

Abstract
Acquired thrombotic thrombocytopenic purpura is primarily caused by the deficiency of plasma ADAMTS13 activity resulting from autoantibodies against ADAMTS13. However, ADAMTS13 deficiency alone is often not sufficient to cause acute thrombotic thrombocytopenic purpura. Infections or systemic inflammation may precede acute bursts of the disease, but the underlying mechanisms are not fully understood. Herein, 52 patients with acquired autoimmune thrombotic thrombocytopenic purpura and 30 blood donor controls were recruited for the study. The plasma levels of human neutrophil peptides 1–3 and complement activation fragments (i.e. Bb, iC3b, C4d, and sC5b-9) were determined by enzyme-linked immunosorbent assays. Univariate analyses were performed to determine the correlation between each biomarker and clinical outcomes. We found that the plasma levels of human neutrophil peptides 1–3 and Bb in patients with acute thrombotic thrombocytopenic purpura were significantly higher than those in the control (P<0.0001). The plasma levels of HNP1-3 correlated with the levels of plasma complement fragment Bb (rho=0.48, P=0.0004) and serum lactate dehydrogenase (rho=0.28, P=0.04); in addition, the plasma levels of Bb correlated with iC3b (rho=0.55, P<0.0001), sC5b-9 (rho=0.63, P<0.0001), serum creatinine (rho=0.42, p=0.0011), and lactate dehydrogenase (rho=0.40, P=0.0034), respectively. Moreover, the plasma levels of iC3b and sC5b-9 were correlated (rho=0.72, P<0.0001), despite no statistically significant difference of the two markers between thrombotic thrombocytopenic purpura patients and the control. We conclude that innate immunity, i.e. neutrophil and complement activation via the alternative pathway, may play a role in the pathogenesis of acute autoimmune thrombotic thrombocytopenic purpura, and a therapy targeted at these pathways may be considered in a subset of these patients.
Introduction
Thrombotic thrombocytopenic purpura (TTP) is characterized by the formation of disseminated microvascular thrombosis in small arterioles and capillaries.1 TTP patients manifest with severe thrombocytopenia (usually less than 20,000/μl of platelet counts), microangiopathic hemolytic anemia with elevated levels of serum lactate dehydrogenase (LDH) and schistocytes on a peripheral blood smear, and signs and symptoms of end-organ dysfunction, including renal failure and/or myocardial or cerebral infarctions.2 Severe deficiency of plasma ADAMTS13 activity (usually <10%), resulting from ADAMTS13 mutations or autoantibodies against ADAMTS13, appears to be the key pathogenic factor of TTP.3,4
ADAMTS13 is a plasma metalloprotease that cleaves von Willebrand factor (VWF) at the Tyr1605-Met1606 bond, thereby regulating hemostasis and preventing thrombosis after vascular injury.4 In patients with hereditary TTP, the lower the plasma ADAMTS13 activity is, the earlier the initial TTP episode occurs,5 suggesting that any residual ADAMTS13 activity may be protective. Treatment with plasma infusion, aimed at increasing plasma ADAMTS13 activity to greater than 5%, is clinically effective in hereditary TTP.6 In patients with acquired TTP, the immunoglobulin (IgG) type of autoantibodies, which bind primarily to the spacer domain of ADAMTS13,7 result in competitive inhibition of plasma ADAMTS13 activity. Therapeutic plasma exchange, often used in combination with immunosuppression, including corticosteroids, vincristine, cyclophosphamide, and rituximab, etc., remains the treatment of choice for acquired TTP with inhibitors.
Clinical observations have also demonstrated that most hereditary TTP patients with plasma ADAMTS13 activity of less than 10% remain asymptomatic for many years before experiencing their first episode.8,9 Patients with acquired autoimmune TTP may achieve clinical remission after therapeutic plasma exchange and other adjunctive therapies, despite ongoing severe deficiency of plasma ADAMTS13 activity and the presence of inhibitors.10 These findings indicate that a triggering event may be necessary to provoke the initial onset of TTP and subsequent recurrent or relapsing episodes. For example, central catheter infection, systemic inflammation, certain medications, and pregnancy are known to be the potential inciting factors for TTP. However, the mechanisms underlying such a triggering event remain poorly understood. Based on our recently published study, wherein we demonstrated that HNP1-3, a group of 29–30 amino acid antimicrobial peptides, potentially released from activated human neutrophils, is a potent inhibitor of ADAMTS13-medidated VWF proteolysis.11 We hypothesize that the locally released HNP1-3 may play a role in the pathogenesis of acute TTP, particularly in those with severely low circulating ADAMTS13 activity.
