

Abstract
Adenylyl cyclases (ACs) generate the second messenger cAMP from ATP. Mammalian cells express nine transmembrane AC (mAC) isoforms (AC1–9) and a soluble AC (sAC, also referred to as AC10). This review will largely focus on mACs. mACs are activated by the G-protein Gαs and regulated by multiple mechanisms. mACs are differentially expressed in tissues and regulate numerous and diverse cell functions. mACs localize in distinct membrane compartments and form signaling complexes. sAC is activated by bicarbonate with physiologic roles first described in testis. Crystal structures of the catalytic core of a hybrid mAC and sAC are available. These structures provide detailed insights into the catalytic mechanism and constitute the basis for the development of isoform-selective activators and inhibitors. Although potent competitive and noncompetitive mAC inhibitors are available, it is challenging to obtain compounds with high isoform selectivity due to the conservation of the catalytic core. Accordingly, caution must be exerted with the interpretation of intact-cell studies. The development of isoform-selective activators, the plant diterpene forskolin being the starting compound, has been equally challenging. There is no known endogenous ligand for the forskolin binding site. Recently, development of selective sAC inhibitors was reported. An emerging field is the association of AC gene polymorphisms with human diseases. For example, mutations in the AC5 gene (ADCY5) cause hyperkinetic extrapyramidal motor disorders. Overall, in contrast to the guanylyl cyclase field, our understanding of the (patho)physiology of AC isoforms and the development of clinically useful drugs targeting ACs is still in its infancy.
I. Introduction
A. Overview of Adenylyl Cyclase Signaling
Adenylyl cyclases (ACs) catalyze the conversion of ATP into the second messenger cAMP. ACs are broadly expressed in the kingdom of life (Linder and Schultz, 2003; Gancedo, 2013). This IUPHAR review exclusively covers mammalian ACs and focuses on the nine membrane AC isoforms, AC1–AC9 (Alexander et al., 2015; http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257). We will discuss in detail structural aspects because knowledge of the molecular architecture of ACs constitutes the rational basis for the development of potent and AC isoform-selective inhibitors and activators.
Certain bacterial AC toxins such as edema factor from Bacillus anthracis and CyaA from Bordetella pertussis constitute potential antibacterial drug targets and are discussed elsewhere (Seifert and Dove, 2012, 2013). ACs from parasites such as Trypanosoma brucei are also drug targets (Seebeck et al., 2004; Makin and Gluenz, 2015).
AC and cAMP were first described by Nobel laureate Earl W. Sutherland (Rall and Sutherland, 1958; Sutherland et al., 1962). The early history of mammalian ACs and cAMP is covered by excellent reviews (Sutherland and Robison, 1966; Hardman et al., 1971). G-protein activation of AC was first described by Pfeuffer (1977) and Gilman (1995). Later, mammalian ACs were found to be under the dual stimulatory and inhibitory control of G-proteins (Rodbell, 1984; Gilman, 1987; Birnbaumer et al., 1990). Moreover, the plant diterpene forskolin (FSK) from Coleus forskohlii (an ancient Indian folk medicine) was identified as an AC activator and has become a very important experimental tool for probing the involvement of ACs in (patho)physiologic processes (Seamon et al., 1981; Daly, 1984; Insel and Ostrom, 2003; Alasbahi and Melzig, 2012). After purification of a mammalian AC and peptide sequencing (Smigel, 1986), subsequent cloning studies revealed a complex transporter-like topology of membrane ACs (mACs) (Krupinski et al., 1989) and the existence of nine isoforms (ACs 1–9) (Sunahara et al., 1996).
In the 1970s, when the field of signal transduction at the membrane was still in its infancy, an AC with unique biochemical properties was described in the soluble fraction of rat testis homogenate (Braun and Dods, 1975; Neer, 1978; Braun, 1991). The initial observation was confirmed by several laboratories (Gordeladze and Hansson, 1981; Gordeladze et al., 1982; Stengel and Hanoune, 1984), but the nature of the enzyme catalyzing this reaction remained elusive for almost three decades. The unique properties of sAC were conclusively elucidated with the cloning of cDNAs derived from mouse testis and human embryonic kidney (HEK) 293 cells (Buck et al., 1999; Geng et al., 2005).
Numerous reviews on the cloning, structure, function, regulation, and pharmacological modulation of mammalian ACs are available (Cooper, 1998; Tang and Hurley, 1998; Defer et al., 2000; Sunahara and Taussig, 2002; Sinha and Sprang, 2006; Willoughby and Cooper, 2007; Sadana and Dessauer, 2009; Pavan et al., 2009; Pierre et al., 2009; Seifert et al., 2012; Steegborn, 2014; Schmid et al., 2014; Levin and Buck, 2015). Methods for the analysis of mammalian ACs have been reviewed in volume 345 of the book series Methods in Enzymology (Iyengar and Hildebrandt, 2002).
The purpose of this review is to bring together leading experts from the AC field and provide a state-of-the-art of the field with a strong focus on mACs 1–9. We discuss (patho)physiologic aspects and implications for drug development. Moreover, we try to integrate a historical perspective and cite representative papers from leading groups over the past six decades. However, we apologize to all colleagues whose works we could not cite due to the focus of this review and limitations in space.
We are fully aware of the current discussion about data reproducibility (Kannt and Wieland, 2016) and positive publication bias (Carvalho et al., 2016). Many “negative” studies trying to confirm published original data were shelved and never published. Confirmatory “positive” papers are similarly discouraged by most scientific journals (Kannt and Wieland, 2016). Therefore and whenever possible, we strived to draw conclusions on the basis of data from independent research groups to obtain a balanced picture, highlight gaps in our knowledge, and point to controversial data. A major problem in the field of pharmacological AC modulation is that the available experimental tools have not always been used critically (Seifert, 2014, 2016; Michel and Seifert, 2015) so that misinterpretations of data are likely to have occurred. Unfortunately, catalogs of chemical companies contribute to the misinformation of researchers because compounds are being advertised as being selective for a specific nucleotidyl cyclase without actually having this property. Thus, researchers should exert great caution when selecting pharmacological tools for their studies, particularly in intact-cell systems.
B. Nomenclature
The enzyme that catalyzes the production of cAMP from ATP was originally named adenyl cyclase [EC 4.6.1.1] (Sutherland et al., 1962). The name adenylate cyclase first appeared in print in 1970 (Stansfield and Franks, 1970), and it did not take long for this alteration in the name to take hold. As the story goes according to Louis Ignarro, over dinner sometime in the late 1970s, many of the leaders in the field all agreed that guanylate was far easier to pronounce than guanylyl. The same logic was true for adenylate, and thus a mixture of names appeared throughout the 1970s and 1980s. However, the name adenylate cyclase does not correctly describe the chemistry of the enzymatic reaction. Because the α,β oxygen leaves ATP upon attack of the O3 oxygen of the ribose ring (Walseth et al., 1981), technically it is an adenylyl moiety that is cyclized, not adenylate (adenylate is equivalent to AMP; if adenylate were cyclized, the reaction would end up with one excess oxygen). The decision by several prominent laboratories to switch to adenylyl cyclase took place slowly in the late 1980s and mid 1990s. Alfred G. Gilman relayed his recollections of events at the 1988 Cold Spring Harbor meeting shortly before his untimely death (and later confirmed by Lutz Birnbaumer). More senior scientists may remember the push for the term N proteins as opposed to G proteins. According to Gilman and in response to a call for clarification of G protein nomenclature,
Lutz proclaimed that he would accept G if we accepted adenylyl. This was a complete surprise to me. I was not familiar with the chemistry, did not realize that there was an argument, did not really give a damn, and quickly said “sold”. For some reason it all happened quickly and painlessly after that.
Thus, despite issues with pronunciation, the name adenylyl cyclase is here to stay. Technically, this should not be a problem because the abbreviation AC for adenylyl cyclase is broadly used and easy to pronounce.
The IUPHAR consortium on ACs discussed nucleotidyl cyclase nomenclature with the IUPHAR consortium on guanylyl cyclases (GCs). The consortia agreed that for chemical reasons, the terms guanylyl cyclase and adenylyl cyclase should be used to reflect the actual biochemistry of NTP cyclization. The International Union of Biochemistry and Molecular Biology still lists adenylate cyclase as the accepted name of AC, mentions a number of alternative names, but the entry into the database is from the year 1972, many years before the nomenclature discussion began. However, it should be kept in mind that specifically in the older literature, the term adenylate cyclase was used commonly. Hence for database searches, both the terms adenylate cyclase and adenylyl cyclase have to be used because with the latter search term, substantially fewer items will be retrieved than with the former.
ACs are encoded by specific genes and divided into five classes, specified with Roman numbers I–V (Linder and Schultz, 2003). All mammalian ACs belong into the class III (Linder and Schultz, 2003; Sinha and Sprang, 2006; Steegborn, 2014). The mammalian AC genes are designated ADCY1-10. These terms are used in genetic studies and are of particular relevance for studies linking specific polymorphisms in AC isoforms to human diseases, an emerging scientific field. In biochemical and pharmacological studies on the actual proteins, the term AC is used. ACs 1–9 are membrane-delimited ACs (mACs) and also referred to as mACs 1–9. They belong to the class IIIa (Linder and Schultz, 2003). The term transmembrane ACs (tmACs) is used less frequently. AC10 corresponds to the soluble AC, sAC. The terms AC10 and sAC are both used in the literature. sAC is a class IIIb AC (Linder and Schultz, 2003). The IUPHAR committee on ACs agreed that the term sAC should be preferred to reflect the initial description of the enzyme as soluble AC. This nomenclature provides an analogy to the GC field, where membrane and soluble GCs (sGCs) are distinguished from each other. In the GC field, the membrane GCs are referred to as particulate GCs (pGCs). Although the terms membrane in the AC field and particulate in the GC field describe identical enzyme anchoring modes, it was decided not to change these terms because they are very well established, specifically in light of the fact that the GC committee introduced a number systems for sGCs (GC1 and GC2) and a capital letter system for pGCs (GC-A–GC-G). With respect to numbering of AC isoforms, both Roman and Arabic numbers are used. The AC committee agreed that for better readability, Arabic numbers should be used.
The term AC implies that the enzyme is capable of producing only cAMP. This assumption is correct for mACs 1-9 (Sunahara et al., 1998; Hasan et al., 2014). However, sAC is more promiscuous in terms of substrate specificity and also synthesizes cGMP and the cyclic pyrimidine nucleotides cCMP and cUMP (Hasan et al., 2014; Kleinboelting et al., 2014b). Accordingly, from a biochemical perspective, sAC is an ATP-preferring nucleotidyl cyclase (Seifert, 2015; Seifert et al., 2015). pGCs are very ineffective at producing cAMP (Beste et al., 2013), whereas sGC is also capable of synthesizing cAMP, cCMP, and cUMP and constitutes a GTP-preferring nucleotidyl cyclase (Beste et al., 2012). The AC committee decided that these additional enzymatic activities of nucleotidyl cyclases will not change the historically established distinction into ACs and GCs. This is also justified in view of the fact that neither sAC nor sGC prefer CTP or UTP relative to ATP or GTP as substrates (Seifert, 2015; Seifert et al., 2015).
With regard to the mAC activator forskolin, both the abbreviations FS or FSK (Fsk) are being used. For literature searches in databases, the term FS is not sufficiently specific. Therefore, we recommend that the term FSK be used.
C. Model Systems for Adenylyl Cyclase Analysis
1. From Organs to Purified Adenylyl Cyclases.
Research on ACs started with crude organ membrane fractions (Rall and Sutherland, 1958; Sutherland et al., 1962) and soluble organ fractions in case of sAC (Braun and Dods, 1975; Neer, 1978; Braun, 1991). Although organ membranes contain numerous AC isoforms, this system is relatively close to the physiologic situation. By combining physiologic and pharmacological activators, inhibitors, RT-PCR analysis of expressed mRNA and, if available, antibodies, analysis of ACs in organ membranes can be quite informative and yield valuable information on functionally dominant AC isoforms (Göttle et al., 2009; Erdorf and Seifert, 2011). Experiments with native organs have been successfully complemented with gene knockout studies and transgene overexpression studies in mice (Ferguson and Storm, 2004; Sadana and Dessauer, 2009).
Studies with membranes from native cells such as platelets (e.g., Wieland et al., 1992) or cultured cells such as neuroblastoma cells (e.g., Sharma et al., 1975a) also yielded most valuable insights into the mechanisms of AC regulation. Of particular importance in this regard are the S49 lymphoma wild-type cells and the Gαs-deficient cyc− cells (named according to the observation that they do not respond to hormonal stimulation in terms of increased cAMP production and erroneously assumed to be AC deficient). These cell lines played a fundamental role in resolving and reconstituting protein components of the G-protein-coupled receptor (GPCR)–G-protein-AC signaling cascade (Shear et al., 1976; Ross et al., 1978; Hildebrandt et al., 1982; Jakobs et al., 1983a,b). Alterations in platelet membrane AC activity have been proposed as biomarkers for depression and alcoholism (Menninger et al., 2000; Hines and Tabakoff, 2005), fostering current studies analyzing polymorphisms in individual ADCY genes (see section II.E).
Although purification of the G-proteins was rather straightforward and the purified Gi- and Gs-proteins were stable (Gilman, 1987; Birnbaumer et al., 1990), purification of functionally and structurally intact GPCRs (Gether et al., 1997) and particularly mACs turned out to be much more difficult because of their very low abundance and conformational and thermal instability under the conditions used for purification (Ross, 1981; Pfeuffer et al., 1985, 1989; Smigel, 1986). However, crude detergent-dispersed AC from rat brain is quite stable and has been used for a number of studies (Johnson and Sutherland, 1973; Desaubry et al., 1996; Desaubry and Johnson, 1998; Gille et al., 2004).
The advent of mAC cloning (Krupinski et al., 1989) enabled studies with recombinantly expressed ACs. ACs can be overexpressed in mammalian cells, HEK293 cells being a very popular model system (Jacobowitz et al., 1994). One should keep in mind that HEK293 cells endogenously express several mACs, providing background activity (Atwood et al., 2011; Yu et al., 2014). ACs can also be effectively overexpressed in Spodoptera frugiperda Sf9 cells (Taussig et al., 1994b). Although few studies have been performed with ACs purified from Sf9 cells (Taussig et al., 1993a,b), most studies are conducted with crude Sf9 membranes overexpressing a given AC isoform, increasing stability of the enzymes and facilitating the execution of extensive pharmacological studies (Pinto et al., 2008, 2011; Brand et al., 2013). Again, the background activity of the endogenous membrane AC of Sf9 cells needs to be properly considered in overexpression studies (Pinto et al., 2008). Like mammalian mACs, the membrane AC of Sf9 cells is potently inhibited by MANT [2′,(3′)-O-(N-methylanthraniloyl)] nucleotides (Gille et al., 2004) and effectively activated by FSK (Pinto et al., 2008).
In the case of GPCRs (Kobilka et al., 1988) and G-protein α-subunits (Woon et al., 1989; Conklin et al., 1993), the approach of constructing and expressing chimeras between various protein isoforms has been successful and provided valuable insights into functional domains. In contrast, in the case of ACs, there are relatively few studies on chimeras of full-length AC isoforms (Levin and Reed, 1995; Crossthwaite et al., 2005; Yoshimura et al., 2006; Diel et al., 2008). A major reason for this situation is the fact that the chimeras are unstable and, hence, functionally inactive (Seebacher et al., 2001). As a result, our current knowledge on the functional relevance of the transmembrane domains is rather poor. It has been proposed that the mammalian AC transmembrane domains may serve in solute transport (Krupinski et al., 1989), as a voltage sensor (Reddy et al., 1995) or dimerization domain (Cooper and Crossthwaite, 2006). Experimental validation of these proposals is still in the very beginning (Beltz et al., 2016).
2. Studies with Purified C1 and C2 Subunits.
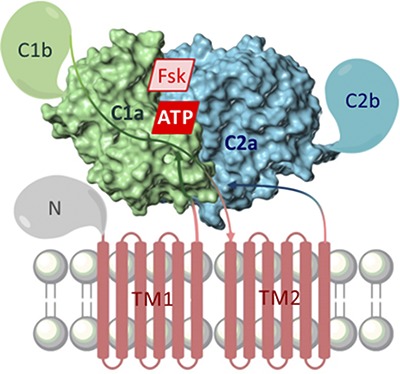
Although the transmembrane domains of mACs constitute a major reason for their extremely difficult biochemical tractability, the cytosolic domains C1 and C2 are much more readily accessible (Fig. 1). Expression in Escherichia coli of a fusion protein of parts of the C1 and C2 domains of ACs 1 and 2, respectively, with complete omission of the transmembrane domains, resulted in a FSK- and Gαs-stimulated enzyme (Tang and Gilman, 1995). This landmark study showed that for the essential aspects of catalysis and regulation, the transmembrane domains are dispensable, raising also the question whether these domains have just a membrane anchor function for the cytosolic domains. The C1:C2 approach has been successfully applied to a number of AC isoforms, expressing either fusion proteins or separate C1 and C2 subunits that form a catalytically active enzyme in solution (Yan et al., 1996, 1997; Whisnant et al., 1996; Scholich et al., 1997; Sunahara et al., 1997; Dessauer et al., 1998; Haunso et al., 2003). In contrast to the C1:C2 heterodimer (Whisnant et al., 1996; Sunahara et al., 1997), the C1:C1 homodimer and the C2:C2 homodimer are catalytically inactive (Whisnant et al., 1996; Zhang et al., 1997; Suryanarayana et al., 2009b).
Fig. 1.

