and Wright C. V. E. (2008). pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev. Biol. 314, 406-417. 10.1016/j.ydbio.2007.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T., Yang C., Pannikar A., Doliba N., Zhang T. et al. (2014). Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 19, 259-271. 10.1016/j.cmet.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler J. G., Zawalich W., Zawalich K., Lakey J. R. T., Stukenbrok H., Milici A. J. and Soeller W. C. (2002). Estrogen can prevent or reverse obesity and diabetes in mice expressing human islet amyloid polypeptide. Diabetes 51, 2158-2169. 10.2337/diabetes.51.7.2158 [DOI] [PubMed] [Google Scholar]
- Gerrish K., Gannon M., Shih D., Henderson E., Stoffel M., Wright C. V. and Stein R. (2000). Pancreatic beta cell-specific transcription of the pdx-1 gene. The role of conserved upstream control regions and their hepatic nuclear factor 3beta sites. J. Biol. Chem. 275, 3485-3492. 10.1074/jbc.275.5.3485 [DOI] [PubMed] [Google Scholar]
- Gerrish K., Cissell M. A. and Stein R. (2001). The role of hepatic nuclear factor 1 alpha and PDX-1 in transcriptional regulation of the pdx-1 gene. J. Biol. Chem. 276, 47775-47784. [DOI] [PubMed] [Google Scholar]
- Gu G., Dubauskaite J. and Melton D. A. (2002). Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447-2457. [DOI] [PubMed] [Google Scholar]
- Hale M. A., Kagami H., Shi L., Holland A. M., Elsässer H.-P., Hammer R. E. and MacDonald R. J. (2005). The homeodomain protein PDX1 is required at mid-pancreatic development for the formation of the exocrine pancreas. Dev. Biol. 286, 225-237. 10.1016/j.ydbio.2005.07.026 [DOI] [PubMed] [Google Scholar]
- Hang Y., Yamamoto T., Benninger R. K. P., Brissova M., Guo M., Bush W., Piston D. W., Powers A. C., Magnuson M., Thurmond D. C. et al. (2014). The MafA transcription factor becomes essential to islet beta-cells soon after birth. Diabetes 63, 1994-2005. 10.2337/db13-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumaitre C., Barbacci E., Jenny M., Ott M. O., Gradwohl G. and Cereghini S. (2005). Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc. Natl. Acad. Sci. USA 102, 1490-1495. 10.1073/pnas.0405776102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemin P., Durviaux S. M., Jensen J., Godfraind C., Gradwohl G., Guillemot F., Madsen O. D., Carmeliet P., Dewerchin M., Collen D. et al. (2000). Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 20, 4445-4454. 10.1128/MCB.20.12.4445-4454.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J., Hara M., Ahlgren U., Sorenson R. L. and Periwal V. (2012). Mathematical models of pancreatic islet size distributions. Islets 4, 10-19. 10.4161/isl.18660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J., Carlsson L., Edlund T. and Edlund H. (1994). Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371, 606-609. 10.1038/371606a0 [DOI] [PubMed] [Google Scholar]
- Kodama S., Nakano Y., Hirata K., Furuyama K., Horiguchi M., Kuhara T., Masui T., Kawaguchi M., Gannon M., Wright C. V. E. et al. (2016). Diabetes caused by elastase-cre-mediated Pdx1 inactivation in mice. Sci. Rep. 6, 21211 10.1038/srep21211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J. L., Dubois C. L., Schaffer A. E., Hao E., Shih H. P., Seymour P. A., Ma J. and Sander M. (2011). Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138, 653-665. 10.1242/dev.056499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. S., Perreault N., Brestelli J. E. and Kaestner K. H. (2002). Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 16, 1488-1497. 10.1101/gad.985002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz G., Yuli M., Donath M. Y., Nesher R., Melloul D., Cerasi E., Gross D. J. and Kaiser N. (2001). beta-cell glucotoxicity in the Psammomys obesus model of type 2 diabetes. Diabetes 50 Suppl. 1, S113-S117. 10.2337/diabetes.50.2007.S113 [DOI] [PubMed] [Google Scholar]