Moreover, several recent studies have indicated that complement activation may be another inciting factor that affects the onset, clinical presentation, and outcome of thrombotic microangiopathy (TMA), including TTP. For instance, serum from patients with TMA caused C3 and membrane attack complex (MAC) deposition on human microvascular endothelial cells (HMEC)-1 and its cytotoxic effect was abolished by complement inhibition.12 Additionally, plasma levels of C3a and C5a were significantly elevated in patients during acute TTP as compared with those in remission.13 Most importantly, complement factor H mutations were identified in 5 out of 6 patients with ticlopidine (anti-platelet drug)-associated TTP with severe deficiency of plasma ADAMTS13 activity.14 Together, these preliminary findings suggest that complement activation via the alternative pathway and severe ADAMTS13 deficiency may play a synergistic role in the pathogenesis of TTP. However, the relationship between the measurement of inflammatory and complement activation markers at the onset of acute TTP and clinical consequences has not been investigated in a large cohort of acquired autoimmune TTP patients.
Methods
Patients
The Institutional Review Board (IRB) of the University of Alabama at Birmingham (UAB), USA, approved the study. TTP was diagnosed in patients with thrombocytopenia (platelet count <150×103/μL) and microangiopathic hemolytic anemia (indicated by reduced hematocrit, increased serum LDH, and the presence of the fragmentation of red blood cells, i.e. schistocytes on the peripheral blood smear), with or without signs and/or symptoms of major organ damage and in the absence of an alternative diagnosis to explain the TMA.15 All patients underwent therapeutic plasma exchange. Blood samples anticoagulated with sodium citrate (0.32%) were obtained prior to the initiation of therapeutic plasma exchange. Control samples were obtained from healthy blood donors whose medical history details were not collected or available for the purpose of this study. Blood was centrifuged at 3,000 rpm for 15 min, and plasma was aspirated from the top and stored in aliquots at −80 °C until assays were performed. All samples were frozen and thawed only once prior to the study.
Assays for plasma ADAMTS13 activity and inhibitors
A commercial FRETS-VWF73 assay in a reference lab (Blood Center of Wisconsin, Milwaukee, WI, USA)16 and a homegrown recombinant FRETS-VWF73 (rF-VWF73) assay determined plasma ADAMTS13 activity as described previously.17 ADAMTS13 inhibitor titers were similarly determined by measuring the residual enzyme activity in normal human plasma (NHP) after being mixed (50:50) and incubated for 30 min with the patient’s plasma (various dilutions) at 37 °C, using the prior mentioned commercial FRETS-VWF73 assay in the reference lab.
Assays for plasma complement activation markers
Enzyme-linked immunosorbent assay (ELISA) kits for complement activation markers, including iC3b, sC5b-9, Bb and C4d, were obtained from a commercial source (MicroVue, San Diego, CA, USA), and the assays were performed on the diluted plasma samples according to the manufacturers’ recommendations.
Statistical analysis
A Nonparametric Mann-Whitney test was used for comparison between the two groups. A nonparametric Spearman rank correlation (rho) was determined between the parameters. The P values <0.05 and <0.01 were considered statistically significant and highly significant, respectively.
Results
Characteristics of TTP patients
A total of 52 patients with acquired TTP were recruited into the study. The detailed demographic information, clinical data, and laboratory results are shown in the Online Supplementary Table S1. Of these, the female to male ratio was 27/25 with 73% being African-American, 25% Caucasian and 2% Hispanic. Comorbidities, including obesity, diabetes mellitus, hypertension, rheumatoid arthritis, systemic lupus erythematosus, etc., were present in 75% of patients. The majority (77%) of patients presented with an initial episode of TTP, and the remainder (23%) presented with a relapse or exacerbation. Nearly 54% of patients had only one episode and 46% had ≥2 episodes. The blood culture performed was positive in 6/26 (23%) of cases (Table 1). Other laboratory tests on admission showed a median platelet count of 14.3 (4.7–108)×103/μL, a median hematocrit of 25% (14–40%), a median white blood cell count of 11.8 (4.5–24.5) ×103/μL, a median serum LDH of 878.6 (226–7,332) U/dL, and a median serum creatinine of 1.3 (0.1–8.5) mg/dL. The median plasma ADAMTS13 activity was 1.2% (0–8%), which was significantly lower than the control levels of 118% (85–226%) (Online Supplementary Table S1, Table 2, and Figure 1A). The mixing study identified inhibitors (>0.4 U/ml) in 90% of TTP patients with a median titer of 2 (0.4 to >8 U/ml) (Online Supplementary Table S1 and Figure 1B). An immunoassay confirmed the presence of anti-ADAMTS13 IgG in all TTP patients including 6 patients with <0.4 U/ml by the mixing assay (Online Supplementary Table S1). These results support the fact that all patients with ADAMTS13 activity <10% in the study cohort had an acquired autoimmune TTP.
Table 1.
Demographic and clinical data of 52 patients with acquired TTP.