Common domain structure of mACs. Components resolved in crystal structures are presented as surface model. Shown is the “ventral” surface of C1a:C2a for the sake of consistency with Figs. 2–4, 6–11, and 14 depicting crystal structures. The real position of the membrane is probably behind the hidden “dorsal” surface. FSK, forskolin site (AC1–8), N-term, N-terminal domain; C2b, C-terminal domain; TM, transmembrane domains; arrow lines, schematic course of the polypeptide chain.
The various C1:C2 systems have yielded valuable insights into the regulatory properties of mACs. The most widely used system in this regard has become the system with the C1 domain from AC5 (5C1) and the C2 domain from AC2 (2C2), referred to as 5C1:2C2 system. Historically, the term VC1:IIC2 has been used frequently. For the sake of consistency with the proposed AC isoform nomenclature, we will use the term 5C1:2C2. The heterodimer allowed molecular insights into the regulation of, and conformational changes in, mACs (Whisnant et al., 1996; Sunahara et al., 1997; Dessauer and Gilman, 1997; Dessauer et al., 1997, 1998, 2002; Mou et al., 2005, 2006, 2009; Suryanarayana et al., 2009a; Pinto et al., 2009, 2011) and the resolution of 17 crystal structures with different ligands in the catalytic site (Table 1). Moreover, this system has been used for extensive pharmacological studies assessing the catalytic site and the allosteric regulatory site (Dessauer, et al., 1997; Gille et al., 2004; Mou et al., 2005, 2006; Suryanarayana et al., 2009a; Pinto et al., 2009, 2011). However, it should be kept in mind that this system constitutes a hybrid with subunits from different AC isoforms so that it can serve just as a model for mACs in general. There are some examples of successful expression of C1:C2 systems from a particular AC isoform, such as ACs 2, 5, 7, and 9 (Scholich et al., 1997; Dessauer et al., 1998; Yan et al., 2001; Haunso et al., 2003; Dokphrom et al., 2011). However, functionality of these systems seems to be very dependent on the specific length of the expressed subunit (Dessauer et al., 1998; Mou et al., 2005). The most widely used purified mAC system is 5C1(dog):2C2(rat) (Table 1). The high conservation of the catalytic domains between dog and rat tempers concerns regarding the use of proteins from different species, but regulation and interactions with the N terminus is missing in this system (Gao et al., 2007).
TABLE 1.
Crystal structures of the catalytic domains of mACs
| PDB ID | C1a sequence | C2a sequence | Metal A Metal Ba | Gαsb | ATP-site | FSK-site | Res.c | Reference |
|---|---|---|---|---|---|---|---|---|
| 1AB8d | — | 2C2 (rat) | — | — | — | FSK | 2.20 | Zhang et al., 1997 |
| 871-1090 | — | |||||||
| 1AZS | 5C1 (dog) | 2C2 (rat) | — | + | — | Methylpiperazino-FSK | 2.30 | Tesmer et al., 1997 |
| 361-575 | 870-1081 | — | GTPγS | |||||
| 1CJK | 5C1 (dog) | 2C2 (rat) | Mg2+ | + | Adenosine-5′-RP-(α-thio-PPP) | FSK | 3.00 | Tesmer et al., 1999 |
| 364-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 1CJT | 5C1 (dog) | 2C2 (rat) | Mg2+ | + | 2’,3′-Dideoxy-ATP | FSK | 2.80 | Tesmer et al., 1999 |
| 364-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 1CJU | 5C1 (dog) | 2C2 (rat) | Mg2+ | + | 2’,3′-Dideoxy-ATP | FSK | 2.80 | Tesmer et al., 1999 |
| 364-580 | 870-1081 | Mg2+ | GTPγS | |||||
| 1CJV | 5C1 (dog) | 2C2 (rat) | Zn2+ | + | 2’,3′-Dideoxy-ATP | FSK | 3.00 | Tesmer et al., 1999 |
| 364-580 | 870-1081 | Mg2+ | GTPγS | |||||
| 1CS4 | 5C1 (dog) | 2C2 (rat) | — | + | 2’-Deoxyadenosine-3′-P + PP | FSK | 2.50 | Tesmer et al., 2000 |
| 361-580 | 870-1081 | Mg2+ | GTPγS | |||||
| 1CUL | 5C1 (dog) | 2C2 (rat) | Mg2+ | + | 2’-Deoxyadenosine-3′-P | FSK | 2.40 | Tesmer et al., 2000 |
| 364-580 | 874-1081 | Mg2+ | GTPγS | |||||
| 1TL7 | 5C1 (dog) | 2C2 (rat) | Mn2+ | + | 3′-MANT-GTP | FSK | 2.80 | Mou et al., 2005 |
| 364-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 1UOH | 5C1 (dog) | 2C2 (rat) | Mg2+ | + | 3′-MANT-GTP | FSK | 2.90 | Mou et al., 2005 |
| 361-580 | 870-1081 | Mg2+ | GTPγS | |||||
| 2GVD | 5C1 (dog) | 2C2 (rat) | Mn2+ | + | TNP-ATP | Methylpiperazino-FSK | 2.90 | Mou et al., 2006 |
| 363-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 2GVZ | 5C1 (dog) | 2C2 (rat) | Mn2+ | + | 3′-MANT-ATP | Methylpiperazino-FSK | 3.27 | Mou et al., 2006 |
| 363-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 3C14 | 5C1 (dog) | 2C2 (rat) | — | + | PP | FSK | 2.68 | Mou et al., 2009 |
| 363-580 | 870-1081 | Ca2+ | GTPγS | |||||
| 3C15 | 5C1 (dog) | 2C2 (rat) | — | + | PP | FSK | 2.78 | Mou et al., 2009 |
| 363-580 | 870-1081 | Mg2+ | GTPγS | |||||
| 3C16 | 5C1 (dog) | 2C2 (rat) | Ca2+ | + | ATP | FSK | 2.87 | Mou et al., 2009 |
| 363-580 | 870-1081 | Ca2+ | GTPγS | |||||
| 3G82 | 5C1 (dog) | 2C2 (rat) | Mn2+ | + | 3′-MANT-ITP | FSK | 3.11 | Hübner et al., 2011a |
| 363-580 | 870-1081 | Mn2+ | GTPγS | |||||
| 3MAA | 5C1 (dog) | 2C2 (rat) | — | + | Adenosine-5′-RP-(α-thio-PPP) | Methylpipera-zino-FSK | 3.00 | Mou et al., 2009 |
| 363-580 | 870-1081 | Ca2+ | GTPγS |
P, monophosphate; PP, pyrophosphate; PPP, triphosphate; 3′-MANT, 3′-O-(N-methylanthraniloyl)-; TNP, 2′,3′-O-(2,4,6-trinitrophenyl)-.
Metal ions A and B located in the nucleotide binding site.
Bovine Gαs not present (−) or present (+) as chain C in the complex together with GTPγS and mostly metal and Cl− ions (not shown).
Resolution [Å].
Homodimer of 2C2.
3. Methodological Aspects.
For the analysis of AC activity in cell-free systems with purified proteins or membranes, reliable and sensitive one- and two-column methods for separation of the substrate [α-32P]ATP and the product [32P]cAMP (eluted from the column) are available (Johnson et al., 1994; Post et al., 2000). For inhibition of cAMP-degrading phosphodiesterases (PDEs), cell-free AC assays often contain the PDE inhibitor isobutylmethylxanthine (IBMX). However, limited water solubility of the inhibitor (unless aqueous solutions are preheated or organic solvents are used) and IBMX resistance of certain PDEs (Bender and Beavo, 2006; Francis et al., 2011) could influence assay results. Other studies included cAMP (see, e.g., Gille et al., 2004), but cAMP at high concentrations can inhibit ACs (Dessauer and Gilman, 1997). A highly important variable in AC assays with cell-free preparations is the choice of the particular divalent cation, Mg2+ or Mn2+. In general, with Mn2+, higher catalytic activities than with Mg2+ are obtained (Gille et al., 2004: Mou et al., 2005), the affinities of inhibitors increase (Gille et al., 2004; Mou et al., 2005) and the pharmacological profile of diterpenes is affected (Erdorf et al., 2011). Moreover, Mn2+ can functionally uncouple AC from Gi-proteins (Hildebrandt et al., 1982; Jakobs et al., 1983a,b; Dessauer et al., 2002; Gille and Seifert, 2003) and alter basal AC activities (Pieroni et al., 1995). Although Mg2+ is usually assumed to be the physiologic cation of catalysis (Ross et al., 1978), indirect evidence points to a role of Mn2+ as cation for catalysis, at least for sGC (Bähre et al., 2014).
In many studies, AC activity is assessed in intact cells. For intact cells, various reliable immuno-based cAMP detection methods are available (Post et al., 2000), and high-throughput screening methods (with varying reliability) have been developed as well (Williams, 2004; Conley et al., 2013). However, formally, intact cell assays do not reflect AC activity assays but constitute cAMP accumulation assays. In intact cell assays, the ATP substrate concentration for ACs cannot be readily controlled. And although PDEs are usually inhibited by IBMX (but see the cautionary note above), it has to be taken into account that cAMP can also be very effectively exported from cells via multidrug resistance associated protein (MRP) isoforms 4 and 5 (Keppler, 2011; Copsel et al., 2011). These transporters are inhibited by numerous compounds such as probenecid (Copsel et al., 2011). Unfortunately, the pharmacological tools used to block PDEs and MRPs are not totally specific. On one hand, methylxanthines can block adenosine receptors and MRPs (Chen et al., 2013b; Xie et al., 2011) and activate ryanodine receptors (Guerreiro et al., 2011). On the other hand, several MRP blockers can also inhibit PDEs (Xie et al., 2011). Thus, caution must be exerted when interpreting the results of intact-cell studies with PDE- and MRP inhibitors.
II. Structures, Expression, Compartmentation, Regulation, and (Patho)physiologic Function of Adenylyl Cyclases
A. Structures of Membrane Adenylyl Cyclases and Comparison with Soluble Adenylyl Cyclase
1. Overall and Crystal Structures of Nucleotidyl Cyclases.
ACs 1–9 differ with respect to sequence and length (between 1080 and 1353 amino acids). ACs 1–9 are single peptide chains consisting of an N-terminal cytosolic domain of varying length, two membrane-spanning domains (TM1, TM2), each with six transmembrane α-helices, and two cytosolic domains C1 and C2 subdivided into catalytic (C1a, C2a) and regulatory (C1b, C2b) subdomains (Fig. 1). ACs 1–9 are activated by Gαs, and ACs 1–8 are also activated by FSK. The interface of C1a and C2a contains the ATP binding site and a pseudosymmetric FSK site. C1a and C2a, each with ∼190 amino acids, are structurally homologous to each other, among mAC isoforms and with corresponding catalytic domains of sAC, sAC-C1 and sAC-C2, and of sGC, sGCαcat and sGCβcat.
Our present knowledge about structure, function, and regulation of the catalytic domains of mammalian mACs is mainly based on X-ray structure analysis of recombinant C1a and C2a originating from dog AC5 and rat AC2, respectively. Until now, 17 crystal structures have been released in the Brookhaven Protein Databank (PDB). Table 1 presents an overview of the structures, varying in cocrystallized metal ions and ligands of the ATP binding site.
C1a and C2a were expressed and purified as 25-kDa peptide chains forming heterodimers with similar AC activity and regulatory properties as the holoenzymes, whereas homodimers of C1a or C2a are catalytically inactive. The heterodimeric construct was termed 5C1:2C2 and in all cases cocrystallized with FSK or its methylpiperazino derivative as well as with recombinant Gαs and GTPγS as Gαs ligand. ATP-site ligands comprise ATP analogs, P-site inhibitors, and 2',3′-substituted purine nucleotide inhibitors.
sAC exhibits an amino acid sequence and domain organization related more closely to cyanobacteria and myxobacteria nucleotidyl cyclases than to mammalian mACs (Buck et al., 1999). In human sAC, the first 470 amino acids comprise a short N-terminal tail as well as C1 and C2 connected by a linker of about 70 residues (Kleinboelting et al., 2014a). The C-terminal domain of variable length (human sAC ∼1140 amino acids) contains a putative autoinhibitory domain, a P-site, and a protein/protein interaction domain. A potential heme-binding domain (sAC-HD) shows some sequence similarities to Per-ARNT-Sim (PAS) domains known as interaction modules in signaling proteins, and more extensive parts of the C-terminal region slightly resemble STAND domains (Signal Transduction ATPases with Numerous Domains) (Middelhaufe et al., 2012; Steegborn, 2014). No clearly defined transmembrane domain is present in the sAC amino acid sequence. A full-length and a short form of sAC were identified by Western blot of testis extracts with the short form encompassing the C1/C2 domain. Initially, the short form was thought to be generated by proteolysis (Buck et al., 1999), this processing being required for expression of catalytic activity. However, it is now established that alternate splicing generates the testis short form (Jaiswal and Conti, 2001), usually termed truncated sAC (tsAC), as well as isoforms reported in other tissues. Some of these variants lack the C1 domain, whereas others have shortened C termini (Geng et al., 2005; Schmid et al., 2007; Chen et al., 2014a).
The catalytic domain structure of human sAC was resolved by X-ray crystallography. At present, the PDB provides 28 sAC crystal structures (Kleinboelting et al., 2014a,b, 2016; Saalau-Bethell et al., 2014; Ramos-Espiritu et al., 2016), including complexes with ATP, α,β-methylene adenosine-5′-triphosphate, cAMP, pyrophosphate, bicarbonate, biselenite, bisulfite, bithionol, and various inhibitors. Comparing structures of 5C1:2C2, of sAC-C1-C2 and of an inactive human sGCαcat:sGCβcat heterodimer (Allerston et al., 2013; Seeger et al., 2014) indicates high structural homology not only of the individual catalytic domains, but also of the whole complexes, and points to a common, well conserved mechanism of cNMP generation facilitated by two metal ions.
2. General Folds and Alignments of Catalytic Domains.
All crystal structures of 5C1:2C2, sGCαcat:sGCβcat, and sAC C1-C2 show a heterodimeric architecture with a pseudo-twofold axis. In Fig. 2, 5C1 and 2C2 are shown in side-by-side comparison denoting secondary structure elements (Zhang et al., 1997), primed in the case of 2C2. A root mean square deviation (rmsd) of only 1.3 Å for 153 structurally equivalent Cα atom pairs indicates the close similarity of 5C1 and 2C2 (Tesmer et al., 1997). More or less analogous fits result from pairwise superposition of 5C1, sGCαcat, and sAC-C1 as well as 2C2, sGCβcat, and sAC-C2, respectively, and of sGCαcat with sGCβcat as well as sAC-C1 with sAC-C2, in particular if only the sections between β1 (β1′) and α5 (α5′) are considered. In contrast, sequence alignments are less similar even if based on structure, but reflect the close evolutionary relatedness of mAC and sGC catalytic domains (amino acid identities: 5C1 versus sGCαcat 29%, 2C2 versus sGCβcat 27%), whereas sAC is quite distant from the other two enzymes (sAC-C1 versus 5C1 14%, versus sGCαcat 17%, sAC-C2 versus 2C2 14%, versus sGCβcat 15%). The amino acid identity between sGCα and sGCβ is high (42%) in contrast to 5C1 and 2C2 (25%) and to the dissimilar sAC-C1 and sAC-C2 domains (12%).
Fig. 2.

5C1 and 2C2 in side-by-side comparison of superimposed monomers. α-Helices, red; β-sheets, blue; loops and turns, yellow. Models are based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999). Secondary structure elements as well as N and C termini are labeled.
Figure 3 presents 5C1:2C2 in complex with Gαs:GTPγS, FSK, ATP, and Mg2+ (Tesmer et al., 1997). The catalytic domains form a “head-to-tail, wreath-like dimer” (Tesmer et al., 1997) with a pseudo-twofold axis. Perpendicular to this axis and in front, the so-called “ventral” surface (Zhang et al., 1997) is visible (see also Fig. 1). Common of each domain are five α-helices surrounding an eight-stranded β-sheet core. Two strands (β1, β4 and β1′, β4′, respectively) are discontinuous. The 5C1-2C2 interface contains a spacious cavity with two pseudosymmetric binding sites for FSK and ATP + 2 Mg2+ ions, respectively. The ligands probably approach from the ventral surface because of extensive 5C1-2C2 contacts at the dorsal side. Gαs interacts nearly exclusively with 2C2 (α1′-α2′ loop, α2′, α3′, α3′-β4′ loop) and projects widely beyond the ventral surface (Fig. 1), suggesting that the dorsal side faces the membrane (Tesmer et al., 1997).
Fig. 3.

5C1:2C2 in complex with FSK, ATP, Gαs, and GTPγS. View along the pseudo-twofold axis. 5C1, green; 2C2, cyan; Gαs helical domain, yellow; ras-like domain, mauve; atom colors: C, gray; N, blue; O, red; P, orange; Mg2+, violet. Models are based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999) with ATP docked instead of 2′,3′-dideoxy-ATP. Secondary structure elements (Gαs only contact sites with 5C1:2C2) as well as N and C termini of 5C1:2C2 are labeled.
The structurally most conserved regions between β1 and β4 as well as β1′ and β4′, including the two metal-ion binding carboxylate residues in 5C1, constitute βαββαβ catalytic cores present also in the Pol 1 family of nucleotide polymerases, which catalyze an attack of a 3′-hydroxyl upon a 5′ phosphate, too (Artymiuk et al., 1997). It may be concluded that mammalian nucleotidyl cyclases have evolved from a very remote ancient nucleotide polymerase ancestor although evolutionary convergence cannot be ruled out.
The architecture of 5C1:2C2 (Fig. 3) is very similar to that of sGCαcat:sGCβcat (Allerston et al., 2013) and, to a lesser extent, sAC-C1-C2 (Kleinboelting et al., 2014a). If only the sequences between β1 and α4 (β1′ and α4′, respectively) are considered, more or less close superpositions are possible (Fig. 4). From these superpositions, a structure-based alignment of the catalytic domains of AC1, AC2, AC5, sAC, and sGC was derived (Fig. 5), being essentially in line with a previous comparison of mACs and sGC, published when only crystal structures of 5C1:2C2 were known (Tesmer et al., 1997), and with a recent alignment of 5C1:2C2, sGCαcat:sGCβcat, and different prokaryotic and eukaryotic sAC-C1-C2 domains (Kleinboelting et al., 2014b). Exact mapping of secondary structure elements was based on 5C1:2C2, but α-helical and β-strand segments between α2 and α4 (α2′ and α4′) are nearly identical in size and position in all cases.
Fig. 4.

Catalytic domains of sGC and sAC aligned with 5C1:2C2. Domains are drawn from β1 to α4 and β1′ to α4′, respectively. Views along the pseudo-twofold axes. sdGCαcat, sAC-C1; 5C1, green; sGCβcat, sAC-C2; 2C2, cyan; atom colors: C, gray; N, blue; O, red; P, orange; Mn2+ and Ca2+, magenta; secondary structure elements as well as N and C termini are labeled. (A) sGCαcat:sGCβcat in complex with 2′-MANT-3′-dATP and Mn2+, model of the intermediate state based on the open state crystal structure PDB 3UVJ (Allerston et al., 2013) and on 5C1:2C2 PDB 1TL7 (Mou et al., 2005) as template (transparent model). (B) sAC in complex with α,β-methylene adenosine-5′-triphosphate, bicarbonate and Ca2+, model based on the crystal structure PDB 4CLK (Kleinboelting et al., 2014a), 5C1:2C2 (transparent model) corresponding to Fig. 3.
Fig. 5.