- Lynn F. C., Smith S. B., Wilson M. E., Yang K. Y., Nekrep N. and German M. S. (2007). Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl. Acad. Sci. USA 104, 10500-10505. 10.1073/pnas.0704054104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna B., Guo M., Reynolds A., Hara M. and Stein R. (2015). Dynamic recruitment of functionally distinct Swi/Snf chromatin remodeling complexes modulates Pdx1 activity in islet beta cells. Cell Rep. 10, 2032-2042. 10.1016/j.celrep.2015.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. B., Schaffer A. E. and Sander M. (2007). The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta-cell fate specification in Pdx1+ pancreatic progenitor cells. Development 134, 2491-2500. 10.1242/dev.002691 [DOI] [PubMed] [Google Scholar]
- Offield M. F., Jetton T. L., Labosky P. A., Ray M., Stein R. W., Magnuson M. A., Hogan B. L. and Wright C. V. (1996). PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983-995. [DOI] [PubMed] [Google Scholar]
- Oliver-Krasinski J. M., Kasner M. T., Yang J., Crutchlow M. F., Rustgi A. K., Kaestner K. H. and Stoffers D. A. (2009). The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J. Clin. Invest. 119, 1888-1898. 10.1172/JCI37028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östenson C. G., Grill V. and Roos M. (1989). Studies on sex dependency of b-cell susceptibility to streptozotocin in a rat model of type-Ii diabetes-mellitus. Exp. Clin. Endocrinol. 93, 241-247. 10.1055/s-0029-1210863 [DOI] [PubMed] [Google Scholar]
- Pan F. C. and Wright C. (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn. 240, 530-565. 10.1002/dvdy.22584 [DOI] [PubMed] [Google Scholar]
- Pan F. C., Brissova M., Powers A. C., Pfaff S. and Wright C. V. (2015). Inactivating the permanent neonatal diabetes gene Mnx1 switches insulin-producing beta-cells to a delta-like fate and reveals a facultative proliferative capacity in aged beta-cells. Development 142, 3637-3648. 10.1242/dev.126011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papantonis A., Kohro T., Baboo S., Larkin J. D., Deng B., Short P., Tsutsumi S., Taylor S., Kanki Y., Kobayashi M. et al. (2012). TNFalpha signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 31, 4404-4414. 10.1038/emboj.2012.288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali L., Gaulton K. J., Rodriguez-Seguí S. A., Mularoni L., Miguel-Escalada I., Akerman İ., Tena J. J., Morán I., Gómez-Marín C., van de Bunt M. et al. (2014). Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 46, 136-143. 10.1038/ng.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raum J. C., Gerrish K., Artner I., Henderson E., Guo M., Sussel L., Schisler J. C., Newgard C. B. and Stein R. (2006). FoxA2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol. Cell. Biol. 26, 5735-5743. 10.1128/MCB.00249-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roat R., Rao V., Doliba N. M., Matschinsky F. M., Tobias J. W., Garcia E., Ahima R. S. and Imai Y. (2014). Alterations of pancreatic islet structure, metabolism and gene expression in diet-induced obese C57BL/6J mice. PLoS ONE 9, e86815 10.1371/journal.pone.0086815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M., Neubüser A., Kalamaras J., Ee H. C., Martin G. R. and German M. S. (1997). Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 11, 1662-1673. 10.1101/gad.11.13.1662 [DOI] [PubMed] [Google Scholar]
- Schaffer A. E., Taylor B. L., Benthuysen J. R., Liu J., Thorel F., Yuan W., Jiao Y., Kaestner K. H., Herrera P. L., Magnuson M. A. et al. (2013). Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic beta cell identity. PLoS Genet. 9, e1003274 10.1371/journal.pgen.1003274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S., Sexton T., Chakalova L., Cope N. F., Horton A., Andrews S., Kurukuti S., Mitchell J. A., Umlauf D., Dimitrova D. S. et al. (2010). Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42, 53-61. 10.1038/ng.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota C., Rocheleau J. V., Shiota M., Piston D. W. and Magnuson M. A. (2005). Impaired glucagon secretory responses in mice lacking the type 1 sulfonylurea receptor. Am. J. Physiol. Endocrinol. Metab. 289, E570-E577. 10.1152/ajpendo.00102.2005 [DOI] [PubMed] [Google Scholar]