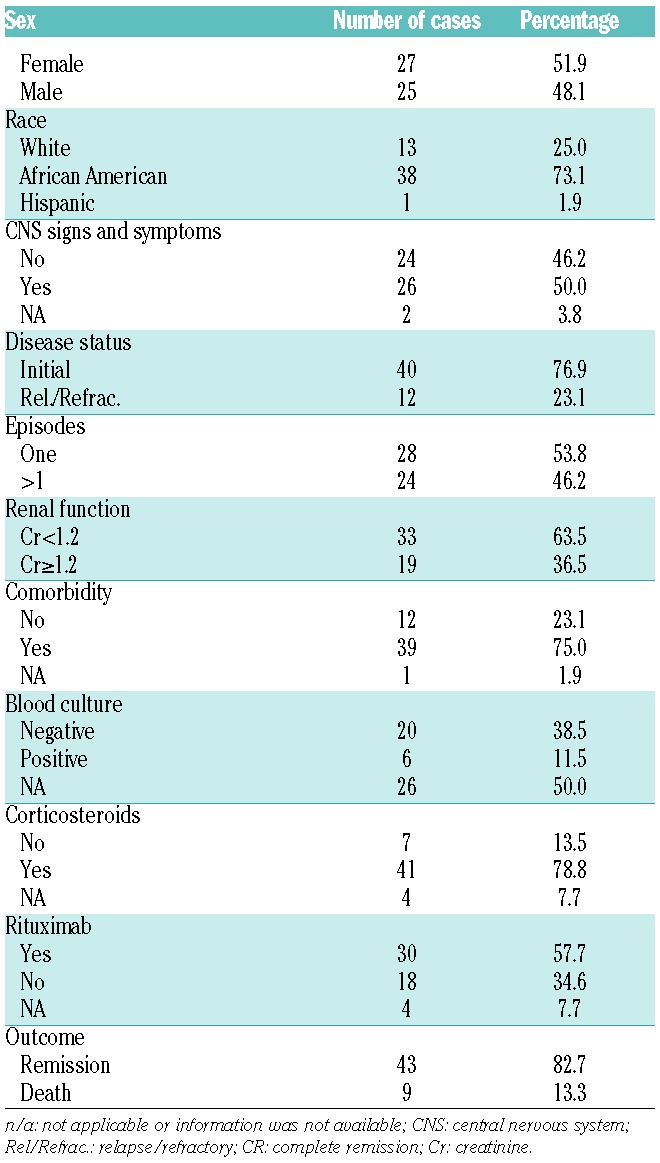
Table 2.
Laboratory results of TTP patients and healthy controls.
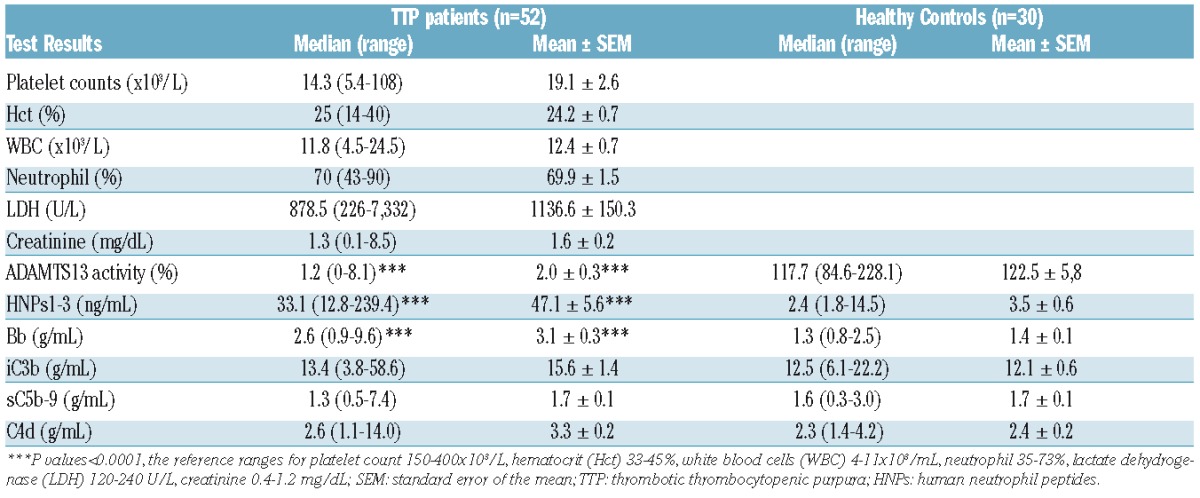
Figure 1.