Structure-based sequence alignment of the catalytic domains of sAC, sGC, AC1, AC2, and AC5. Secondary structure elements are primarily derived from 5C1 (green) and 2C2 (cyan), after α5 of sAC1, 5C1 and sGCα, only β7 and β8 are common. The sequence numbers above and below the alignment correspond to 5C1 and 2C2, respectively. Amino acids in contact with Gαs are highlighted in yellow, underscored residues form the C1-C2 interface of 5C1:2C2 and sAC. Bold-colored amino acids belong to the FSK (5C1:2C2) or bicarbonate (sAC) and nucleotide binding sites and interact with FSK or bicarbonate (magenta), metal ions (red), triphosphate (brown), the ribosyl moiety (green), and the nucleobase (blue), respectively. Amino acids in italics contribute via backbone, hydrophobic, or van der Waals interactions to nucleotide binding. Other interactions are electrostatic (including ionic, hydrogen bonds) and via side chains.
Differences are obvious between lengths of various loops and between secondary structure elements in the N- and C-terminal regions. The exceptionally long insert in 2C2 between β3′ and α3′ is not fully resolved in 5C1:2C2 structures and probably involved in interactions with Gβγ (Chen et al., 1995), a characteristic modulator of AC2. The extended β2′-β3′ loop of sAC-C2 contributes to the conversion of the mAC-FSK site into a binding site for the sAC activator bicarbonate (Kleinboelting et al., 2014a). The α1-α2 and α1′-α2′ regions vary within and between the three structures. In 5C1:2C2, α1 is one helical turn longer than α1′ and connected by a hairpin turn with α2. The long, protruding α1′-α2′ loop is in keeping with its role as part of the Gαs binding site. By contrast, α1 and α1′ are of similar length (two turns) in sGCαcat and sGCβcat, respectively. Helix α1′ is not present in sAC-C2.
The most striking differences between the sequences and structures appear in the C-terminal regions (after α5 and α5′, respectively). In 5C1, a short β7-strand is connected with β8 by a long Ω loop including two short α-helices which are not present in sGCαcat and sAC-C1. Nevertheless, the course of sGCαcat in this region roughly resembles that of 5C1. The β7-strand of sAC-C1 is longer, and the course between β7 and β8 differs compared with 5C1. The β7′-β8′ segments of the three structures are more similar. Two extended β-strands are connected by a reverse turn directed to the triphosphate group of the nucleotide.
3. Interaction of C1 Catalytic Domain of AC5:C2 Catalytic Domain of AC2 with α-Subunit of the Stimulatory G-protein of Adenylyl Cyclase Gs.
The interface between 5C1:2C2 and Gαs is only about half as large as that between 5C1 and 2C2 (Tesmer et al., 1997), and only a few structural elements are involved in interactions (Figs. 3, 5, and 6). The vast majority of contacts are formed by 2C2. The switch II helix of Gαs is embedded into a groove enclosed by the α1′-α2′ loop, α2′, and the α3′-β4′ loop, which, together with α3′, also contacts α3 and the α3-β5 loop of Gαs. 5C1 contributes to mAC-Gαs interactions only at the N terminus (before β1) approaching Gαs at the switch II helix and the α3-β5 loop. It has been suggested that Gαs binding (insertion of the switch II helix between the α1′-α2′ loop and the α3′ helix) is necessary for the transition between an inactive, “open” into an active, “closed” 5C1:2C2 state. As a result of this activating conformational change, the β2-β3 loop is slightly shifted toward nucleotide binding elements in β5′ and α4’, leading to proper alignment of the catalytic residues with the ATP substrate (Tesmer et al., 1997).
Fig. 6.

Detailed 5C1:2C2-Gαs interactions. 5C1, green; 2C2, cyan; Gαs, mauve; heteroatom colors: N, blue; O, red; S, yellow. Hydrogen bonds are drawn as red dashed lines. Model based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999). Contacting amino acids (distance <3 Å) and secondary structure elements are labeled.
Seven polar interactions predominate among AC-Gαs contacts, namely hydrogen bonds of side chains of N905 (α1′-α2′ loop) and C911 (α2′) with Q236 (Gαs switch II) and of E917 (α2′) with W281 (Gαs α3-β5 loop), a hydrogen bond of the backbone oxygen of H989 (α3′-β4′ loop) with the terminal NH2 group of N279 (Gαs α3-β5 loop), a salt bridge between D921 (α2′) and R280 (Gαs α3-β5 loop), and contacts of the side chains of R913 (α2′) and N992 (α3′-β4′ loop) with N239 and R232 (Gαs switch II), respectively. The bottom of the binding groove for switch II is crossed by a hydrophobic patch consisting of M378, F379 (before β1), F898 (α1′-α2′ loop) and L914 (α2′). In particular, I235 and F238 of the switch II helix interact with this patch. At the α3-β5 loop of Gαs, W281 is stacked with 5C1 F379, and L282 contacts 5C1 M378. A second center of hydrophobic interactions emanates from F991 (α3′-β4′ loop) approaching W234 (switch II), L272, and I276 (α3) of Gαs. This pattern of AC-Gαs interactions is common for mAC isoforms, because all respective mAC amino acids are conserved (Fig. 5).
Gαi inhibits AC5 and AC6 noncompetitively with respect to Gαs (Taussig et al., 1994a), binding at a site that is opposite to that for Gαs, i.e., between the α1-α2 loop and the α3 helix in the 5C1 domain (Tesmer et al., 1997; Dessauer et al., 1998).
4. C1 Catalytic Domain of AC5:C2 Catalytic Domain of AC2 Interface.
The interface reflects the pseudosymmetry of 5C1 and 2C2. Figure 7 shows the secondary structure segments and amino acids involved in polar and hydrophobic 5C1-2C2 interactions. Contacts (interatomic distances < 4Å) are formed by 28 amino acids of 5C1 and 33 of 2C2 (Tesmer et al., 1997), from which 17 are located at equivalent domain positions and 7 are identical (Fig. 5).
Fig. 7.

5C1-2C2 interface. Cα-atoms of all amino acids involved in 5C1-2C2 interactions (distance <4 Å, see Fig. 5) are drawn as balls, hydrogen bonds, and salt bridges of labeled residues as sticks. Colors: H-acceptor/acidic side chain, red; backbone O as H-acceptor, orange; H-donor/basic side chain, dark blue; backbone NH as H-donor, light blue; other contacting amino acids and framework of 5C1, green, of 2C2, cyan.
The rough symmetry of interacting secondary structure elements becomes obvious: contacts of α1-α2 (α1′-α2′) with β1′, β4′ and the β5′-α4′ turn (β1, β4, β5-α4 turn) as well as of the C-terminal α2 (α2′) section and β2 (β2′) with the β4′-β5′ (β4-β5) loop. However, explicit interactions of amino acids are rather asymmetric. Only two charge-assisted hydrogen bonds between D424 (α2) and the backbone NH moieties of A1012 and Q1013 (β4′-β5′ loop) are reflected by positionally identical hydrogen bonds of D923 (α2′) with L499 and R500 (β4-β5 loop). The asymmetry of the contacts correlates with the different shape of the FSK and the ATP binding site. The “upper righthand” interface region is characterized by extensive contacts and additionally stabilized by FSK binding. A hydrogen bond between K436 (β2) and the backbone oxygen of T939 (β2′) may be regarded as borderline between this region and the “lower lefthand” interface region with fewer contacts and a larger cavity involving the ATP and metal binding site. Characteristic of the lower region is a hydrogen bond between T401 and K1065, which keeps the β7′-β8′ loop in an “open” position with respect to α1. This contact is broken on nucleotide binding. Gαs and FSK are suggested to activate mACs by changing the relative orientation of the catalytic domains while preserving nearly all interfacial contacts (Tesmer et al., 1997). Conformational switches are mainly based on a few alternate interactions of flexible segments (loops) of one domain with core components (helices, strands) of the other domain.
The interface in the structure of apo sAC-C1-C2 (Kleinboelting et al., 2014a) corresponds largely to that in 5C1:2C2 (Fig. 5). Thirty-two amino acids of sAC-C1 and thirty amino acids of sAC-C2 participate in contacts. Nineteen residues, five of which are identical, are located at equivalent domain positions. Interactions between secondary structure elements show the same rough symmetry as in case of 5C1:2C2 (see above and Figs. 4B and 7), and also the asymmetry of contacts is similar. The C1-C2 interface of sAC is stabilized by ten hydrogen bonds. Among them, the following five hydrogen bonds overlap with hydrogen or ionic bonds between 5C1 and 2C2 (Fig. 7): Y38 (β1)-E308 (α2′) with K386-E907, E84 (α2)-H398 (β4′-β5′ loop) with D424-A1012, F96 (β2)-K334 (β2′) with K436-T939, G158 (β4-β5 loop)-Q315 (α2′) with G498 - N916 and D159 (β4-β5 loop)-Y318 (α2′) with L499-D923. The structure of apo sAC-C1-C2 resembles the “closed” state of 5C1:2C2 with no contact between α1 and the β7′-β8′ loop.
5. Forskolin Binding Site.
FSK binds in the “upper” pseudosymmetric region of the ventral cleft close to Gαs (Fig. 3). The binding site is located between β1, β3, and the β5-α4 loop of 5C1 on one hand and α1′ and the β2′-β3′ loop of 2C2 on the other hand. Figure 8 presents the binding mode of FSK in more detail. The backbone oxygen of V506 (β5), the side chain of T512 (α4), and the backbone NH moieties of S508 (β5-α4 loop) and S942 (β2′-β3′ loop) form four direct hydrogen bonds with hydroxy and carboxy groups, respectively, of FSK. The side chain of S942 is further involved in a water-mediated H-bond network bridging the O1 and O4 atoms in FSK. Hydrophobic contacts with V511 (α4), K896 (α1′), I940, G941 (β2′-β3′ loop) and a couple of aromatic amino acids, namely F394 (β1), Y443 (β3), W507 (β5), F895 (α1′), and Y899 (α1′-α2′ loop) complete the binding mode. Whereas FSK stabilizes 5C1-2C2 interactions by one order of magnitude (Sunahara et al., 1997), it does not affect the Km value of ATP (Dessauer et al., 1997). However, Vmax increases 60-fold (Sunahara et al., 1997), indicating that FSK induces a conformational change of the ATP site leading to more efficient cAMP synthesis. Probably contacts between the β2-β3 and the β2′-β3′ loop link amino acids of the FSK site with residues contributing to catalysis (Tesmer et al., 1997).
Fig. 8.

Detailed 5C1:2C2-FSK interactions. 5C1, green; 2C2, cyan; FSK and water, gray; heteroatom colors: N, blue; O, red. Hydrogen bonds are drawn as red dashed lines. Model based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999). Contacting amino acids (distance < 3.5 Å) and secondary structure elements are labeled.
The bicarbonate binding site of sAC overlaps with that of FSK at 5C1:2C2 (see Figs. 4B and 5). According to the structure of human sAC in complex with bicarbonate (Kleinboelting et al., 2014a), the bicarbonate ion forms charge-assisted hydrogen bonds with the backbone NH moieties of V167 (β5) and M337 (β2′-β3′ loop) as well as salt bridges with K95 (β2) and R176 (α4). The bicarbonate binding site of sAC is too small to accommodate FSK (Kleinboelting et al., 2014a).
6. Nucleotide Binding Site.
The nucleotide binding site in the “lower” pseudosymmetric region of the ventral cleft is more spacious than the FSK site, and interfacial contacts are weaker than in the “upper” region. The resulting high conformational flexibility facilitates ATP binding, catalysis, regulation, as well as cAMP and pyrophosphate dissociation. Moreover, not only Gαs but also ATP binding is necessary to transfer the inactive open into the active closed 5C1:2C2 state. In Fig. 9, an apo-AC 5C1:2C2 structure is aligned with an ATP bound structure (Tesmer et al., 1997). The closed state of the latter results from translocation and 7°-rotation of 5C1 around the center of the heterodimer, closing the catalytic site by moving α1 toward α4′ and the β7′-β8′ loop of 2C2. Nevertheless, all interfacial contacts except two between α1 and the β7′-β8′ loop are conserved in both states. With two inhibitory Ca2+ ions instead of Mg2+, 5C1:2C2 binds ATP without switching into a closed state (Mou et al., 2009). The substituents of 2′,(3′)-O-(N-methylanthraniloyl) nucleoside 5′-triphosphates and 2′,3′-O-(2,4,6-trinitrophenyl-NTPs bind between α1 and the β7′-β8′ loop, preventing full closure of the catalytic site (Mou et al., 2005, 2006). As a consequence, the respective complexes represent intermediate (between closed and open) states of 5C1:2C2. In case of the sGCαcat:sGCβcat heterodimer, similar differences of an open and a GTP-bound closed conformation have been suggested (Allerston et al., 2013).
Fig. 9.

Closed and open 5C1:2C2 conformations. Alignment of the complex with ATP, Mg2+, and FSK with the open inactive state of apo-5C1:2C2. View along the pseudo-twofold axes. Closed state 5C1, green; 2C2, cyan; open state 5C1, yellow; 2C2, purple; atom colors: C, gray; N, blue; O, red; P, orange; Mg2+, violet. Models are based on the crystal structures PDB 1CJU with ATP docked instead of 2′,3′-dideoxy-ATP and 1AZS, respectively (Tesmer et al., 1997, 1999). Unfitting secondary structure elements as well as N and C termini are labeled.
The structural similarity between mammalian mACs, sAC, and sGC in the nucleotide binding regions is exceptionally high. On one hand, common binding modes of ATP, GTP, and metal ions as well as common interactions and structural transformations indicate an evolutionarily well conserved catalytic mechanism of cNMP generation. On the other hand, differences observed in crystal structures result from substrate and inhibitor selectivity and from the flexibility of the ribosyl moiety and the triphosphate chain. The common nucleotide binding region of mACs, sAC and sGC and raw positions of aligned amino acids interacting with ATP and GTP, respectively, are shown in Fig. 10.
Fig. 10.

Common nucleotide binding site of mAC, sGC and sAC. Essential amino acids are drawn as space fill models of Cα and Cβ atoms; catalytic domain 1, green; catalytic domain 2, cyan; metals A and B, red balls; interactions of metal ions, red; triphosphate, orange; ribosyl moiety, green; nucleobase, blue. The model is based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999) with ATP docked instead of 2′,3′-dideoxy-ATP. Common secondary structure elements and amino acids of the binding site are labeled.
Metal-, triphosphate-, ribosyl-, and nucleobase-cyclase interactions are clearly distinguishable (Fig. 5). Two aspartates in β1 and the β2-β3 loop, respectively, coordinate metal ions A and B (Mn2+ or Mg2+ in native enzymes). The three phosphate moieties interact with conserved basic residues (Arg, Lys) in β4, α4′ and the β7′-β8′ loop. A relatively spacious cavity with the β2-β3 loop as well as β5′ and α4′ as lateral margins and with β2′ and the β2′-β3′ loop as roof accommodates the entire nucleoside moiety. Only two replacements between mACs and sGC, namely Lys by Glu (β2′) and Asp by Cys (β5′), are required to reverse adenine into guanine selectivity (Sunahara et al., 1998; Tucker et al., 1998).
Figure 11A shows detailed interactions between ATP and 5C1:2C2. Both Mg2+ ions are coordinated with D396 and D440, MgA additionally with the α-phosphate and the 3′-OH group of ATP, MgB with all phosphate moieties and with the backbone oxygen of I397. The three phosphates are integrated in a compact network of charge-assisted hydrogen bonds and salt bridges: the position and charge of the α-phosphate is stabilized by R1029 (α4′), the β-phosphate interacts with the backbone NH functions of F400 and T401 (α1), and the γ-phosphate with G399 (backbone NH, β1-α1 loop), R484 (β4), and K1065 (β7′-β8′ loop). The ribosyl moiety forms two hydrogen bonds between the ring oxygen and N1025 (α4′) and between the 2′-OH group and S1028 (α4′).
Fig. 11.