- Stoffers D. A., Ferrer J., Clarke W. L. and Habener J. F. (1997a). Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17, 138-139. 10.1038/ng1097-138 [DOI] [PubMed] [Google Scholar]
- Stoffers D. A., Thomas M. K. and Habener J. F. (1997b). Homeodomain protein IDX-1: a master regulator of pancreas development and insulin gene expression. Trends Endocrinol. Metab. 8, 145-151. 10.1016/S1043-2760(97)00008-8 [DOI] [PubMed] [Google Scholar]
- St-Onge L., Sosa-Pineda B., Chowdhury K., Mansouri A. and Gruss P. (1997). Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 387, 406-409. 10.1038/387406a0 [DOI] [PubMed] [Google Scholar]
- Sugiyama T., Rodriguez R. T., McLean G. W. and Kim S. K. (2007). Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc. Natl. Acad. Sci. USA 104, 175-180. 10.1073/pnas.0609490104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sund N. J., Vatamaniuk M. Z., Casey M., Ang S. L., Magnuson M. A., Stoffers D. A., Matschinsky F. M. and Kaestner K. H. (2001). Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev. 15, 1706-1715. 10.1101/gad.901601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift G. H., Liu Y., Rose S. D., Bischof L. J., Steelman S., Buchberg A. M., Wright C. V. E. and MacDonald R. J. (1998). An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2). Mol. Cell. Biol. 18, 5109-5120. 10.1128/MCB.18.9.5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo A. K. K., Tsuneyoshi N., Hoon S., Tan E. K., Stanton L. W., Wright C. V. E. and Dunn N. R. (2015). PDX1 binds and represses hepatic genes to ensure robust pancreatic commitment in differentiating human embryonic stem cells. Stem Cell Rep. 4, 578-590. 10.1016/j.stemcr.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J., García-Hurtado J., Moran I., Maestro M. A., Xu X., Van De Casteele M., Skoudy A. L., Palassini M., Heimberg H. and Ferrer J. (2010). Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 20, 722-732. 10.1101/gr.101709.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoose A. M., Samaras S., Artner I., Henderson E., Hang Y. and Stein R. (2008). MafA and MafB regulate Pdx1 transcription through the Area II control region in pancreatic beta cells. J. Biol. Chem. 283, 22612-22619. 10.1074/jbc.M802902200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Velkinburgh J. C., Samaras S. E., Gerrish K., Artner I. and Stein R. (2005). Interactions between areas I and II direct pdx-1 expression specifically to islet cell types of the mature and developing pancreas. J. Biol. Chem. 280, 38438-38444. 10.1074/jbc.M508594200 [DOI] [PubMed] [Google Scholar]
- Wiebe P. O., Kormish J. D., Roper V. T., Fujitani Y., Alston N. I., Zaret K. S., Wright C. V. E., Stein R. W. and Gannon M. (2007). Ptf1a binds to and activates area III, a highly conserved region of the Pdx1 promoter that mediates early pancreas-wide Pdx1 expression. Mol. Cell. Biol. 27, 4093-4104. 10.1128/MCB.01978-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R., Everett L. J., Lim H.-W., Patel N. A., Schug J., Kroon E., Kelly O. G., Wang A., D'Amour K. A., Robins A. J. et al. (2013). Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 12, 224-237. 10.1016/j.stem.2012.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.-R., Cole P. A., Meyers D. J., Kormish J., Dent S. and Zaret K. S. (2011). Chromatin “prepattern” and histone modifiers in a fate choice for liver and pancreas. Science 332, 963-966. 10.1126/science.1202845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.-P., Thorel F., Boyer D. F., Herrera P. L. and Wright C. V. E. (2011). Context-specific {alpha}-to-{beta}-cell reprogramming by forced Pdx1 expression. Genes Dev. 25, 1680-1685. 10.1101/gad.16875711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M., Kayo T., Ikeda T. and Koizumi A. (1997). A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46, 887-894. 10.2337/diab.46.5.887 [DOI] [PubMed] [Google Scholar]
- Zhang H., Ables E. T., Pope C. F., Washington M. K., Hipkens S., Means A. L., Path G., Seufert J., Costa R. H., Leiter A. B. et al. (2009). Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech. Dev. 126, 958-973. 10.1016/j.mod.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]