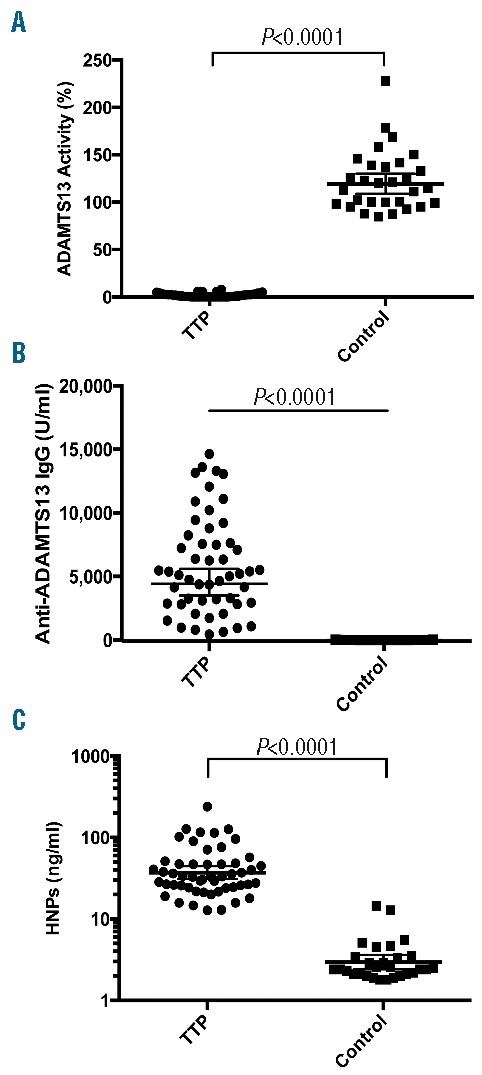
Plasma ADAMTS13 activity and HNP1-3 in patients with TTP and controls. Plasma ADAMTS13 activity (A), anti-ADAMTS13 IgG (B), and HNP1-3 (C) in TTP patients and controls were determined by the cleavage of rF-VWF73 and commercial ELISAs according to manufacturer’s recommendation. A nonparametric Mann-Whitney U test was performed. The P values <0.0001 are highly statistically significant. The horizontal lines within the dots represent the geometric mean ± 95% confidential interval; TTP: thrombotic thrombocytopenic purpura; HNP: human neutrophil peptide; ELISA: enzyme-linked immunosorbent assay. IgG; immunoglobin G.
Therapeutic plasma exchange, corticosteroids, and rituximab were offered on admission to 100%, 79%, and 58% of patients, respectively. Follow-up results indicated that the mortality rate was 19.9%, with all but one patient dying within 30 days of admission (Online Supplementary Table S1 and Table 1), consistent with results reported by other groups.18,19
Plasma HNP1-3 in TTP patients and controls
Neutrophil activation is common in patients with acute TTP.20,21 To determine if HNP1-3 are released from activated neutrophils, we measured their plasma concentrations in patients with acute TTP by an immunoassay. As shown in Table 2 and Figure 1C, the median plasma level of HNP1-3 in acute TTP patients was 33.1 (12.8–239.4) ng/ml, significantly higher than the median level of 2.4 (1.8–14.5) in the blood donor controls (P<0.0001). Plasma HNP1-3 was independent of total white blood cell counts (rho=0.10, P=0.47), the percentage of neutrophils (rho=0.13, P=0.36), and absolute neutrophil counts (rho=0.15, P=0.28) (Online Supplementary Table S1). These results suggest that neutrophil activation rather than neutrophil counts contribute to the increased levels of plasma HNP in patients with acute TTP.
Plasma complement activation fragments in TTP and controls
Complement activation is the primary cause of atypical hemolytic uremic syndrome (aHUS),22 but it may also be a risk factor for the development of acquired TTP. Plasma complement fragments generated as a consequence of activation via the classical pathway (C4d), the alternative pathway (Bb), both the pathways of (iC3b), and the formations of the terminal complexes (sC5b-9)22 were determined by immunoassays in 52 TTP plasma samples. The results showed that the median level of plasma Bb in TTP patients was 2.6 (0.9–9.6) μg/mL, significantly higher than the median level of 1.3 (0.8–2.5) μg/mL in the blood donor controls (P<0.0001) (Table 2 and Figure 2A). A small fraction of TTP patients exhibited high levels of plasma C4d (Figure 2B), iC3b (Figure 2C), and sC5b-9 (Figure 2D), but there was no statistically significant difference of these fragments in TTP samples as a group when compared with those in the controls (Table 2 and Figure 2). These results indicate that complement activation via the alternative pathway is present in a subset of patients with acute autoimmune TTP.
Figure 2.
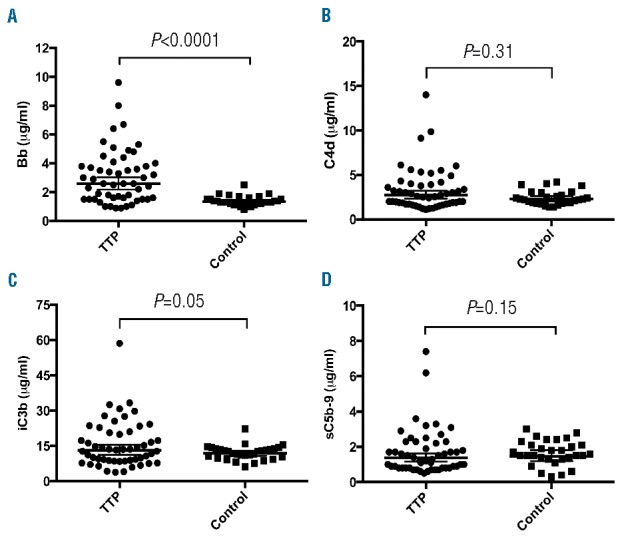
Plasma levels of complement fragments Bb, C4d, iC3b, and sC5b-9 in TTP patients and controls. Commercial ELISAs were used to determine plasma levels of Bb (A), C4d (B), iC3b (C), and sC5b-9 complexes (D) according to the manufacturer’s protocol. The horizontal lines within the dots represent the geometric means ± 95% confidential intervals. A Mann-Whitney U test was performed and the P value (<0.0001) in panel A is statistically highly significant. The P values in all other panels are greater than 0.05, not statistically significant. TTP: thrombotic thrombocytopenic purpura; ELISA: enzyme-linked immunosorbent assay.