Detailed interactions of mACs and sAC with ATP. C1, green; C2, cyan; ATP, gray; heteroatom colors: N, blue; O, red; P, orange;, hydrogen bonds are drawn as red dashed lines, contacting amino acids (distance <3.5 Å) and secondary structure elements are labeled. (A) 5C1:2C2-ATP interactions, Mg2+, violet, model based on the crystal structure PDB 1CJU (Tesmer et al., 1997, 1999) with ATP docked instead of 2′,3′-dideoxy-ATP. (B) sAC-ATP interactions, Na+, violet; w, water oxygen; model based on the crystal structure PDB 4USW (Kleinboelting et al., 2014b).
A two-metal-ion catalytic mechanism, previously derived from crystal structures in complex with ATP analogs and various divalent cations (Tesmer et al., 1999), was recently corroborated by density functional theory calculations of 5C1:2C2-ATP complexes (Hahn et al., 2015). Metal A acts as Lewis acid facilitating deprotonation of the 3′-OH group. Water-mediated transfer of the proton to the γ-phosphate is followed by changes in the coordination of both metal ions and in the conformation of ATP. The resulting shorter distance facilitates the nucleophilic attack of the 3′-oxygen on the α-phosphorus atom. The subsequent, concerted phosphoryl transfer step is rate-limiting and includes a transition state tightened by R1029. K1065 contributes to shifting the proton transfer equilibrium.
Binding of the nucleobase to mACs is relatively strong compared with sAC and sGC. The adenine moiety is involved in hydrogen bonds of the N1 nitrogen with K938 (β2′) and of the 6-NH2 group with D1018 and the backbone oxygen of I1019 (β5′). Van der Waals contacts of the purine nucleus with L438 and G439 (β2-β3 loop) and with F889 (β1′) align the nucleobase between both domains.
The spacious nucleotide binding site of mACs is promiscuous for different nucleobases and accommodates various ligands, e.g., P-site inhibitors, 2′- and 3′-MANT as well as TNP [2′,3′-O-(2,4,6-trinitrophenyl)]-substituted nucleotides, with often high affinity (Seifert et al., 2012). The general binding mode of these inhibitors corresponds to that of ATP, but flexible fit of the phosphate and ribosyl moieties (ring pucker) leads to slightly different and specific interactions. However, NTPs other than ATP do not act as mAC substrates (Sunahara et al., 1998; Hasan et al., 2014), because coherent dynamic transformations rather than tight interactions account for kinetic processes. The high substrate selectivity of mACs with respect to variation of the nucleobase and the triphosphate moiety depends on specific stable and/or transient hydrogen bonds and on distinct flexibility compatible with a concerted catalytic mechanism.
Figure 11B demonstrates similarities and differences of the binding mode of ATP at sAC compared with that at mACs. The structure of the complex (Kleinboelting et al., 2014b) contains only one Na+ (or Ca2+) ion located somewhat between the reference positions of metals A and B in 5C1:2C2. The triphosphate moiety adopts a specific conformation with partially different interactions than in the case of 5C1:2C2. The α-phosphate interacts with R416 (α4′) and K451 (β7′-β8′ loop); the β-phosphate with S49 (β1), K144 (β4), and Na+; and the γ-phosphate forms three hydrogen bonds with G50 (backbone NH, α1), T52 (α1), and N412 (α4′) and contacts also Na+. R176 (α4), in fact pointing to the regulatory bicarbonate site, was modeled as in the sAC complex with α,β-methylene-ATP (Kleinboelting et al., 2014a), because this amino acid together with R416 (α4′) probably takes part in catalysis. The adenine base adopts a syn conformation in contrast to the anti conformation in most other mammalian nucleotidyl cyclase structures resolved so far. The 6-NH2 group is involved in a hydrogen bond with the backbone oxygen of V406 (β5′) and in a water-mediated interaction with K334 (β2′). During catalysis, a transient direct contact between K334 and the 6-NH2 substituent may be formed (Kleinboelting et al., 2014a). The catalytic constants of ATP and GTP as sAC substrates indicate that base discrimination takes place during turnover rather than by substrate binding (Kleinboelting et al., 2014b). Km values for ATP for sAC are ∼20 times higher than those for mACs (Kleinboelting et al., 2014a). Figure 11 provides possible reasons: lower affinity of ATP at sAC may be due to fewer explicit hydrogen bonds compared with 5C1:2C2. Specifically, the ribosyl moiety forms no hydrogen bonds corresponding to those with N1025 (sAC N412) and S1028 (sAC A415) in the case of 5C1:2C2.
B. Expression of Adenylyl Cyclases
Understanding the specific role of AC isoform in cellular signaling has been hampered by the complex and heterogeneous expression pattern of ACs in nearly all cell types (Defer et al., 2000; Willoughby and Cooper, 2007; Sadana and Dessauer, 2009; Sanabra and Mengod, 2011). The specific signaling roles of each AC isoform are further complicated by their unique, but often overlapping, regulatory properties. Much of our understanding on the specific roles of individual AC isoforms in signaling pathways has been derived from studies with gene knockout and transgenic mice.
Description of the cellular expression patterns for each AC provides important information in delineating their signaling roles. However, identifying AC isoform distribution remains a challenge (Antoni et al., 2006). First and foremost, there is a dearth of well-characterized isoform-specific antibodies (Iwamoto et al., 2003; Antoni et al., 2006; Sadana and Dessauer, 2009; Göttle et al., 2009). In some cases, antibodies have been used along with other molecular (mRNA measures) or biochemical (functional assays) approaches revealing partially consistent results (Ostrom et al., 2003; Johnston et al., 2004; Liu et al., 2004; Göttle et al., 2009). A second factor concerning quantitative expression studies is the generally low levels of AC protein expression (Alousi et al., 1991; Seifert et al., 1998). For the reasons discussed above, most studies have used mRNA quantification methods to gain insight into the potential expression pattern of ACs. These approaches include RT-PCR analysis, Northern analysis, and in situ hybridization (Defer et al., 2000; Willoughby and Cooper, 2007; Sadana and Dessauer, 2009; Sanabra and Mengod, 2011). There is also an emerging body of information on mRNA expression using microarray analysis and next generation sequencing technology (Atwood et al., 2011). Despite the number of available data sets, the generally low correlation between mRNA and protein expression highlights the need for caution in using mRNA as a surrogate for protein expression (Maier et al., 2009; Ostlund and Sonnhammer, 2012; Werner et al., 2016). The reasons for the lack of correlation presumably reflect the complex processes ultimately contributing to protein expression including transcription, translation, and protein stability/turnover. Although meaningful comparisons of the mRNA expression levels between individual isoforms are discouraged, comparisons of a single isoform across tissues or after perturbation may be informative given that appropriate experimental controls are met.
The general expression patterns for AC mRNA have been expertly described in several previous reviews, and the major sites of robust mRNA expression for each isoform have been specified (Defer et al., 2000; Willoughby and Cooper, 2007; Sadana and Dessauer, 2009). These reports used information from a variety of species including human, rat, mouse, and chicken. Much of the expression work has been supported by studies with transgenic or knockout mice. Perhaps the most thoroughly studied examples involve AC1 and AC8. Both isoforms are highly expressed in brain regions associated with learning and memory, synaptic plasticity, and opioid withdrawal, and experiments with single and double knockout animals support these implications (Ferguson and Storm, 2004; Sadana and Dessauer, 2009).
Rieg and Kohan (2014) described the expression pattern of AC isoforms within the nephron. This carefully written review focused primarily on mRNA studies and generally used antibody studies as supporting data. There was a notable absence of AC1 and AC8, consistent expression for AC6 and AC9, and a more mixed expression pattern of the other AC isoforms throughout the nephron (Rieg and Kohan, 2014). The distribution of AC isoforms in the digestive system was also reviewed (Sabbatini et al., 2014). Similar to the other organs/cells reported, mRNA for multiple ACs were found in hepatocytes and cholangiocytes (Strazzabosco et al., 2009), as well as in the pancreatic acini and duct (Sabbatini et al., 2013). Another paper describing the cAMP signaling elements in lung reported the AC protein/mRNA expression patterns in several tissues and cells including bronchial epithelium, vascular endothelium, airway smooth muscle cells, vascular smooth muscle cells, pulmonary fibroblasts (WI-38 cells), and inflammatory cells (Oldenburger et al., 2012). Although multiple AC isoforms were found in all cells, the number of AC isoforms expressed in the vascular endothelium appears to be more limited (e.g., four isoforms by antibodies) when compared with other cells including airway or vascular smooth muscle cells that appear to express mRNA for seven or all nine ACs, respectively (Xu et al., 2001; Jourdan et al., 2001; El-Haroun et al., 2004). It is likely that RT-PCR approaches would have identified more ACs in the vascular endothelium because of its enhanced sensitivity. The expression of multiple ACs is common in most if not all tissues/cells, including myometrium (Price and Bernal, 2001), adipocytes (Serazin-Leroy et al., 2001), skeletal muscle (Suzuki et al., 1998), and adrenal gland (Shen et al., 1997).
The heart also expresses multiple AC isoforms (Manolopoulos et al., 1995; Göttle et al., 2009). Differential expression of AC4–AC7 mRNA in various regions of the heart and vascular system of humans has been described (Wang and Brown, 2004). There are also cell-type expression differences with AC5 and AC6, proposed to be the most abundant AC isoforms in cardiomyocytes (Yu et al., 1995; Li et al., 2012). However, there is controversy with regard to the relative expression of AC5 versus AC6 in the heart as well as the changes in expression that occur during development or stress (Tobise et al., 1994; Espinasse et al., 1995, 1999; Scarpace et al., 1996; Wang and Brown, 2004; Hu et al., 2009; Braeunig et al., 2013). AC1 mRNA is found only in the sinoatrial node (Mattick et al., 2007). Cardiac fibroblasts express ACs 2–8 with differential patterns in caveolar versus non-caveolar regions (Ostrom et al., 2003).
A number of studies examined AC isoform expression across the central nervous system (Matsuoka et al., 1997; Antoni et al., 1998; Nicol et al., 2005). In situ hybridization was used to examine the AC expression patterns in the rodent brain (Visel et al., 2006; Sanabra and Mengod, 2011). Differences in AC isoform expression between glutamatergic, GABAergic, and cholinergic neurons were revealed (Visel et al., 2006; Sanabra and Mengod, 2011). High levels of mRNA for AC1 and AC8 are found in areas important for learning and memory and high levels of AC5 mRNA in the striatum (Sanabra and Mengod, 2011). AC2 was found in limbic as well as in other areas. AC3 and AC6 were found widespread throughout the brain as was AC9 with enhanced expression in the neocortex and hippocampus (Sanabra and Mengod, 2011). There was limited expression of AC7 in thalamic and hypothalamic nuclei and AC4 was found only in blood vessels (Visel et al., 2006; Sanabra and Mengod, 2011). The reviews and reports highlighted above support a very broad distribution of ACs with multiple isoforms being expressed in any given tissue as well as cell type.
Studies with human tissues and cells are somewhat limited but also suggest that the mRNAs of many AC isoforms are broadly expressed across multiple tissues (Xu et al., 2001; Ludwig and Seuwen, 2002; Wang and Brown, 2004; Pinto et al., 2012). Of particular note is the study of the expression of mRNA for ACs 1–9 in 16 human tissues (Ludwig and Seuwen, 2002). These investigators attempted to visually quantify the RT-PCR products’ band intensity and compare expression levels of individual isoforms across tissues (Table 2). Multiple AC isoforms ranging from three isoforms in skeletal muscle to all nine mAC isoforms in the testis were found. The data also point to differences between human and mouse expression patterns in certain tissues including skeletal muscle (Suzuki et al., 1998; Ludwig and Seuwen, 2002). In addition to the brain, there is significant AC1 expression in heart, skeletal muscle, kidney, pancreas, spleen, testis, and ovary (Ludwig and Seuwen, 2002). AC8 showed the most restricted expression, being detected in the brain and testis. The generally broad AC expression patterns in humans as well as other species often contrast with the initial cloning studies where mRNA expression was examined at the whole tissue level. The discrepancies may be explained when one considers potential differences in methodology (Northern analysis versus RT-PCR) as well as when comparing whole tissue extracts to cell preparations where AC mRNAs may show selective expression in certain cell types.
TABLE 2.
Expression of mAC isoforms at the mRNA level
Expression pattern of AC isoforms in rodent brain and human tissue. Sanabra and Mengod (2011) used in situ hybridization to examine and compare the AC isoform expression in rodent brain tissues. Semiquantitative RT-PCR was used to examine and compare the AC isoform expressions in 16 human tissues (Ludwig and Seuwen, 2002).
| AC Isoform | Rodent CNS Expression (Sanabra and Mengod, 2011) | Relative Human Tissue Abundance (Ludwig and Seuwen, 2002) |
|---|---|---|
| AC1 | Hippocampus, cerebellum, cortex, and thalamus | Peripheral blood leukocytes > heart ∼ skeletal muscle ∼ kidney ∼ pancreas ∼ spleen ∼ testis ∼ ovary |
| AC2 | Limbic and widespread | Skeletal muscle ∼ testis > heart ∼ prostate ∼ ovary ∼ colon |
| AC3 | Widespread | Placenta ∼ testis ∼ ovary ∼ colon > lung ∼ spleen ∼ thymus |
| AC4 | Not detected | Heart ∼ placenta ∼ lung ∼ spleen ∼ testis ∼ ovary ∼ small intestine |
| AC5 | Caudate putamen and olfactory tubercle | Testis ∼ heart ∼ prostate ∼ ovary ∼ small intestine ∼ colon |
| AC6 | Widespread | Heart ∼ kidney ∼ prostate ∼ testis ∼ ovary ∼ small intestine ∼ colon |
| AC7 | Thalamus and hypothalamus | Peripheral blood leukocytes > placenta ∼ lung ∼ spleen ∼ thymus > heart ∼ liver ∼ ovary ∼ colon |
| AC8 | Hippocampus, cerebellum, cortex, thalamus and hypothalamus | Testis (limited detection) |
| AC9 | Cortex and hippocampus | Heart ∼ placenta ∼ lung ∼ liver ∼skeletal muscle ∼ kidney ∼ pancreas ∼ testis ∼ ovary ∼ peripheral blood leukocytes |
In addition to knowing tissue expression patterns, investigators are interested in the AC isoform expression in relevant cell lines or primary cells. The majority of studies employed a RT-PCR approach to assess mRNA levels (Varga et al., 1998; Johnston et al., 2004; Liu et al., 2004), but microarray approaches have been used as well (Atwood et al., 2011; Zhu and Oxford, 2011). HEK293 cells are often used for examining both endogenous and exogenous AC signaling. Reports on AC mRNA expression in HEK293 cells indicate the presence of three to seven AC isoforms (Hellevuo et al., 1993; Premont, 1994; Ludwig and Seuwen, 2002; Lefkimmiatis et al., 2009). Consensus analysis of these studies suggest that HEK293 cells express mRNA for AC1, AC3, AC5 (weak), AC6, AC7 (weak), and AC9. A similar pattern in HEK293 cells was found using microarray, with the exception that AC5 and AC7 were reported at similar levels to AC1, AC3, AC6, and AC9 (Atwood et al., 2011). The reported discrepancies in expression patterns of the same cell model could reflect any of the numerous experimental or methodological variations including differences in cell harvesting/preparation, cell growth/maintenance conditions, RT-PCR approach/conditions, or primer design.
Much remains unknown regarding the actual expression patterns of individual AC isoforms. The use of mRNA as a surrogate for protein expression has its limitations, but provides a starting point into understanding signaling pathways. The astute scientist is encouraged to gather converging pieces of evidence to establish a link between mRNA abundance with AC function. One example for such an approach is the elaboration of the FSK-insensitive AC9 as the functionally predominant AC isoform in human myeloid cells (Mahadeo et al., 2007; Brunskole Hummel et al., 2013; Werner et al., 2014, 2016). Combining mRNA studies with additional data from the use of well-characterized antibodies (when available) and gene silencing approaches like shRNA and siRNA (Fan et al., 2009; Emery et al., 2015) can further connect AC isoform expression to cellular signaling.
It is firmly established that sAC is expressed at high levels in the testis of all mammalian species studied so far including human, and its expression is confined to postmeiotic germ cells (Schmid et al., 2014; Levin and Buck, 2015). Two major protein variants are retrieved from testis and spermatozoa; i.e., a protein of 187 kDa corresponds to the full-length enzyme and a 48–50 kDa splicing tsAC variant containing only the C1/C2 domains. Both molecular species are catalytically active. Lower level expression has been reported in a wide range of additional tissues and cells (Schmid et al., 2014; Levin and Buck, 2015).
C. Compartmentation of Adenylyl Cyclases
1. Cellular Localization.
Although many signaling processes can differently alter activity of each AC isoform, there is a single, primary signal output by ACs 1–10, i.e., production of the second messenger cAMP. This simple fact belies both the intuitive notion and the experimental observation that not all AC isoforms regulate identical cellular functions. To explain this complication, one must evoke the concept of cAMP signaling compartmentation (Ostrom et al., 2012; Cooper and Tabbasum, 2014). Indeed, specific AC isoforms localize in different cellular compartments where they associate with a subset of GPCRs and form distinct complexes with downstream effectors. ACs 3, 5, 6, and 8 associate with caveolar and lipid raft microdomains, whereas the other mACs are localized in nonraft plasma membrane domains (Schwencke et al., 1999; Fagan et al., 2000; Ostrom et al., 2000, 2002; Smith et al., 2002). This isoform-specific localization is remarkably stable across different cell types, species, and activation states (Ostrom and Insel, 2004). ACs partner with GPCRs and downstream effectors that are similarly localized, creating discrete signaling complexes. It is attractive to imagine that these complexes allow the common second messenger, cAMP, to generate a wide array of specific responses, even in a single cell. This is especially true when one considers the large number of different GPCR expressed in a single cell that use cAMP as their primary intracellular signal.
GPCRs display some AC-specific coupling that reflects the colocalization of receptors and AC isoforms in lipid raft or nonraft plasma membranes. By using overexpression or knockdown of an AC isoform, specific GPCR-AC coupling was unmasked. AC6 overexpression in a number of cells, including airway smooth muscle, lung fibroblasts, and neonatal myocytes, enhances signaling by β-adrenoceptors (βARs) without affecting signaling by a number of other Gαs-coupled receptors (Ostrom et al., 2000, 2002; Liu et al., 2008; Bogard et al., 2011). In each case, receptor coupling could be explained by the colocalization or exclusion of the receptor in lipid rafts (where AC6 resides). This concept of colocalization can be even more complex in cardiac myocytes. Because of specialized T-tubule and sarcolemmal membrane environments in these specialized cells, AC and GPCR localization is even more specific. AC5 is localized primarily in T-tubules, whereas AC6 resides in sarcolemmal membranes (Timofeyev et al., 2013). The β1-adrenoceptor (β1AR), but not the β2-adrenoceptor (β2AR), couples effectively to AC5 (Tsunematsu et al., 2015). This mirrors the differential distribution of the β1AR and β2AR in T-tubules (Cros and Brette, 2013).
In airway smooth muscle cells, β2AR signaling is selectively increased when AC6 is overexpressed and these components colocalize in lipid rafts (Bogard et al., 2011). In contrast, prostanoid EP receptors couple only to AC2 in nonraft domains. Unique cellular responses can be mapped to individual AC isoforms in these cells. By examining gene expression after FSK stimulation in cells overexpressing either AC2 or AC6, interleukin 6 expression is increased by AC2-produced cAMP, whereas somatostatin expression was regulated by cAMP emanating from AC6 (Bogard et al., 2014). Studies using Förster resonance energy transfer-based biosensors to detect cAMP in different subcellular domains have provided direct evidence of these signaling compartments (Agarwal et al., 2011, 2014). These findings show that cAMP is generated in discrete places by individual ACs and that the response machinery in these locations differs enough to yield contrasting regulation in the nucleus.
Specific examples of receptor regulation of AC activity depending on colocalization in membrane domains continue to emerge. Dopamine D1 receptors preferentially regulate AC3, AC5, and AC6 activity and this depends on interaction with lipid rafts, whereas dopamine D5 receptors are more nonraft localized and couple to just AC5 (Yu et al., 2014). Family B GPCRs also seem to depend upon colocalization of receptor and AC, as evidenced by the observation that the pituitary AC-activating polypeptide type 1 receptor was unable to signal to downstream effectors when AC6 (but not AC7) expression was knocked down (Emery et al., 2015). Prostacyclin receptor inhibition of platelet activation depends upon intact lipid rafts (Raslan and Naseem, 2015), which is a known location of both prostacyclin receptor and AC5/6 expression (Liu et al., 2008).
Coupling of GPCRs to specific AC isoforms is highly regulated. In early pregnancy, α2-adrenoceptors inhibit uterine smooth muscle AC activity but then switch to being stimulatory during midterm pregnancy (Zhou et al., 2000). An upregulation of uterine AC2 expression at about the same time renders it likely that the switch in α2 adrenoceptor signaling is due to the altered AC isoform expression (Zhou et al., 2007). Signaling by a coexpressed receptor can even dictate coupling between a specific GPCR and AC. Activation of prostaglandin EP1 receptors facilitates dopamine D1 receptor activation of cAMP production (Ehrlich et al., 2013). This effect depends on Gβγ signaling and expression of AC7, a Gβγ-sensitive isoform, and may explain how prostaglandin E2 can regulate neuronal dopamine signaling.
Lipid rafts exert a more complex function than just providing an assembly point for GPCR and ACs. AC regulation by capacitive calcium entry depends on intact lipid rafts and is specific to raft-localized AC isoforms (Cooper et al., 1995; Fagan et al., 1998, 2000; Smith et al., 2002). Disruption of lipid rafts causes a loss of coupling between capacitive calcium entry and calcium-regulated AC isoforms, AC6 and AC8. Thus, lipid rafts facilitate the juxtaposition of calcium-sensitive AC isoforms and capacitive calcium entry channels.
Given that AC isoforms are either lipid raft localized or not, a structural component must exist in all ACs to confer an affinity for one domain or the other. Intuition leads one to consider sequence-specific protein-protein or protein-lipid interactions and posttranslational modifications as likely mechanisms. Evidence of sequence-specific interactions come from studies making chimeras between raft and nonraft isoforms (Crossthwaite et al., 2005) as well as reductionist approaches examining fragments of ACs (Thangavel et al., 2009). Both of these approaches found evidence for roles of the intracellular C1 and C2 domains in targeting ACs to lipid rafts. These studies imply that AC isoforms have key protein-protein interactions that confer their lipid raft localization. However, protein-lipid interactions may be just as important. Mutation of three N-linked glycosylation sites on an extracellular loop of AC8 causes this lipid raft-localized isoform to appear in nonraft membranes (Pagano et al., 2009).
With intracellular protein-protein interactions facilitating lipid raft localization of AC, it is tempting to implicate a known scaffolding protein and resident of these domains, caveolin. However, caveolins are not responsible for anchoring ACs in lipid rafts. Specifically, AC isoforms have the same expression and localization in cardiac fibroblasts isolated from both caveolin-1 knockout and wild-type mice (Thangavel et al., 2009). Additionally, cell lines that naturally lack caveolin expression still display lipid raft-localized AC isoforms (Thangavel et al., 2009).
sAC has been detected in a variety of organelles including nuclei, microtubules, cilia, and mitochondria (Zippin et al., 2003). Functional data supporting the expression of an active enzyme are available particularly for mitochondria and cilia of the airway epithelium. Intramitochondrial sAC activation is associated with an increase in cAMP measured by an immunodetection assay (Acin-Perez et al., 2009) or by monitoring Förster resonance energy transfer of mitochondria-targeted cAMP sensors (Di Benedetto et al., 2013). The increase in cAMP leads to PKA-mediated phosphorylation of respiratory chain enzymes (Acin-Perez et al., 2009; Di Benedetto et al., 2013) and an increase in ATP.
2. Organization of Adenylyl Cyclases with Downstream Effectors.
A long-standing idea is that cAMP signaling is “tailored” in various cell types via the combinatorial expression of AC and PDE isoforms (Houslay and Milligan, 1997). However, a greater level of organization of signaling is achieved by A kinase anchoring proteins (AKAPs). Via their defining PKA binding motif, AKAPs organize cAMP-dependent protein kinase (PKA) signaling to a wide variety of other proteins (Colledge and Scott, 1999). They scaffold the kinase and its substrates and bring together a diverse array of other proteins involved in various signaling pathways. Several diverse members of the AKAP family interact with ACs in an isoform-specific manner and regulate cAMP signaling (Bauman et al., 2006). To date, the specific AKAP-AC interactions that have been defined are AKAP9/Yotiao with ACs 1, 2, 3, and 9 (Piggott et al., 2008), mAKAP with ACs 2 and 5 (Kapiloff et al., 2009) and AKAP79/150 with ACs 2, 3, 5, 6, 8, and 9 (Efendiev et al., 2010; Delint-Ramirez et al., 2011; Shen and Cooper, 2013).
The role of specific AC-AKAP associations in signal transduction was reviewed by Dessauer (2009). AKAP9/Yotiao has complex effects on AC regulation that depend on the AC isoform. AKAP9 has a pure scaffolding function, with no alteration of enzyme activity, when combined with AC1 and AC9 (Piggott et al., 2008). By contrast, this same AKAP inhibits the activity of AC2 and AC3 when these components interact. AC8 forms a complex with AKAP5 (AKAP79/150) and the effect appears to be a decrease in AC8 sensitivity to Ca2+ (Willoughby et al., 2010). But many more elements of the cAMP signaling cascade can be localized to a given site of signal generation. AKAP5 also interacts with the β2AR and AC5 and AC6 (in addition, of course, to binding PKA) (Bauman et al., 2006). With this arrangement, PKA rapidly phosphorylates the β2AR to initiate desensitization, receptor translocation and even G-protein switching (Daaka et al., 1997), but the G-protein switch is discussed controversially (Friedman et al., 2002). PKA also inhibits AC5 and AC6 via phosphorylation (Beazely and Watts, 2006) and alters AC8 sensitivity to store-operated calcium entry; regulation by PKA is facilitated by AKAP5 (Delint-Ramirez et al., 2011). In each case, the AKAP-AC interaction establishes critical feedback regulation in the signaling cascade.
AKAPs can also regulate other ACs in an isoform-specific manner via targeting protein kinase C (PKC) phosphorylation. PKC increases activity of certain ACs (ACs 1, 2, 3, 5, 7) but inhibits the activity of others (AC6) (Sunahara et al., 1996; Lai et al., 1997). AKAP5 orchestrates muscarinic receptor activation of cAMP production by recruitment of PKC to a complex containing the PKC-stimulated AC2 (Shen and Cooper, 2013). PDE4 and PKA are also key elements in the complex. βAR regulation of postsynaptic AMPA-type glutamate receptors in mouse forebrain slices depends upon AKAP5 anchoring both PKA and AC (Zhang et al., 2013). Although the AC isoform in this model was not defined, these studies demonstrate that long-term potentiation is lost without this AKAP5-mediated organization.
AKAP-AC interactions have other pathophysiological implications (Efendiev and Dessauer, 2011). Mutations of the potassium channel KCNQ1 subunit interrupt its binding to Yotiao, causing a loss of PKA regulation of the channel, alteration in the action potential, and arrhythmias (Marx et al., 2002). Moreover, AKAP-AC complexes serve to sensitize bound PKA effectors to the effects of cAMP, shifting the concentration/response curves by two orders of magnitude (Efendiev et al., 2010). Furthermore, PKA/PDE4D feedback contributes to local regulation of ACs (Mika and Conti, 2016).
D. Regulation of Adenylyl Cyclases
Table 3 summarizes regulation of mAC isoforms. Isoform-specific regulation of AC isoforms has been well described and extensively reviewed (Tang and Gilman, 1992; Sunahara et al., 1996; Hanoune and Defer, 2001; Patel et al., 2001; Cooper, 2003). All mACs are activated by Gαs (Tang and Gilman, 1992; Iyengar, 1993; Jones et al., 1990), although differences in AC activation may exist (Jones et al., 1990; Seifert, et al., 1998; Chan et al., 2011). Only ACs 1, 5, and 6 are directly inhibited by Gαi (Federman et al., 1992; Taussig et al., 1993a, 1994; Chen and Iyengar, 1993).
TABLE 3.
Regulation of mAC isoforms
This table focuses on known direct regulators of AC, although there are indirect routes to altering AC activity. Some of the effects listed have been clearly demonstrated as “direct” using purified, cell free components, whereas others have only been shown in intact cells. In the case of the latter, one must consider indirect mechanisms as well as the requirement for other factors for the effect to be realized. This table is adapted and updated from that published by Ostrom et al. (2012).
Gβγ subunits increase the activity of ACs 2 and 4 (Gao and Gilman, 1991; Tang and Gilman, 1991; Taussig et al., 1993b). The stimulatory effects of Gβγ typically require the presence of another activator such as Gαs or FSK (Tang and Gilman, 1991). When overexpressed, Gβγ subunits have been reported to inhibit ACs 5 and 6 (Bayewitch et al., 1998). However, cellular and in vitro assays using purified proteins display a conditional stimulation of AC5 and AC6 by Gβγ (Gao et al., 2007; Thomas and Hoffman, 1996) that involves different structural determinants in AC5 compared with AC6 (Brand et al., 2015).
Although all ACs are inhibited by high concentrations of Ca2+, AC5 and AC6 are inhibited by submicromolar concentrations of Ca2+; AC1 and AC8, and to a lesser extent AC3, are stimulated by low concentrations of Ca2+ via CaM (Tang et al., 1991; Choi et al., 1992; Katsushika et al., 1992; Yoshimura and Cooper, 1992; Cali et al., 1994). Expression of AC5 and/or AC6 along with L-type Ca2+ channels allows an oscillatory regulation between Ca2+ and cAMP, because cAMP stimulates Ca2+ influx via the L type Ca2+ channel and the subsequent Ca2+ influx inhibits AC activity (Cooper et al., 1995). AC1 activation by Ca2+/CaM is sensitive to oxidation of specific methionine residues in the N- and C-terminal domains of CaM (Lübker et al., 2015a,b). The oxidation-dependent CaM-regulation of AC1 may be relevant in ageing processes (Lübker et al., 2015a,b). The structural basis for AC inhibition by Ca2+ has been resolved (Mou et al., 2009). Transient receptor potential channels contribute to regulation of Ca2+-sensitive ACs (Willoughby et al., 2014). In intact cell experiments, the Ca2+ ionophore A23187 is useful as an experimental tool to stimulate Ca2+-dependent ACs (Vortherms et al., 2006). PKC can also stimulate many AC isoforms (AC1, AC2, AC3, AC5, and AC7) but reduces activity of AC6 (Jacobowitz et al., 1993; Kawabe et al., 1994; Lai et al., 1997). Phorbol esters are often used as experimental tools to modulate AC function via PKC activation (Olianas and Onali, 1986; Freyaldenhoven et al., 1992; Lin and Chen, 1997). Different PKC isoforms have different effects on AC activity, but there are no direct comparisons of multiple PKC isoforms on activity of individual AC isoforms. Regulator of G-protein signaling proteins (RGS proteins) modulate activity of some AC isoforms as well. RGS2 reduces AC3, AC5, and AC6 activity (Sinnarajah et al., 2001; Salim et al., 2003; Salim and Dessauer, 2004). RGS9-2 suppresses basal activity and modulates Gβγ activation of AC5 (Xie et al., 2012). Annexin A4 reduces AC activity and downstream cAMP signaling in HEK-293 cells and cardiac myocytes (Heinick et al., 2015). Coimmunoprecipitation of annexin A4 and AC5 was shown, but evidence that the observed effects are specific to this AC isoform is missing.
Several AC isoforms are also subject to a paradoxical increase in activity after persistent activation of Gi/Go-linked receptors. This process is referred to as heterologous sensitization, supersensitization, superactivation, or cAMP overshoot (Sharma et al., 1975b). Persistent activation of opioid receptors on neuronal cells led to a marked increase in AC activity (Sharma et al., 1975b). This phenomenon has been demonstrated with a number of Gi/Go-linked receptors and multiple AC isoforms (Watts and Neve, 2005). Moreover, this adaptive response has been observed in vivo after chronic administration of opioids or D2 dopamine receptor agonists (Brust et al., 2015).
The importance of the different regulatory properties of individual ACs cannot be overstated. Cells tailor their cAMP production characteristics in part by expressing different cohorts of AC isoforms. This was perhaps most clearly demonstrated by Henry Bourne’s group (Federman et al., 1992) when exogenous expression of AC2 (an isoform stimulated by Gβγ) along with a constitutively active Gαs, converted Gαi-coupled receptor inhibition of cAMP to a stimulation of cAMP production. Therefore, it is imperative to know the contribution of each AC isoform to a cell’s total cAMP production capacity. Defining this is a challenge given that Mg2+ and Mn2+ levels cause sizable changes in AC activity and that isoforms may have varying sensitivities to both Gαs and FSK. AC isoform knockout serves as the most reliable tool for understanding the contribution of individual ACs to a given cell’s signaling repertoire.
Regulation differences aside, mAC isoforms also differ in their basal and maximal activities. For example AC2 has a 10- to 15-fold higher basal activity compared with AC6, although FSK-stimulated activities only differ twofold (Pieroni et al., 1995). Differences in maximal catalytic activity (kcat or turnover number) are difficult to quantify and require purified AC isoforms or other means to accurately quantitate enzyme levels. Studies with purified AC1 and AC2 suggest FSK-stimulated AC1 activity is ∼3.5 times higher than AC2, with a turnover number of ∼15 per second (Taussig et al., 1993b). AC5 has even higher activity with a FSK/PKC-stimulated turnover number of 59 second−1 (Kawabe et al., 1994). This compares favorably with the Gαs- and Gαs /FSK-stimulated 5C1:2C2 turnover numbers of 30 and 100 second−1, respectively (Dessauer et al., 1997). Thus in addition to regulatory control of ACs, relative intrinsic differences in basal and maximal AC activity may also influence their physiologic roles, as discussed below.