Correlations between HNP1-3 or Bb and other key clinical and laboratory parameters
To determine the relationship between inflammation, complement activation and end-organ damage during acute episodes, Spearman rank correlation coefficients (rho) were determined between HNP1-3 or Bb and various other biomarkers and clinical parameters. Our results showed that in TTP patients their plasma levels of HNP1-3 correlated with plasma levels of Bb (rho=0.48, P<0.001) (Figure 3A) and serum LDH (rho=0.28, P<0.05) (Figure 3B), but not with the inhibitor titer (P>0.05) (Figure 3C) and serum creatinine (P>0.05) (Figure 3D).
Figure 3.
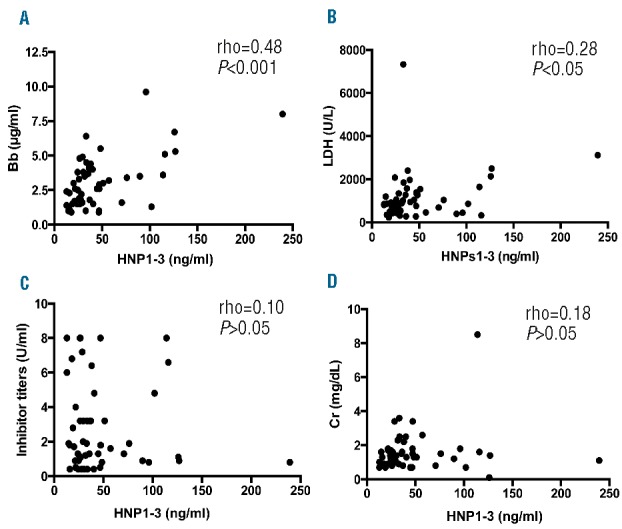
Correlations between HNP1-3 and the other key laboratory parameters. Nonparametric Spearman rank correlation coefficient (rho) tests were performed to determine the correlations between plasma levels of HNP1-3 and Bb (A), serum LDH (B), anti-ADAMTS13 inhibitor titer (C), and serum creatinine (D), respectively, in patients with acquired TTP. The P values in panel A (P<0.001) and panel B (P<0.05) are considered to be statistically highly significant and significant, respectively. The P values in panels C and D (≥ 0.05) are not statistically significant. HNP: human neutrophil peptide; LDH: lactate dehydrogenase; rho: Spearman rank correlation coefficient.
As expected, plasma Bb in these patients significantly correlated with iC3b (rho=0.55, P<0.0001) (Figure 4A), sC5b-9 (r=0.64, P<0.0001) (Figure 4B), serum LDH (rho=0.40, P=0.003) (Figure 4C), and serum creatinine (rho=0.42, P=0.0011) (Figure 4D), respectively. In addition, plasma levels of iC3b in TTP patients highly correlated with plasma sC5b-9 (rho=0.72, P<0.0001) (Figure 4E), but not with serum creatinine (Figure 4F) and LDH (Figure 4G). No statistically significant correlation was observed between plasma levels of sC5b-9 and serum creatinine (Figure 4H). None of these biomarkers measured in this cohort of acute TTP samples predicted the relapse and mortality rate (data not shown). Nevertheless, these results suggest that innate immunity, including neutrophil activation and complement activation via an alternative pathway, may participate in the onset and progress of the disease in a subset of TTP patients.
Figure 4.
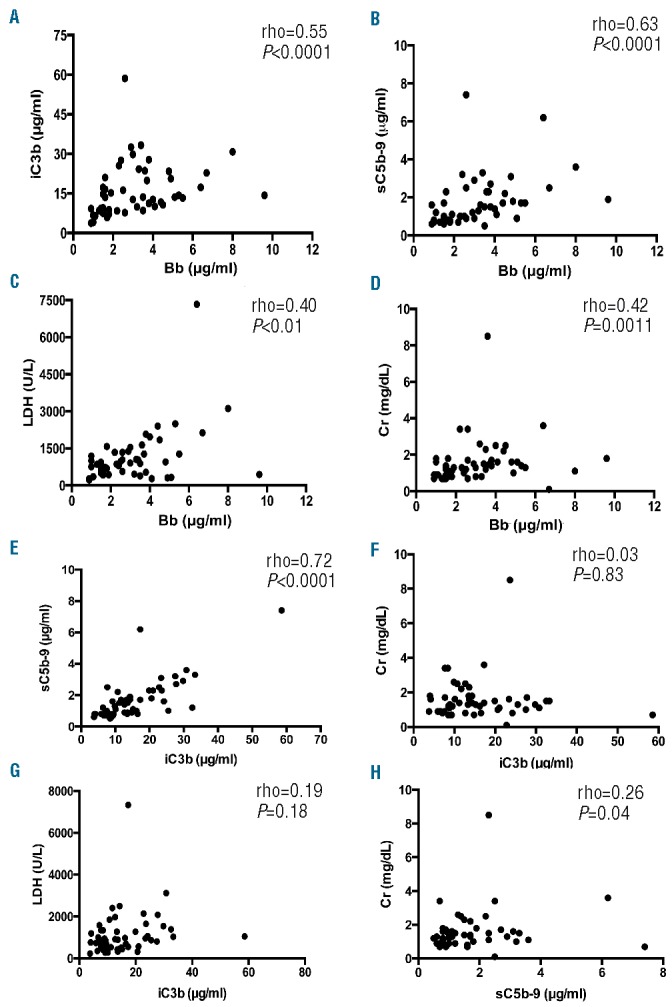
Correlations between complement fragments and the severity of end-organ damage. Nonparametric Spearman rank correlation coefficients (rho) were determined between plasma Bb and iC3b (A), between Bb and sC5b-9 (B), between Bb and serum LDH (C), and between Bb and creatinine (D). Also, the Spearman rank correlation coefficients between iC3b and sC5b-9 (E), between iC3b and serum creatinine (F), between iC3b and serum LDH (G), and between sC5b-9 and creatinine (H) were determined. The P values < 0.05 and < 0.01 are considered to be statistically significant and highly significant, respectively. LDH: lactate dehydrogenase; rho; Spearman rank correlation coefficient; Cr: creatinine.
Discussion
The present study demonstrates the significant increase in plasma HNP1-3 and Bb in patients with acute autoimmune TTP when compared with the control; the increased plasma levels of HNP1-3 correlate with Bb, which, in turn, correlates with iC3b and sC5b-9, as well as serum LDH and/or creatinine. This is, to our knowledge, by far the largest and the most comprehensive analysis of the relationship between the innate immunity (i.e. neutrophil activation and complement activation) and the disease onset, progression, and long-term outcome in patients with TTP.
How could HNP1-3 contribute to the onset of TTP? Nearly all acquired TTP with severe deficiency of plasma ADAMTS13 activity (< 10% of normal) is caused by autoantibodies against ADAMTS13.23,24 The autoantibodies bind to the spacer domain of ADAMTS13, which may physically block its substrate recognition.7 The inability to cleave newly released ultralarge (UL) VWF results in the accumulation of ULVWF polymers on endothelial cells25 and in the circulation.26 Consequently, the ULVWF multimers serve as templates for rapidly recruiting platelets,27,28 neutrophils,28,29 and complement components30 from the circulation to the sites of vascular injury. Platelets play critical roles in both hemostasis and inflammation. Studies have shown that platelet surface glycoprotein (GP) Ib-VI31 and p-selectin32 or ADP,33 released from activated platelets, may provide receptors or signal for recruiting neutrophils and monocytes. The accumulation and activation/degranulation of neutrophils may result in the massive release of granular contents, including neutrophil extracellular traps (NETs)20 and HNP1-334 at the sites of vascular injury. HNP1-3 is highly cationic and has hydrophobic peptides, which can bind to various plasma proteins and cellular components. Only a small fraction of HNP1-3 may be able to escape from the sites of their release and circulate in plasma. Therefore, 7–10 fold increases of plasma HNP1-3 in our cohort of TTP patients may reflect the massive local release of HNP1-3 in situ from activated and accumulated neutrophils.
HNP1-3 exhibits a broad-spectrum of bactericidal activity contributing to human innate immunity. The positive charge of amino acid side chains is responsible for the initial interaction with negatively charged bacterial membranes, resulting in cytotoxicity. HNP1-3 may also have a variety of other biological functions including activation of platelets,35 stimulation of inflammatory responses and cytokine release,36 and inhibition of fibrinolysis.37 In addition, HNP1-3 has been shown to cause endothelial dysfunction and increased endothelial permeability.38 More recently, we demonstrated that HNP1-3 inhibits proteolytic cleavage of VWF by ADAMTS13.11 Dramatic increases in plasma HNP1-3 are also reported in patients with myocardial infarction,39,40 systemic lupus erythematosus,41 and septic meningitis,42 suggesting that HNP may also play a role in the pathogenesis of other inflammatory and thrombotic disorders. In TTP patients, we speculate that the massive release of HNP1-3 at the sites of vascular injury may be extremely detrimental when the circulating ADAMTS13 is limited. The finding that plasma HNP1-3 correlates with patient’s serum LDH, indicative of tissue ischemia in TTP,43 supports the potential involvement of HNP in the pathogenesis of TTP.