sAC is insensitive to G-protein regulation and FSK; it is instead activated by bicarbonate (Chen et al., 2000) and by Ca2+(Jaiswal and Conti, 2003; Litvin et al., 2003). In contrast, mACs are not activated by bicarbonate (Xie et al., 2006). The crystal structure of human sAC indicates that binding of bicarbonate disrupts a salt bridge involving an aspartate residue that is involved in the substrate binding site (Kleinboelting et al., 2014a). Additional major allosteric changes induced by bicarbonate binding affecting the active site have been described in structural studies of a bicarbonate-regulated, cyanobacterial sAC (Kleinboelting et al., 2014b). Given the low affinity of this enzyme for ATP, sAC activity is sensitive to the ATP levels in the cell (Buck et al., 1999; Jaiswal and Conti, 2003). The removal of bicarbonate from the extracellular medium may be used to probe the involvement of sAC in a given physiologic process (Baudouin-Legros et al., 2008; Choi et al., 2012; Hasan et al., 2014).
Both the activities of sAC and mACs are pH dependent (Xie et al., 2006). At pH 7.4, basal activities of sAC and mACs are low and continuously increase until a plateau at pH 8.6–9.0 (Xie et al., 2006). The effect of pH on basal activity of mACs is more prominent than on sAC.
E. (Patho)physiologic Functions of Adenylyl Cyclase Isoforms
As described above, most cells and tissues express multiple AC isoforms. Therefore it has been difficult to tease apart the physiologic role for any given AC isoform. In view of the lack of isoform-specific inhibitors and isoform-specific antibodies, knockout and overexpression studies with mice have driven most of our current understanding (Table 4). Several reviews delve more deeply into the exact roles for different AC isoforms in a variety of systems (Sadana and Dessauer, 2009; Xia and Storm, 2012; Guellich et al., 2014; Nicol and Gaspar, 2014; Rieg and Kohan, 2014; Sabbatini et al., 2014; Lee, 2015). Overall, our knowledge on the role of specific AC isoforms in human pathophysiology is still very limited. For some isoforms such as ACs 1 and 5, quite a lot of information is available, whereas other isoforms, particularly AC4, are rather unexplored. Localization of AC isoforms in “interesting” or “important” locations such as brain and heart seems to play a role in the extent it has been studied.
TABLE 4.
Knockout and transgenic (overexpression) studies with mice: (patho)physiologic functions of mAC isoforms
Because of the lack of selective mAC inhibitors and antibodies, advances in our knowledge on the functions of mACs isoforms largely depended on knockout and overexpression studies in mice. Table updated from Sadana and Dessauer (2009). With few exceptions, the table does not consider associations of ADCY gene polymorphisms with diseases in humans.
| AC Isoform | Availability of |
(Patho)physiologic Functions (Mice and Humans) | |
|---|---|---|---|
| Knockout (mice) | Overexpression (mice) | ||
| AC1 | Yes | Yesa | Mice: Learning, memory, synaptic plasticity, opiate withdrawal and cocaine sensitization, behavioral inhibition, brain development, pain |
| AC2 | No | No | Mice: No data available |
| Humans: AC2 impairment in Lesch-Nyhan syndrome (Kinast et al., 2012)? | |||
| AC3 | Yes | No | Mice: Olfactory and pheromone responses, olfactory dependent and contextual memory, sperm function, diet-induced obesity, maternal behavior, inter-male aggressiveness |
| AC4 | Yesb | Mice: No phenotype | |
| AC5 | Yes | Yesa | Mice: Cardiac contraction, motor coordination, striatum-dependent learning, opiate and ethanol dependency, pain responses, diet-induced obesity, renin secretion, stress responses. |
| Humans: AC5 gain of function in human movement disorders associated with dystonia, myokymia and chorea (Chen et al., 2012c, 2014b) | |||
| AC6 | Yes | Yesa | Mice: cardiac contraction and calcium sensitivity, polycystic kidney disease and renal function including mild Bartter syndrome, sympathetic tone, bone adaptations, pancreatic fluid secretion |
| AC7 | Yes | Yesa | Mice: Ethanol dependency, depression, immune function |
| AC8 | Yes | Yesa | Mice: Learning, memory, synaptic plasticity, opiate withdrawal, mood disorders, anxiety behavior, glucose homeostasis |
| AC9 | Yesc | No | Mice: Immune response in monocytes, possible links to sepsis and cardiac repolarization |
Cardiac or brain directed overexpression.
Tissue-specific expression in kidney collecting duct (Kittikulsuth et al., 2014a).
Unpublished results and International Mouse Phenotyping Consortium (http://www.mousephenotype.org/).
1. Group I (Adenylyl Cyclases 1, 8, and 3).
a. Adenylyl cyclase 1.
The first insights into roles for AC1 came from studies in Drosophila melanogaster and Aplysia californica. The rutabaga gene in flies is most closely related to AC1 and was named for its deficits in learning and memory (Tempel et al., 1983; Levin et al., 1992). Deletion of AC1 in invertebrates or mice leads to a reduction of long-term potentiation (Levin et al., 1992; Wu et al., 1995) and impairments in numerous learning tasks, including spatial learning and learning to avoid a neutral odor (Sadana and Dessauer, 2009; Xia and Storm, 2012; Lee, 2015). Mice with AC1 deletion have deficits in neural wiring during brain development, including enhanced projections and/or branching patterns in the somatosensory system, the visual system, and in the peripheral nervous system (Abdel-Majid et al., 1998; Nicol et al., 2006; Dhande et al., 2012; Nicol and Gaspar, 2014; Suzuki et al., 2015). These developmental patterns are unique to AC1 knockout mice and are consistent with the higher levels of AC1 during development compared with AC8.
AC1 is also required for homeostatic plasticity during activity deprivation in mice (Gong et al., 2007) and sleep plasticity in flies (Donlea et al., 2009). Mice with overexpression of AC1 show improved recognition memory (Wang et al., 2004) but impaired spatial memory in older animals (Garelick et al., 2009) along with deficits in behavioral inhibition (Chen et al., 2015a). Finally, mice with deleted AC1 show decreased susceptibility to proteinuria (Xiao et al., 2011) and reductions in pain (Vadakkan et al., 2006). We are not aware of studies linking polymorphisms in the ADCY1 gene with specific human diseases. An involvement of AC1 in the pathophysiology of alcoholism and Alzheimer’s disease was proposed (Sohma et al., 1999; Yamamoto et al., 2000, 2001), but these studies hinged on the use of commercial antibodies with questionable specificity (Göttle et al., 2009).
b. Adenylyl cyclase 1 and adenylyl cyclase 8 redundancies.
AC1 has many overlapping functions with AC8 that are often revealed in AC1/AC8 double knockouts. These include a reduction in neural responses to ethanol (Maas et al., 2005a,b), cocaine sensitization (DiRocco et al., 2009), and opiate withdrawal behaviors, including lowered morphine-induced hyperlocomotion and loss of cAMP-dependent transcription factor activation in the reward response circuitry of the brain (Li et al., 2006; Zachariou et al., 2008). In addition, double knockouts of AC1 and AC8 show deficits in memory acquisition, memory retention (Zhang et al., 2011), and fear memory (Sindreu et al., 2007; Wieczorek et al., 2010). Administration of FSK to double knockout mice can restore normal long-term memory (Wong et al., 1999), suggesting that stimulation of other AC isoforms is sufficient to overcome loss of AC1 and AC8.
Ca2+-stimulated AC activity and AC1 and AC8 transcripts are also present in sinoatrial nodal cells of the heart. One or both of these AC isoforms appear to be important in sympathetic regulation of pacemaker current in guinea pig, mouse, and rat sinoatrial node cells (Mattick et al., 2007). The exact isoform involved in this process awaits confirmation at the whole animal level using knockouts of AC1 and/or AC8.
c. Adenylyl cyclase 8.
AC8 is expressed at high levels in adult brain, and the AC8 knockout shows significant impairment in short-term plasticity and abnormal anxiety behavior under stress (Schaefer et al., 2000; Wang et al., 2003), in addition to the overlapping functions with AC1 discussed above. Lack of AC8 confers a hyperactive phenotype and an increased risk-taking behavior, similar to certain mood disorders (Razzoli et al., 2010). AC8 is also required for glucose homeostasis and hypothalamic adaptation to high-fat diet (Raoux et al., 2015). ADCY8 gene polymorphisms may be linked to neuropsychiatric diseases, such as bipolar disorder, depression, and alcoholism (de Mooij-van Malsen et al., 2009; Zhang et al., 2010; Procopio et al., 2013; Wolf et al., 2014).
d. Adenylyl cyclase 3.
AC3 is the predominant isoform in the olfactory epithelium and is largely responsible for odorant and pheromone detection in the olfactory bulb (reviewed in Wang et al., 2007). Odorants interact with GPCRs to stimulate AC via activation of the G-protein Gαolf. All components of this signaling system, including AC3, are located within olfactory cilia (Wang and Storm, 2003). Deletion of AC3 leads to major defects in odorant-induced signaling, olfactory-dependent learning, olfaction-based behavioral tests, olfactory axonal guidance processes, and mechanical force of airflow across olfactory cilia (Wong et al., 2000; Col et al., 2007; Chen et al., 2012b). Mice lacking AC3 are unable to detect mouse urine or pheromones and show a reduction in maternal behavior, inter-male aggressiveness, male sexual behavior, spermatozoa function, and male fertility (Livera et al., 2005; Wang et al., 2006; Wang and Storm, 2011). AC3 is also expressed in oocytes (Horner et al., 2003), and AC3 knockout mice show premature oocyte maturation (Horner et al., 2003).
Although the highest levels of expression of AC3 are present in olfactory tissue, AC3 is also found in the brain, hypothalamus, and at much lower levels in other tissues. Loss of AC3 in mice is associated with impaired short-term memory for novel objects and failure to exhibit extinction of contextual fear conditioning (Wang et al., 2011b), altered sleep patterns, and depression-like phenotypes (Chen et al., 2016). Key components of the olfactory signal transduction machinery (olfactory receptors, AC3, and Gαolf) are expressed in the distal nephron (Pluznick et al., 2009). However, there is no effect on renal sodium or water excretion after selective deletion of AC3 in collecting ducts, suggesting compensation by other ACs (Kittikulsuth et al., 2014b). ADCY3 gene polymorphisms have been associated with obesity (Nordman et al., 2008; Wang et al., 2009, 2010; Wen et al., 2012; Stergiakouli et al., 2014; Warrington et al., 2015), depression (Wray et al., 2012), and inflammatory bowel disease (Hulur et al., 2015).
1. Group II (Adenylyl Cyclases 2, 4, and 7).
a. Adenylyl cyclase 2.
Reports of knockout phenotypes for AC2 are lacking. AC2 selectively couples to prostanoid EP2 receptors in smooth muscle cells, pointing to a potential role in regulating smooth muscle tone in response to prostaglandins (Bogard et al., 2012). AC2 and AC4 are the predominantly expressed AC isoforms in human airway smooth muscle cells, but AC2 expression is lacking in mouse bronchial smooth muscle cells, making confirmation of its function difficult (Bogard et al., 2011, 2012). Development of AC2-selective inhibitors may overcome the difficulties to elucidate the (patho)physiologic functions of AC2 (Pinto et al., 2008; Erdorf et al., 2011; Kinast et al., 2012; Conley et al., 2013). ADCY2 polymorphisms may be related to neuropsychiatric (Mühleisen et al., 2014; Suarez-Rama et al., 2015) and pulmonary diseases (Hardin et al., 2012; Panasevich et al., 2013). Pharmacological studies point to a role of AC2 in the pathogenesis of Lesch-Nyhan disease, a neuropsychiatric disorder with severe self-injurious behavior (Kinast et al., 2012).
b. Adenylyl cyclase 4.
A tissue-specific knockout of AC4 has been generated for loss of expression in kidney collecting duct principal cells. AC4 does not appear to play a role in water and sodium handling in kidney (Kittikulsuth et al., 2014a). Phenotypes from other tissues have not yet been reported. We are not aware of studies linking ADCY4 gene polymorphisms to specific human diseases.
c. Adenylyl cyclase 7.
AC7 is a major contributor to cAMP synthesis in B and T lymphocytes. Mice with AC7 deletion produce fewer leukocytes and have a high mortality upon bacterial infections, producing less antigen-specific antibodies and displaying prolonged inflammation when challenged with zymosan (Duan et al., 2010; Jiang et al., 2013). Downregulation of AC7 is observed in monocytes of young men and is associated with significantly increased tumor necrosis factor-α release (Risoe et al., 2015).
AC7 also may play a role in pathways contributing to alcohol consumption. Starting from the early observation that ethanol at clinically relevant concentrations increases AC activity in mouse striatal membranes (Hoffman and Tabakoff, 1982), subsequent studies revealed that AC7 is particularly sensitive to stimulation by ethanol and that the effects of ethanol are mediated via the cytosolic domains (Yoshimura and Tabakoff, 1999; Yoshimura et al., 2006; Kou and Yoshimura, 2007; Dokphrom et al., 2011). Female mice with reduced AC7 levels show increased alcohol consumption, whereas increased AC7 levels lead to elevated ethanol-induced adrenocorticotropin and corticosterone levels (Pronko et al., 2010; Desrivieres et al., 2011). In humans, ACDY7 polymorphisms have been linked to alcoholism (Hellevuo et al., 1997) and autoimmune diseases (Li et al., 2015). Human polymorphisms of ACDY7 are associated with depression, supported by depressive phenotypes in AC7 knockout mice (Hines et al., 2006; Joeyen-Waldorf et al., 2012).
3. Group III (Adenylyl Cyclases 5 and 6).
a. Adenylyl cyclase 5.
AC5 has received significant attention for its roles in heart and brain. Loss of AC5 results in improved basal left ventricular function but decreased isoproterenol-stimulated left ventricular ejection fraction, elimination of parasympathetic control of cAMP levels, and attenuation of baroreflexes that maintain blood pressure (Okumura et al., 2003a; Tang et al., 2006). Deletion of AC5 protects the heart against chronic βAR stimulation and chronic pressure overload by attenuating the decline in cardiac function and increased apoptosis (Okumura et al., 2003b, 2007). AC5 disruption is also protective against age-related cardiac myopathy and gives rise to an increased life span compared with wild-type animals (Yan et al., 2007). AC5 knockout mice show decreased body weight on standard and high-fat diet, with improved glucose tolerance and increased insulin sensitivity (Ho et al., 2015). However, loss of AC5 does not rescue the cardiomyopathy associated with overexpression of Gαq (Timofeyev et al., 2010). AC5 gain-of-function mutations may be associated with heart failure (Chen et al., 2012c).
In contrast to the possible protection against cardiac and metabolic diseases, AC5 deletion has detrimental consequences for central nervous system (CNS) function. Specifically, AC5 knockout results in poor stress-coping mechanisms, presumably due to high AC5 expression levels in the reward and stress-related circuits of the dorsal striatum and nucleus accumbens (Kim and Han, 2009). Striatum-dependent learning and corticostriatal plasticity is also impaired upon loss of AC5 (Kheirbek et al., 2009). Most strikingly, AC5 knockout mice show a Parkinson-related motor disorder with impaired coordination and bradykinesia (Iwamoto et al., 2003). This phenotype can be partially rescued by dopamine D1- and D2-receptor stimulation (Iwamoto et al., 2003). Dopamine D1 receptor and D2 receptor signaling to AC is abolished in striatum upon knockout of AC5; the loss of D2 receptor function is consistent with a loss of motor activity responses to the antipsychotic drugs haloperidol and sulpiride (Lee et al., 2002). In addition, the dyskinesia induced by treatment with dopamine precursors, often observed in Parkinson’s patients, is greatly reduced upon knockout of AC5 (Park et al., 2014). Finally, loss of AC5 leads to increased ethanol consumption and reduced sensitivity to high-dose ethanol (Kim et al., 2011a, 2012).
Initial reports suggested a role for AC5 in renin secretion (Ortiz-Capisano et al., 2007), but more recent studies show that these effects are largely due to AC6, with AC5 playing only a minor role in isoproterenol-stimulated plasma renin concentration (Aldehni et al., 2011).
Numerous studies have linked ADCY5 gene polymorphisms to altered glucose metabolism, diabetes, and obesity (Barker et al., 2011; Rees et al., 2011; Wagner et al., 2011; Windholz et al., 2011; Holstein et al., 2013; Ng et al., 2013; Hodson et al., 2014; Knigge et al., 2015). ADCY5 gene polymorphisms have also been linked to various neuropsychiatric diseases (Chen et al., 2012c; Procopio et al., 2013). Most notably, two nonconserved point mutations in ADCY5 (R418W localized in the region between TM1 and C1a and A726T localized in the C1b region) have been associated with familial dyskinesia with facial myokymia (FDFM) (Chen et al., 2012c, 2014b). FDFM is a rare autosomal-dominant disease, characterized by paroxysmal and involuntary dystonic and choreiform movements of limb and facial muscles (Chen et al., 2012c, 2014b). Expression of the two AC5 mutants in intact HEK293 cells massively increased cAMP accumulation compared with wild-type AC5 upon stimulation with the βAR agonist isoproterenol (Chen et al., 2014b). These data indicate a gain-of-function in the nonconservative R418W and A726T mutants at the molecular level and are consistent with a gain-of-function on the extrapyramidal movement level (Chen et al., 2012c, 2014b; Chang et al., 2016). The AC regions in which the mutations are localized have not yet been resolved crystallographically. Other nonconservative mutations in the ADCY5 gene (R418G and R418Q) are associated with dystonia, myoclonus, and choreoathetosis, too (Chen et al., 2015b; Chang et al., 2016). In line with these data, the R418W mutation may cause benign hereditary chorea (Mencacci et al., 2015). The R418W and A726T mutants may also be associated with chronic heart failure (Chen et al., 2012c). Mutations of the ADCY5 gene resulting in RNA instability can cause chorea and dystonia as well (Carapito et al., 2015). Finally, the nonconservative M1029K mutation in the ADCY5 gene, localized in the C2a domain, has been linked to dystonia, hypotonia, and chorea (Chen et al., 2015c). Taken together, these data provide convincing evidence for an important role of AC5 in regulating the extrapyramidal motor system. It should be noted though, that presently, the characterization of the biochemical and pharmacological properties of disease-causing AC5 mutants is still very incomplete. It also remains to be determined whether loss-of-function mutations in the ADCY5 gene cause Parkinson’s disease in humans.
b. Adenylyl cyclase 6.
AC6 is widely expressed with documented roles in heart, kidney, pancreas, bone, and brain. In heart, deletion of AC6 results in no change in basal cardiac cAMP levels but greatly reduced βAR-stimulated cAMP levels that may be due to a reduction in AC5 levels, in addition to loss of AC6 (Tang et al., 2008). AC6 knockout animals show greatly reduced βAR-stimulated left ventricular contractile function and reduced calcium transporter SERCA2a calcium affinity and calcium transients. Levels of AC6, but not AC5, appear to limit βAR signaling in heart (Gao et al., 1998; Tepe et al., 1999). Overexpression of AC6 in some heart failure models improves markers of heart failure, increasing cardiac contractility, cardiac responsiveness, and attenuating deleterious left ventricular remodeling (Roth et al., 1999, 2002; Lai et al., 2000, 2004). However, no improvement in heart function was observed in mouse chronic pressure overload models upon AC6 overexpression (Guellich et al., 2010). The differences between AC5 and AC6 roles in heart may be due to distinct compartmentalization of the two enzymes and the influence of local PDE pools in ventricular myocytes, with AC5 localized largely in the T-tubular region, whereas AC6 is found outside the T-tubules (Timofeyev et al., 2013).
In kidney, AC6 is localized in the tubules, collecting ducts, and juxtaglomerular cells, contributing to water homeostasis and renin secretion (Chien et al., 2010; Aldehni et al., 2011; Rieg and Kohan, 2014). Mice lacking AC6 have increased urine output, decreased urine osmolality, reduced responsiveness to arginine-vasopressin (AVP) receptor V2, and a mild Bartter syndrome-like phenotype (Rieg et al., 2010, 2013). Mice with a specific knockout of AC6 in collecting ducts also show a urinary concentrating defect and lack AVP‐stimulated epithelial sodium channel activity (Roos et al., 2012, 2013). The phenotypes associated with loss of AC6 are similar to that of nephrogenic diabetes insipidus and are consistent with a loss of vasopressin-induced cAMP in the inner medullary collecting ducts of the kidney. AVP-cAMP signaling within primary cilium directly regulates cyst growth in autosomal dominant polycystic kidney disease, the most common genetic cause of kidney failure (Rieg and Kohan, 2014). In a mouse model of polycystic kidney disease, loss of AC6 markedly reduced cyst formation and renal injury (Rees et al., 2014), consistent with AC6 association with an AKAP-scaffolded complex containing polycystin-2 in primary cilium (Choi et al., 2011). The localization of AC6 in osteocyte cilia likely contributes to its role in loading-induced bone adaptation (Lee et al., 2014). Other functions of AC6 include the regulation of basal sympathetic nerve discharge or sympathetic tone within the brain stem (Chien et al., 2013) and regulation of cAMP-dependent pancreatic amylase and fluid secretion (Sabbatini et al., 2013). In humans, the ADCY6 S674 variant results in a hyperfunctional AC6 protein (Gros et al., 2007), but follow up studies are required. ADCY6 gene polymorphisms have been linked to altered adhesion of red sickle cells (Eyler et al., 2008), but despite the importance of AC6 for heart function, no association of ADCY6 gene polymorphisms with cardiovascular disease has been found in the study by Ikoma et al. (2003).
4. Group IV (Adenylyl Cyclase 9).
Reports for AC9 knockout phenotypes are lacking. However, microRNAs targeting AC9 (miR-142-3p, miR-181a, and miR-181b) point to a decrease in AC9 expression in sepsis (Risoe et al., 2011) and potential roles in regulating cell proliferation in cervical cancer cells and differentiation of promyelocytic leukemia (Yang et al., 2014; Zhuang et al., 2014). Several reports suggest roles for AC9 in immune function. miR-142-3p restricts cAMP production in regulatory T cells (Huang et al., 2009), whereas knockdown or overexpression of AC9 in human neutrophils alters chemotaxis (Liu et al., 2010). In newly diagnosed patients with acute myelogenous leukemia and in those with a disease relapse, AC9 expression is lower than in cases of remission (Zhuang et al., 2014), suggesting that AC9 could be used as biomarker for diagnosis and clinical course of certain leukemia forms. In cervical carcinoma cells, miR-181b downregulates AC9 and promotes proliferation (Yang et al., 2014), pointing to a more general role of AC9 in cancer biology. AC9 is the functionally predominant AC isoform in neutrophils and monocytes (Mahadeo et al., 2007; Brunskole Hummel et al., 2013; Werner et al., 2014, 2016). Finally, association of AC9 with Yotiao and certain potassium channels in heart may point to a role in cardiac repolarization and certain forms of long-QT syndrome (Li et al., 2012). ADCY9 gene polymorphisms are linked to different diseases including asthma (Small et al., 2003; Kim et al., 2011b), pancreatic cancer (Lee and Gyu Song, 2015), mood disorders (Toyota et al., 2002), body weight (Berndt et al., 2013), and susceptibility to malaria (Manjurano et al., 2012; Maiga et al., 2013). ADCY9 gene polymorphisms determine the efficacy of dalcetrapib, increasing high-density lipoprotein, on atherosclerosis (Tardif et al., 2015).
5. Soluble Adenylyl Cyclase (Adenylyl Cyclase 10).
Two mouse knockout lines for this gene have been generated [deletion of the first exon of the sAC gene, which prevents the expression of the C1 domain (C1KO)] (Esposito et al., 2004) and deletion of exons 12–14 that code for the C2 domain (C2KO) (Chen et al., 2013a). Both lines lead to similar phenotypes in the testis, including a complete loss of bicarbonate-sensitive AC activity in the testis and spermatozoa and undetectable cAMP levels in isolated spermatozoa, giving rise to infertility because of defective sperm motility (Esposito et al., 2004; Xie et al., 2006; Chen et al., 2013a). Additional phenotypes of these mouse lines have been reviewed elsewhere (Schmid et al., 2014; Levin and Buck, 2015). Reed et al. (2002) identified 18 base substitutions in the human sAC gene in a population of patients with absorptive hypercalciuria and increased propensity to kidney stones. Additional ADCY10 gene polymorphisms suggest association with oligoazoospermia, asthenozoospermia, osteporosis, and pancreatic cancer (Herrinton et al., 2011).
IV. Pharmacological Modulation of Adenylyl Cyclases
A. General Aspects
Given the highly differential expression, regulation and function of mAC isoforms discussed in the preceding text, ACs, in principle, constitute an excellent drug target. However, in marked contrast to the progress made in these basic science fields, to this end, translation of this knowledge into clinically useful drugs is still very limited. In contrast to ACs, the development of drugs targeting sGC and pGCs is much more advanced (Friebe et al., 2015). Specifically, the exploration of allosteric regulatory sites in sGC resulted in the development of potent NO-independent sGC activators (cinaciguat being the prototype) and NO-dependent sGC stimulators (riociguat being the prototype) with many clinical applications (Stasch et al., 2011, 2015).
Like GCs, ACs can be targeted at the catalytic site and allosteric regulatory sites, i.e., the FSK binding site in case of mACs and the bicarbonate binding site in case of sAC, and other sites distinct from these locations (Seifert and Beste, 2012). In case of mACs, it should also be possible to target the nonconserved transmembrane domains, but this field of pharmacology is still in its infancy (Beltz et al., 2016). The long-term goal in the field of AC pharmacology is to obtain stimulatory and inhibitory compounds with high affinity and selectivity for a given AC isoform. So far, this goal has not been met.
Bacterial toxins constitute most valuable tools for the analysis of signal transduction processes in general and in many cases constitute pathogenic factors for disease development (Moss and Vaughan, 1989; Seifert and Dove, 2012, 2013; Simon et al., 2014; Seifert et al., 2015). Several bacterial toxins, specifically CyaA from Bordetella pertussis, edema factor from Bacillus anthracis and ExoY from Pseudomonas aeruginosa possess nucleotidyl cyclase activity and flood target cells with cAMP (and other cNMPs) (Seifert and Dove, 2012, 2013; Seifert et al., 2015). There is no known bacterial toxin that covalently modifies mACs. However, cholera toxin from Vibrio cholera ADP-ribosylates Gαs and, thereby, blocks its GTPase activity (Cassel and Selinger, 1977; Moss and Vaughan, 1979). As a result, Gαs is persistently activated and strongly activates mACs, resulting in massive cAMP production in enterocytes and, ultimately, profuse secretion with subsequent dehydration, the major clinical symptoms of cholera. Because ADP-ribosylation of Gαs is irreversible, symptomatic fluid and electrolyte substitution is crucial for successful recovery of patients suffering from cholera (Moss and Vaughan, 1989). Pertussis toxin from Bordetella pertussis ADP-ribosylates Gαi and, hence, uncouples GPCRs from inhibition of AC (Katada et al., 1982, 1984).
Currently, there is no drug on the market that exerts its main therapeutic effects via inhibition of mACs, although many patents have been filed (Seifert et al., 2012). However, as an off-target effect, certain antiviral compounds such as foscarnet and acyclic nucleoside phosphonates may inhibit mACs under clinical conditions (Shoshani et al., 1999; Kudlacek et al., 2001). It has been proposed that lithium and carbamazepine, mood stabilizers used for the treatment of bipolar disorder (Harwood and Agam, 2003), may act via preferential inhibition of AC5 (Mann et al., 2009). However, these data have not yet been confirmed independently, and mood stabilizers, most notably lithium, are known to act via multiple mechanisms (Harwood and Agam, 2003).
One important parameter when evaluating reports of AC-specific modulators is the system that was used. As noted in section I.C.3, a variety of methodologies are routinely used to assess AC activity, and two common approaches use either cellular membranes or intact cell preparations from cells overexpressing individual AC isoforms. The assays are biologically distinct (see section I.C.3) and have advantages and disadvantages that are important to consider when evaluating published reports. Studies with cell membranes can provide important mechanistic insight (i.e., direct versus indirect, competitive versus noncompetitive) and AC isoform specificity information (particularly when Sf9 membranes expressing a given AC isoform); however, the assays are generally of low throughput (Brand et al., 2013). Intact cell studies are well suited for high-throughput studies but provide only limited information regarding modulator mechanism. For example, NB001 is reported to be a selective AC1 inhibitor; however, it is inactive in cell membrane preparations expressing AC1 (Brand et al., 2013). The use of intact cells for inhibitor studies also requires selective modulation of recombinant AC isoform of interest. For example, cells expressing AC1 and AC8 show robust cAMP responses to the Ca2+ ionophore A23187, and cells expressing AC2 are robustly activated by the phorbol ester PMA (Cumbay and Watts 2001). Although no selective activators of other AC isoform are available, overexpression allows for AC5 and others to be robustly activated by concentrations of FSK, showing little activity in wild-type cells (Cumbay and Watts 2001). Advantages of intact cell studies include a more native cellular environment, and presumably only cell permeable modulators will be active. However, counterscreening against other AC isoforms and studies examining the potential role of G proteins (e.g., pertussis toxin treatment and sequestering Gβγ subunits) are important considerations for intact cell studies. The results of intact cell studies can further be strengthened and supported by additional studies using cell membranes (Conley et al., 2013). Ultimately, one would like confirmation that pharmacological inhibition and genetic inhibition give rise to similar results. The considerations noted above should assist investigators when evaluating the strengths and weaknesses of reports examining AC isoform specific modulators.
B. Membrane Adenylyl Cyclase Activators
1. General Aspects.
The FSK binding site in mACs constitutes the classic target for stimulatory drugs. Initially, FSK was characterized in membrane preparations containing a mixture of various mAC isoforms (Daly, 1984). ACs 1–8, but not AC9, are activated by FSK (Premont et al., 1996; Yan et al., 1998; Onda et al., 2001; Cumbay and Watts, 2004) (Table 3). There is marked synergism between Gαs and FSK with respect to increasing Vmax of catalysis, characterized in detail with the 5C1:2C2 system (Sunahara et al., 1997; Mou et al., 2005). For identification of small molecule AC activators, low basal catalytic activity is acceptable.
The lack of FSK-activation of AC9 has been used to identify this isoform as functionally dominant AC in human myeloid cells (Mahadeo et al., 2007). Specifically, human monocytes and neutrophils express AC activity that is effectively stimulated by the β2AR and histamine H2 receptor, whereas FSK is almost ineffective at increasing cAMP levels in these cell types (Brunskole Hummel et al., 2013; Werner et al., 2014, 2016). At the mRNA level, several AC isoforms including AC9 are expressed in myeloid cells (Mahadeo et al., 2007; Brunskole Hummel et al., 2013; Werner et al., 2014, 2016). The molecular basis for the FSK resistance of AC9 is attributable to the presence of a tyrosine at position 1082, corresponding to a leucine in other AC isoforms (Yan et al., 1998). FSK can also be coupled to agarose and used as an affinity ligand for purification of mACs (Pfeuffer et al., 1985, 1989; Pfeuffer, 1991).
Despite intense efforts in the AC research community, no endogenous ligand for the FSK binding site has been identified so far (Laurenza et al., 1989; Seifert et al., 2012). Reports on the occurrence of an endogenous FSK-like molecule have not yet been confirmed (Putnam et al., 2007). Thus, the physiologic relevance of the FSK binding site is still elusive.
2. Structure-activity Relationships of Diterpenes.
A limited set of systematic structure-activity relationship studies for FSK was performed at recombinant ACs 1, 2, and 5, representatives of the three major AC families, and the purified catalytic mAC subunits 5C1:2C2 (Table 5). Figure 12 shows the structures of the compounds examined. In the presence of Mn2+, which is very often used in AC assays because it confers higher catalytic activity to these systems (Gille et al., 2004; Mou et al., 2005), FSK is the most potent and most efficacious diterpene at all ACs studied (Pinto et al., 2008). AC1 is the most sensitive isoform, but the difference in potency to the least sensitive AC isoform, AC2, is just 10-fold. In general, modifications such as the introduction of a hydrophilic N-methylpiperaziono-γ-butyryloxy group, the removal of the 7-acetyl group or 7,6-isomerization of the acetyl group result in losses in potency and efficacy. Loss of the 9-hydroxyl group in FSK results in decreases in potency and efficacy, particularly for AC2. The removal of the 1-hydroxyl group in FSK is detrimental for AC activation, pointing to the crucial importance of a hydrogen bond between the 1-hydroxyl group and Val506 for activation (Pinto et al., 2008, 2009). Some degree of AC isoform selectivity of FSK and FSK derivatives was also reported in other studies (Sutkowski et al., 1994; Onda et al., 2001), but all available studies are incomplete to some extent, i.e., not all AC isoforms were studied, and complete concentration-response curves were not routinely generated.
TABLE 5.
Interaction of prototypical diterpenes with ACs 1, 2 and 5 and 5C1:2C2
AC activities were determined as described in the presence of Mn2+. Reaction mixtures contained diterpenes at concentrations from 100 nM–300 μM. Data were analyzed by nonlinear regression to determine the EC50 values. For recombinant holo-ACs, the efficacy for each analog was determined by dividing the maximal stimulation obtained for the analog by the maximum stimulation obtained by treatment with 300 μM FSK expressed in percent. In case of 5C1:2C2, the efficacy for each analog was determined by dividing the maximal stimulation obtained for the analog by the maximum stimulation obtained by treatment with 100 μM FSK expressed in percent. Data for holo-ACs were adapted from Erdorf et al. (2011), and data for 5C1:2C2 were taken from Pinto et al. (2009).
| Diterpene | AC1 |
AC2 |
AC5 |
5C1:2C2 |
||||
|---|---|---|---|---|---|---|---|---|
| EC50 | Efficacy | EC50 | Efficacy | EC50 | Efficacy | EC50 | Efficacy | |
| μM | % | μM | % | μM | % | μM | % | |
| FSK | 3.3 | 100 | 38 | 100 | 18 | 100 | 7.4 | 100 |
| DMB-FSK | 2.8 | 65 | 50 | 46 | 11 | 81 | 28 | 41 |
| 6A7DA-FSK | 3.0 | 110 | 17 | 70 | 13 | 70 | 2.6 | 83 |
| 7DA-FSK | 9.3 | 109 | 81 | 62 | 38 | 124 | 3.1 | 86 |
| 9d-FSK | 17 | 98 | 127 | 30 | 61 | 108 | 20 | 79 |
| 1d-FSK | N.D. | 3 | N.D. | 0.4 | N.D. | 0.7 | ine. | ine. |
| BODIPY-FSK | 1.2 | 73 | 0.2 | −19 | 2.7 | 69 | N.D. | N.D. |
N.D., not determined. Ine., ineffective. Structures of diterpenes are shown in Fig. 12. FSK, forskolin; DMB, 7-deacetyl-7-(N-methylpiperazino-butyryloxy); 6A7DA, 6-acetyl-7-deacetyl; 7DA, 7-deacetyl; 9d, 9-deoxy; 1d, 1-deoxy; BODIPY, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene.
Fig. 12.