How may HNP and complement activation be related? HNP1 has been shown to inhibit the classical and lectin pathways of complement activation.44 However, its relationship with the alternative pathway is not known. The levels of plasma HNP1-3 appear to correlate with plasma Bb, suggesting that the HNP1-3 may activate complement or vice versa. While our results did not find a statistically significant increase in iC3b and sC5b-9 complexes in TTP patients when compared with healthy controls, other studies have demonstrated the increased levels of C3a and sC5b-9 during acute TTP.45
Moreover, the increased levels of Bb, C3a, and C5a, and sC5b-9 appear to correlate with a worse outcome in TTP.46 We did not find an association between any of these markers we measured and the relapse and mortality rate in our cohort of patients. The reasons for the discrepant results of plasma C5b-9 compared to the literature may be multifactorial, including sample collection and storage (freeze and thaw), assay methodology (dilution factor), and patient population (African vs. European ancestry). However, we do demonstrate the significant association between plasma levels of Bb and the evidence of organ tissue ischemia (i.e. serum LDH and creatinine), although the levels of plasma complement activation components are variable in TTP patients. Such a variation may be partially attributed to the comorbidity, but perhaps to other unknown genetic predispositions in a subset of patients. More interestingly, our unpublished preliminary results demonstrated that an administration of anti-complement factor H IgG (mAb7.1) in Adamts13−/− and wild-type mice, which provokes complement activation via the alternative pathway, resulted in more severe thrombocytopenia in Adamts13−/− mice than in wild-type mice, but a similar degree of renal insufficiency in an otherwise TMA-resistant mouse strain (C57BL/6),1 suggesting a potential causative role of complement activation rather than merely a biomarker of acute TTP.
In conclusion, we found significantly increased levels of plasma HNP1-3 and Bb in patients with acute autoimmune TTP; plasma levels of HNP1-3 and/or Bb correlated with the downstream complement activation markers and organ tissue ischemia in TTP patients. Our findings may provide the molecular basis for additional targeted therapies, including the blockage of neutrophil activation and degranulation with colchicine and complement activation with eculizumab, in a subset of patients with acquired autoimmune TTP.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/11/1319
Funding
This work is partially supported by Answering T.T.P. foundation, Institutional Adam’s resident research grant, and NIH-R01HL115187-01A1.
References
- 1.Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127(7):834–839. [DOI] [PubMed] [Google Scholar]
- 2.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398–403. [DOI] [PubMed] [Google Scholar]
- 3.Tsai HM. Thrombotic thrombocytopenic purpura: a thrombotic disorder caused by ADAMTS13 deficiency. Hematol Oncol Clin North Am. 2007;21(4):609–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng XL. ADAMTS13 and von Willebrand Factor in Thrombotic Thrombocytopenic Purpura Annu Rev Med. 2015;66:211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lotta LA, Wu HM, Mackie IJ, et al. Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura. Blood. 2012;120(2):440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbot J, Costa E, Guerra M, et al. Ten years of prophylactic treatment with fresh-frozen plasma in a child with chronic relapsing thrombotic thrombocytopenic purpura as a result of a congenital deficiency of von Willebrand factor-cleaving protease. Br J Haematol. 2001;113(3):649–651. [DOI] [PubMed] [Google Scholar]
- 7.Casina V, Hu WB, Mao JH, et al. High Resolution Epitope Mapping by HX MS Reveals the Pathogenic Mechanism and a Possible Therapy for Autoimmune TTP Syndrome. PNAS. 2015;112(31):9620–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mise K, Ubara Y, Matsumoto M, et al. Long term follow up of congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome) on hemodialysis for 19 years: a case report. BMC Nephrol. 2013;14:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9 Suppl 1:283–301. [DOI] [PubMed] [Google Scholar]
- 10.Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillai VG, Bao J, Zander CB, et al. Human neutrophil peptides inhibit cleavage of von Willebrand factor by ADAMTS13: a potential link of inflammation to TTP. Blood. 2016;128(1):110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz-Torres MP, Casiraghi F, Galbusera M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemost. 2005;93(3):443–452. [DOI] [PubMed] [Google Scholar]
- 13.Westwood JP, Langley K, Heelas E, Machin SJ, Scully M. Complement and cytokine response in acute Thrombotic Thrombocytopenic Purpura. Br J Haematol. 2014;164(6):858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapin J, Eyler S, Smith R, Tsai HM, Laurence J. Complement factor H mutations are present in ADAMTS13-deficient, ticlopidine-associated thrombotic microangiopathies. Blood. 2013;121(19):4012–4013. [DOI] [PubMed] [Google Scholar]
- 15.George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116(20):4060–4069. [DOI] [PubMed] [Google Scholar]
- 16.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129(1):93–100. [DOI] [PubMed] [Google Scholar]
- 17.Raife TJ, Cao W, Atkinson BS, et al. Leukocyte proteases cleave von Willebrand factor at or near the ADAMTS13 cleavage site. Blood. 2009;114(8):1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deford CC, Reese JA, Schwartz LH, et al. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood. 2013;122(12):2023–2029; quiz 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korkmaz S, Keklik M, Sivgin S, et al. Therapeutic plasma exchange in patients with thrombotic thrombocytopenic purpura: a retrospective multicenter study. Transfus Apher Sci. 2013;48(3):353–358. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012; 120(6):1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikes B, Sinkovits G, Farkas P, et al. Elevated plasma neutrophil elastase concentration is associated with disease activity in patients with thrombotic thrombocytopenic purpura. Thromb Res. 2014;133(4):616–621. [DOI] [PubMed] [Google Scholar]
- 22.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8(11):622–633. [DOI] [PubMed] [Google Scholar]
- 23.Zheng X, Pallera AM, Goodnough LT, Sadler JE, Blinder MA. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med. 2003;138(2):105–108. [DOI] [PubMed] [Google Scholar]
- 24.Li D, Xiao J, Paessler M, Zheng XL. Novel recombinant glycosylphosphatidylinositol (GPI)-anchored ADAMTS13 and variants for assessment of anti-ADAMTS13 autoantibodies in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;106(5):947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. [DOI] [PubMed] [Google Scholar]
- 26.Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307(23):1432–1435. [DOI] [PubMed] [Google Scholar]
- 27.Bernardo A, Ball C, Nolasco L, et al. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562–570. [DOI] [PubMed] [Google Scholar]
- 28.Chauhan AK, Motto DG, Lamb CB, et al. Systemic antithrombotic effects of ADAMTS13. J Exp Med. 2006;203(3):767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gandhi C, Khan MM, Lentz SR, Chauhan AK. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood. 2012;119(10):2385–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner NA, Moake J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PloS one. 2013;8(3):e59372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corken A, Russell S, Dent J, Post SR, Ware J. Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol. 2014;34(5):996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Downing LJ, Wakefield TW, Strieter RM, et al. Anti-P-selectin antibody decreases inflammation and thrombus formation in venous thrombosis. J Vasc Surg. 1997;25(5):816–827. [DOI] [PubMed] [Google Scholar]
- 33.Song ZF, Chen DY, Du B, Ji XP. Poly (ADP-ribose) polymerase inhibitor reduces heart ischaemia/reperfusion injury via inflammation and Akt signalling in rats. Chin Med J (Engl). 2013;126(10):1913–1917. [PubMed] [Google Scholar]
- 34.Zhao L, Lu W. Defensins in innate immunity. Curr Opin Hematol. 2014;21(1):37–42. [DOI] [PubMed] [Google Scholar]
- 35.Horn M, Bertling A, Brodde MF, et al. Human neutrophil alpha-defensins induce formation of fibrinogen and thrombospondin-1 amyloid-like structures and activate platelets via glycoprotein IIb/IIIa. J Thromb Haemost. 2012;10(4):647–661. [DOI] [PubMed] [Google Scholar]
- 36.Chaly YV, Paleolog EM, Kolesnikova TS, et al. Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur Cytokine Netw. 2000;11(2):257–266. [PubMed] [Google Scholar]
- 37.Higazi AA, Ganz T, Kariko K, Cines DB. Defensin modulates tissue-type plasminogen activator and plasminogen binding to fibrin and endothelial cells. J Biol Chem. 1996;271(30):17650–17655. [DOI] [PubMed] [Google Scholar]
- 38.Kougias P, Chai H, Lin PH, et al. Neutrophil antimicrobial peptide alpha-defensin causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2006;43(2):357–363. [DOI] [PubMed] [Google Scholar]
- 39.Quinn K, Henriques M, Parker T, Slutsky AS, Zhang H. Human neutrophil peptides: a novel potential mediator of inflammatory cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2008;295(5):H1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Yan H, Yamashita S, et al. Acute ST-segment elevation myocardial infarction is associated with decreased human antimicrobial peptide LL-37 and increased human neutrophil peptide-1 to 3 in plasma. J Atheroscler Thromb. 2012;19(4):357–368. [DOI] [PubMed] [Google Scholar]
- 41.Sthoeger ZM, Bezalel S, Chapnik N, Asher I, Froy O. High alpha-defensin levels in patients with systemic lupus erythematosus. Immunology. 2009;127(1):116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panyutich AV, Panyutich EA, Krapivin VA, Baturevich EA, Ganz T. Plasma defensin concentrations are elevated in patients with septicemia or bacterial meningitis. J Lab Clin Med. 1993;122(2):202–207. [PubMed] [Google Scholar]
- 43.Cohen JA, Brecher ME, Bandarenko N. Cellular source of serum lactate dehydrogenase elevation in patients with thrombotic thrombocytopenic purpura. J Clin Apher. 1998;13(1):16–19. [DOI] [PubMed] [Google Scholar]
- 44.Groeneveld TW, Ramwadhdoebe TH, Trouw LA, et al. Human neutrophil peptide-1 inhibits both the classical and the lectin pathway of complement activation. Mol Immunol. 2007;44(14):3608–3614. [DOI] [PubMed] [Google Scholar]
- 45.Reti M, Farkas P, Csuka D, et al. Complement activation in thrombotic thrombocytopenic purpura. J Thromb Haemost. 2012;10(5):791–798. [DOI] [PubMed] [Google Scholar]
- 46.Wu TC, Yang S, Haven S, et al. Complement activation and mortality during an acute episode of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2013;11(10):1925–1927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.