Structures of diterpenes. Pharmacological data for selected diterpenes are listed in Table 5. FSK, forskolin; DMB, 7-deacetyl-7-(N-methylpiperazino-butyryloxy); 6A7DA, 6-acetyl-7-deacetyl; 7DA, 7-deacetyl; 9d, 9-deoxy; 1d, 1-deoxy; BODIPY, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene. Diterpenes are highly lipophilic. To increase water solubility, hydrophilc substituents were introduced at the 6′-position (colforsin daropate) and the 7′-position (DMB-FSK) of the diterpene ring. In Japan, colforsin daropate is used clinically for treatment of heart failure. BODIPY-FSK is a fluorescent FSK derivative that shows inverse agonist activity at AC2 and partial agonist activity at ACs 1 and 5.
The poor water solubility constitutes a serious problem in the pharmacological analysis of diterpenes, requiring dimethyl sulfoxide concentrations of up to 3% (w/v) to fully solubilize compounds (Pinto et al., 2008, 2009). Although such high solvent concentrations may be compatible with cell-free AC activity assays, experiments with intact cells generally do not tolerate dimethylsulfoxide concentrations higher than 1% (Brunskole Hummel et al., 2013). The high hydrophobicity of FSK also delays its dissociation from ACs and potentially prolongs drug action (Pinto et al., 2009). One strategy to circumvent these problems is the introduction of water-soluble FSK derivatives (Laurenza et al., 1987; Onda et al., 2001) (Fig. 12).
Not surprisingly, potencies and efficacies of ligands differ to some extent between 5C1:2C2 on one hand and ACs 1, 2, and 5 on the other, but the critical importance of the 1-hydroxyl group for activation was retained (Pinto et al., 2009). Interestingly, fluorescence spectroscopy studies with MANT-nucleotides as sensors revealed that FSK derivatives without the 1-hydroxyl group can still bind to AC (Pinto et al., 2009), suggesting that formation of the hydrogen bond between the 1-hydroxyl group of FSK and Val506 is important for the actual activation step of the enzyme. 1-Deoxy-FSK prevents activation of 5C1:2C2 by FSK, supporting the view that the former compound still binds to the FSK binding site.
The most notable selectivity among FSK derivatives for AC isoforms was observed for BODIPY-FSK, characterized by a large bulky fluorescent substituent at the 7′-position of the diterpene ring. Although BODIPY-FSK is a partial activator of AC1 and AC5, BODIPY-FSK decreases the basal activity of AC2 (Pinto et al., 2008). AC2 is characterized by a high degree of ligand-independent constitutive activity (Pieroni et al., 1995; Pinto et al., 2008). By analogy to the situation described for GPCRs (Seifert and Wenzel-Seifert, 2002), BODIPY-FSK could be considered as inverse agonist at AC2. When Mn2+ is substituted by Mg2+, potencies and efficacies change, the most remarkable difference being a profound increase in inverse agonistic efficacy of BODIPY-FSK at AC2 (Erdorf et al., 2011). These data also raise the question about the possible physiologic relevance of the high constitutive activity of AC2. The selectivity of BODIPY-FSK for AC2 inhibition, in conjunction with RT-PCR studies and analysis of MANT-nucleotides, was used to define AC2 as the functionally predominant AC isoform in rat B103 neuroblastoma cells (Kinast et al., 2012). Intriguingly, upon loss of hypoxanthine phosphoribosyl transferase, the causative enzyme defect of Lesch-Nyhan disease (Fu et al., 2014), AC2 function is substantially impaired (Kinast et al., 2012).
3. Off-target Effects of Diterpenes.
Thus, although the selectivity of currently available FSK derivatives for defined AC isoforms is far from satisfying, proof of principle has been provided that ligand selectivity could be obtained, particularly by the introduction of large substituents. However, it should be kept in mind that FSK does not only bind to mACs but also to other important pharmacological targets such as glucose transporters, P-glycoprotein multidrug resistance transporters, ion channels, and nuclear receptors such as pregnane X receptor (Joost et al., 1988; Morris et al., 1991; Ding and Staudinger, 2005; Pavlikova et al., 2010; Kinast et al., 2012; Angel-Chavez et al., 2015). Such interactions of FSK are of concern for clinical application, specifically for long-term systemic treatment in conjunction with other drugs, increasing the risk of drug/drug interactions. Large dissociations between Kd values for [3H]FSK binding and EC50 values of FSK for AC activation support the notion that multiple FSK targets exist (Seamon et al., 1984). Limited structure-activity relationship studies have been undertaken to dissociate interaction of FSK analogs with ACs and off-target sites (Joost et al., 1988; Pfeuffer and Pfeuffer, 1989; Appel et al., 1992). When using BODIPY-FSK as a tool, nonspecific effects of the compound should be kept in mind as well (Kinast et al., 2012).
4. Clinical Applications of Diterpenes.
A water-soluble FSK derivative, colforsin daropate hydrochloride, also referred to as NKH477 (Onda et al., 2001), is used for treatment of acute heart failure in Japan (Hosono, 1999) (Fig. 12). Colforsin exhibits moderate selectivity for AC5 relative to ACs 2 and 3, but its complete pharmacological profile at all mAC isoforms is not known (Toya et al., 1998; Onda et al., 2001). However, the primary goal of colforsin development was not as an AC5-selective inhibitor but rather to obtain a compound with high water solubility to prevent unwanted effects in the CNS. Colforsin daropate hydrochloride may also be useful for the treatment of septic syndrome and subarachnoid hemorrhage (Oishi et al., 2012; Suzuki et al., 2012). The local use of FSK for treatment of glaucoma is being explored, too (Majeed et al., 2015). Moreover, the dermatological application of FSK to protect the skin against ultraviolet light and induce persistent tanning is under investigation (Spry et al., 2009; Scott et al., 2012; Sikora et al., 2015). Furthermore, FSK derivatives are explored as anticancer compounds (Ponnam et al., 2014). Lastly, Coleus forskohlii extracts with varying and undefined FSK content are widely and uncritically used as dietary supplement for weight reduction (Rios-Hoyo and Gutierrez-Salmean, 2016). The available clinical evidence (Godard et al., 2005) is insufficient to recommend the use of FSK for treatment of obesity.
C. Adenylyl Cyclase Inhibitors
1. General Aspects.
Most inhibitory compounds bind to the catalytic site. Such compounds may act either as noncompetitive, activity-dependent inhibitors, so-called P-site inhibitors, or as competitive inhibitors that are actually also activity-dependent (Seifert et al., 2012). Figure 13 shows representative competitive mAC inhibitors [(M)ANT- and TNP-nucleotides] and noncompetitive P-site inhibitors. For more structures and detailed descriptions of individual inhibitor profiles, the reader is referred to Seifert et al. (2012). So far, there is no known selective inhibitor for a given mAC isoform with clinical relevance. In contrast to the development of AC activators (see section B.1), the development of AC inhibitors requires a reasonably high catalytic activity to begin with, because otherwise inhibition cannot be detected analytically. Therefore, all typical AC inhibitor studies are conducted under optimal stimulatory AC assay conditions. Should an AC isoform-selective inhibitor be identified, it would have to be verified that the inhibitor and the corresponding ADCY gene knockout show very similar effects. Conceptually, this is an important control to exclude off-target effects of small molecule inhibitors.
Fig. 13.

Structures of representative inhibitors targeting the catalytic site of mACs. Pharmacological data for competitive inhibitors are listed in Table 6, and pharmacological data for noncompetitive P-site inhibitors are listed in Table 7. The (M)ANT group can spontaneously isomerize between the 2′- and 3′-ribosyl position, provided that there is only substituent and a free hydroxyl group. The different bases (A, adenine; G, guanine; I, hypoxanthine; X, xanthine; C, cytosine; U, uracil) constitute substituent Y at the ribosyl moiety. The different phosphate chains (mono-, di-, and triphosphate and hydrolysis-resistant phosphorothioates or β,γ-imidophosphates) constitute substituent X at the ribosyl moiety. (M)ANT- and TNP-nucleotides do not cross the plasma membrane and can only be used in cell-free systems or in electrophysiological studies in which compounds are injected into the cell via the patch pipette. Experimental problems with the membrane-permeable P-site inhibitors are the relatively low potency, insufficient selectivity and high lipophilicity, requiring the use of high concentrations of organic solvents.
The catalytic site possesses three pharmacophoric sites, i.e., the polyphosphate-binding site, the base-binding domain, and a hydrophobic pocket between the latter two sites (Mou et al., 2005, 2006; Hübner et al., 2011). In terms of inhibitor affinity, the hydrophobic pocket exhibits the largest impact on inhibitor affinity, followed by the phosphate-binding domain and the base-binding domain (Seifert et al., 2012). Accordingly, the catalytic site of ACs exhibits a broad base specificity and accepts numerous purine and pyrimidine bases, xanthine being an exception for extremely poor binding (Mou et al., 2006).
2. P-site Inhibitors.
The first identified mAC inhibitors were P-site inhibitors; adenosine analogs that are typically noncompetitive or uncompetitive with respect to substrate ATP (Londos and Wolff, 1977). These compounds were characterized in the classic cell-free AC assay with [α-32]ATP as substrate. Representative P-site inhibitors, ordered by potency, include: 2′,5′-dideoxyadenosine-3′-tetraphosphate > 2′,5′-dideoxy-3′-ATP > 2′,5′-dideoxy-3′-ADP > 2′,5′-dideoxy-3′-AMP > 2′-deoxy-3′-AMP > 3′-AMP > 2′-deoxyadenosine > adenosine (Johnson et al., 1989; Desaubry et al., 1996; Dessauer et al., 1999; Seifert et al., 2012). P-site inhibitors containing a phosphate residue are too hydrophilic to cross the plasma membrane and cannot be used in intact-cell studies. The requirement for an intact adenine ring reflects the fact that P-site inhibitors bind within the same enzyme pocket as substrate ATP (Tesmer et al., 1997; Tesmer et al., 2000). These inhibitors have revealed important insights into the mechanism of AC catalytic activity. Kinetic analysis of the forward and reverse (i.e., formation of ATP from cAMP and PPi) reactions of mAC show that the cAMP product is preferentially released from the enzyme before PPi (Dessauer and Gilman, 1997). In the basal state or under low levels of activity, the slow step of the reaction is dictated primarily by the cyclization of ATP to cAMP. But upon activation of the enzyme, the catalytic rate speeds up and the release of PPi becomes the slow step in the reaction. P-site inhibitors bind to the catalytic pocket of AC and mimic a product-bound transition state of the enzyme that often includes bound PPi. In the basal state, the catalytic site is generally open. Activators of the enzyme (e.g., Gαs and FSK) increase the affinity between the C1 and C2 domains and result in a more closed conformation (Whisnant et al., 1996; Yan et al., 1996; Tesmer and Sprang, 1998). Structures of AC with bound P-site inhibitors exhibit an even further closed conformation. This explains the uncompetitive nature of their inhibition; they require substrate binding and catalysis to form an enzyme-PPi-inhibitor dead-end complex. Moreover, P-site inhibitors are far more potent on stimulated forms of mAC, where release of PPi is the slow step and the concentration of steady-state enzyme-PPi complex increases (Dessauer and Gilman, 1997).
P-site inhibitors that have 3′-polyphosphate moieties, such as 2′,5′-dideoxy-3′-ATP, bind in the absence of PPi but still display noncompetitive kinetics (Desaubry et al., 1996; Desaubry and Johnson, 1998; Tesmer et al., 2000). The 3′-polyphosphates bind in the pyrophosphate pocket of the catalytic site and serve to increase the potency of these molecules by 100–1,000-fold. Because these inhibitors do not require bound PPi, one might expect a competitive pattern of inhibition, as is observed for ATP analogs (Gille and Seifert, 2003). However, P-site inhibitors still require for binding a conformation that resembles a product-like transition state and thus are more potent on the active forms of the enzyme. The affinity of mAC for all P-site inhibitors increases in the presence of Mn2+ versus Mg2+, similar to the reduction of the Km for ATP with Mn2+. The tight binding of these inhibitors at the active site renders these important tools for understanding the function of these enzymes and the development of future cell-permeable inhibitors (Seifert et al., 2012).
3. Structure-activity Relationships and Binding Modes of 2′,(3′)-O-(N-Methylanthraniloyl)- and 2′,3′-O-(2,4,6-Trinitrophenyl)-nucleotides.
MANT- and TNP-nucleotides explore all three pharmacophoric domains in ACs and are competitive inhibitors (Gille and Seifert, 2003). Some of the compounds studied exhibit nanomolar affinities that enabled their use for crystallographic studies (Mou et al., 2005, 2006; Hübner et al., 2011) and fluorescence spectroscopy studies monitoring ligand-specific conformational changes (Pinto et al., 2009, 2011; Suryanarayana et al., 2009a) with purified 5C1:2C2. Unfortunately, because of the fact that MANT-nucleotides bind to many proteins with a nucleotide binding site (see, e.g., Remmers et al., 1994; Franks-Skiba and Cooke, 1995; Scheidig et al., 1995; Dayan et al., 1997; Hiratsuka, 2003; Zielke et al., 2015), their potential as starting point for the development of AC isoform-selective drugs is very limited.
The binding modes of 2′- and 3′-MANT as well as TNP-substituted nucleotides at mACs are roughly similar to the binding mode of ATP (Fig. 11). In the complex of 5C1:2C2 with 3′-MANT-GTP (Mou et al., 2005), the ribosyl moiety adopts a 2′-endo conformation with an axial orientation of the MANT group (Fig. 14A). Its wedge-like fit between α1, α2, and the β5′-α4′ loop prevents the formation of the fully closed conformation. The phenyl ring is involved in hydrophobic interactions with L412 (α2) and van der Waals contacts with the backbone of W1020, G1021, and N1022 (β5′-α4′ loop). With a 3′-endo conformation of the ribosyl moiety corresponding to an equatorial 3′- and an axial 2′-orientation, 2′-MANT, and even 2′,3′-bis-MANT nucleotides may bind in a similar mode.
Fig. 14.

Interactions of 5C1:2C2 with selected MANT- and TNP-nucleotides. 5C1, green; 2C2, cyan; heteroatom colors: N, blue; O, red; P, orange; Mn2+, violet. Nucleobase-2C2 hydrogen bonds are drawn as red dashed lines, contacting amino acids (distance < 3.5 Å) and secondary structure elements are labeled. (A) Binding mode of 3′-MANT-GTP, model based on the crystal structure PDB 1TL7 (Mou et al., 2005) C and selected H atoms of 3′-MANT-GTP, gray. (B) Binding mode of TNP-ATP, model based on the crystal structure PDB 2GVD (Mou et al., 2006), C and selected H atoms of TNP-ATP, mauve. The gray shadow image shows 3′-MANT-GTP from a superposition of both binding sites (root-mean-square deviation of Cα atoms, 0.44 Å).
The triphosphate conformation in 3′-MANT-GTP differs from that in ATP. In particular, α- and β-phosphates interact only with metal ions. The γ-phosphate forms three hydrogen bonds with the backbone NH functions of F400 and T401 (α1) and with K1065 (β7′-β8′ loop), but does not contact R484 in contrast to ATP. The ribosyl moieties of 3′−MANT-GTP and ATP also interact differently with 5C1:2C2. In case of 3′-MANT-GTP, a weak hydrogen bond of the 2′-OH group with N1025 (α4′) and distant van der Waals contacts with the side chains of F400 (α1) and V1024 (α4′) are formed. The hydrogen bonds of the adenylyl moiety in the 5C1:2C2-ATP complex with K938 (β2′) and D1018 (β5′) are reproduced by the oxygen, the N1 atom and the 2-NH2 group, respectively, of the guanine base. In both cases, the base adopts an anti conformation. In contrast, a syn position of the adenylyl moiety and no specific enzyme-base interactions are characteristic for the binding mode of 3′-MANT-ATP in complex with 5C1:2C2 (Mou et al., 2006).
TNP-ATP binds roughly similar to 3′-MANT-GTP (Mou et al., 2006), i.e., the pockets occupied by the three components nucleobase, triphosphate, and substituent are common (Fig. 14B). The fit of the trinitrophenyl ring between L412 (α2) and the backbone of W1020, G1021, and N1022 (β5′-α4′ loop) corresponds to that of the 3′-MANT substituent. The 2-nitro group is involved in a hydrogen bond with the side chain amide of N1022. N1025 (α4′) also interacts with the 2-nitro group and not with the ribosyl fragment, which is almost planar with two axial oxygens in 2′- and 3′-position. Interactions and conformation of the triphosphate moiety of TNP-ATP differ from those of 3′-MANT-GTP with respect to the β- and γ-phosphate. In case of TNP-ATP, the β-phosphate forms two hydrogen bonds with the backbone NH of F400 (α1), and the γ-phosphate interacts with R484 (β4) and K1065 (β7′-β8′ loop). In contrast to 3′-MANT-ATP, the adenine base in TNP-ATP adopts an anti position. A single hydrogen bond is formed between the 6-NH2 group and the side chain of S1028 (α4′). Taken together, Fig. 14B is suggestive of flexible fits of MANT- and TNP-nucleotides to partially different components of the binding pocket as major cause of mAC promiscuity for competitive inhibitors.
Table 6 lists the structure-activity relationships for selected (M)ANT- and TNP-nucleotides at ACs 1, 2, and 5 as representatives of the three major mAC families, purified catalytic subunits 5C1:2C2, sAC and purified sGCα1β1, also referred to as GC1. In general, AC2 is the least sensitive mAC isoform toward TNP- and (M)ANT-nucleotides, but selectivity for ACs 1 and 5 is at best 10-fold (Gille et al., 2004; Suryanarayana et al., 2009a; Pinto et al., 2011). In addition, TNP- and (M)ANT-nucleotides are very potent sGC inhibitors (Gille et al., 2004; Suryanarayana et al., 2009a; Dove et al., 2014). The potent sGC inhibition by (M)ANT-nucleotides is not surprising in light of the high conservation of the catalytic domains of these enzymes (Sunahara et al., 1998; Dove et al., 2014). Given the beneficial clinical effects of allosteric sGC activators and sGC stimulators (Stasch et al., 2011, 2015), sGC inhibition would be clearly an off-target effect of mAC inhibitors (Dove et al., 2014). However, there is evidence for pharmacological distinction between mACs and sGC with (M)ANT- and TNP-nucleotides (Dove et al., 2014) (Table 6).
TABLE 6.
Inhibition of mACs, sAC, and sGC by (M)ANT- and TNP-nucleotides
Nucleotidyl cyclase activity was determined in the presence of Mn2+. Reaction mixtures contained nucleotides analogs at concentrations from 10 nM to 100 μM. Inhibition curves were analyzed by nonlinear regression and were best-fitted to monophasic sigmoidal curves. pKi values are listed in the table. The structures of the inhibitors examined are shown in Fig. 13. Compounds 1, 3, 7, 10, 11, and 20 were studied at partially purified sAC from rat testis; compounds 22–25 were studied at the purified catalytic domain of human sAC. The Table has been modified from Table 1 in Dove et al. (2014). Data for mACs 1, 2, and 5 and 5C1:2C2 were taken from the literature as noted by the footnotes.
| Cpd. | Inhibitor | AC1 | AC2 | AC5 | 5C1:2C2 | sAC | sGC α1β1 (GC1a) |
|---|---|---|---|---|---|---|---|
| 1 | MANT-ATP | 6.82b | 6.48b | 7.00b | 7.80b | 5.25c | 6.77 |
| 2 | MANT-ITP | 8.55b | 7.85b | 8.92b | 9.15b | N.D. | 6.31 |
| 3 | MANT-GTP | 7.05b | 6.21b | 7.28b | 7.74b | >4.00c | 6.06 |
| 4 | MANT-XTP | 5.96b | 5.52b | 5.89b | 5.92b | N.D. | 6.95 |
| 5 | MANT-CTP | 6.82b | 6.16b | 6.82b | 8.04b | N.D. | 6.28 |
| 6 | MANT-UTP | 7.34b | 6.34b | 7.49b | 8.21b | N.D. | 6.30 |
| 7 | 2’-MANT-3′-dATP | 6.33b | 6.27b | 7.19b | 7.05b | 4.60c | 7.78 |
| 8 | 3′-MANT-2’-dATP | 6.49b | 5.32b | 6.44b | 6.72b | N.D. | 6.13 |
| 9 | MANT-ATPγS | 6.80d | 6.42d | 7.11d | N.D. | N.D. | 6.88 |
| 10 | MANT-ITPγS | 7.39c | 6.92c | 7.51c | 7.72b | >4.00c | 6.03 |
| 11 | MANT-GTPγS | 7.21c | 6.44c | 7.46c | 7.62b | >4.00c | 6.33 |
| 12 | ANT-ATP | 6.89b | 6.19b | 6.92b | 7.77b | N.D. | 6.63 |
| 13 | Cl-ANT-ATP | 7.10e | 6.31e | 7.35e | N.D. | N.D. | 7.33 |
| 14 | Cl-ANT-ITP | 8.48e | 8.10e | 8.70e | N.D. | N.D. | 5.92 |
| 15 | Br-ANT-ITP | 8.15e | 7.66e | 8.34e | N.D. | N.D. | 6.22 |
| 16 | Pr-ANT-ATP | 6.36e | 5.96e | 6.44e | N.D. | N.D. | 6.80 |
| 17 | Pr-ANT-ITP | 7.66e | 7.17e | 8.00e | N.D. | N.D. | 6.41 |
| 18 | AcNH-ANT-ATP | 5.17e | 4.96e | 5.47e | N.D. | N.D. | 6.11 |
| 19 | AcNH-ANT-ITP | 6.85e | 6.41e | 7.43e | N.D. | N.D. | 6.22 |
| 20 | MANT-AppNHp | 6.80c | 6.31c | 6.92c | 7.70c | 5.72c | 6.77 |
| 21 | MANT-GppNHp | 6.96c | 6.18c | 7.01c | 7.47c | N.D. | 5.56 |
| 22 | TNP-ATP | 8.05f | 7.00f | 8.43f | 7.09g | 6.19f | 7.67 |
| 23 | TNP-GTP | 7.64f | 6.66f | 7.57f | 7.08g | 5.14f | 7.97 |
| 24 | TNP-CTP | 7.62f | 6.96f | 7.51f | 6.51g | 5.29f | 7.01 |
| 25 | TNP-UTP | 8.15f | 7.62f | 7.82f | 7.04g | 5.51f | 7.29 |
| 26 | Bis-MANT-ATP | 6.15e | 5.68e | 6.37e | N.D. | N.D. | 6.23 |
| 27 | Bis-MANT-ITP | 6.51e | 5.96e | 6.85e | N.D. | N.D. | 6.46 |
| 28 | Bis-MANT-CTP | 6.21e | 5.11e | 6.12e | N.D. | N.D. | 6.10 |
| 29 | Bis-MANT-IDP | 6.00e | 5.85e | 6.15e | N.D. | N.D. | 5.72 |
| 30 | Bis-MANT-IMP | <3e | <3e | <3e | N.D. | N.D. | <3 |
| 31 | Bis-Cl-ANT-ATP | 5.77e | 5.62e | 5.80e | N.D. | N.D. | 6.28 |
| 32 | Bis-Cl-ANT-ITP | 7.18e | 6.70e | 7.19e | N.D. | N.D. | 6.08 |
| 33 | Bis-Br-ANT-ATP | N.D. | N.D. | N.D. | N.D. | N.D. | 6.37 |
| 34 | Bis-Br-ANT-ITP | 7.68e | 7.15e | 7.82e | N.D. | N.D. | 6.20 |
| 35 | Bis-Pr-ANT-ATP | 4.74e | 4.44e | 4.74e | N.D. | N.D. | 5.44 |
| 36 | Bis-Pr-ANT-ITP | 6.36e | 5.85e | 6.60e | N.D. | N.D. | 5.91 |
| 37 | Bis-AcNH-ANT-ATP | 4.66e | 5.15e | 5.21e | N.D. | N.D. | 5.41 |
| 38 | Bis-AcNH-ANT-ITP | 5.77e | 5.25e | 6.32e | N.D. | N.D. | 5.97 |
N.D., not determined.
In addition to nucleotidyl cyclases, (M)ANT- and TNP-nucleotides bind to many other nucleotide-binding enzymes and signal-transducing guanine nucleotide-binding proteins (see e.g., Remmers et al., 1994; Franks-Skiba and Cooke, 1995; Scheidig et al., 1995; Dayan et al., 1997; Hiratsuka, 2003; Zielke et al., 2015), rendering AC specificity difficult. Application of (M)ANT- and TNP-nucleotides in intact cells is also impossible because the compounds are too hydrophilic to penetrate the plasma membrane. Electrophysiological experiments with intracellularly injected MANT-GTP pointed to a role of AC5 in the regulation of voltage-dependent calcium channels in cardiomyocytes (Rottlaender et al., 2007), but studies with the structurally closely related nucleotide MANT-ITP revealed unexpected off-target effects (Hübner et al., 2011b), although MANT-ITP is the most potent mAC inhibitor known so far (Hübner et al., 2011a). Intriguingly, bulky bis-MANT-nucleotides exhibit strong preference for the Bordetella pertussis AC toxin CyaA relative to mACs, constituting the basis for the development of bacterial toxin inhibitors (Geduhn et al., 2011) (Table 6).
4. Membrane-permeable Membrane Adenylyl Cyclase Inhibitors.
The issues with impermeability of the plasma membrane for nucleotides can be avoided by applying membrane-permeable nucleotide prodrugs (Laux et al., 2004), but this strategy has not yet found broad application in the field, leaving many of the most potent AC inhibitors with little cellular utility. As an alternative strategy, membrane-permeable and hence lipophilic nucleoside analogs can be used in intact cell studies. In general, these compounds are much less potent than MANT/TNP-nucleotides because they only explore the base-binding domain of ACs (Seifert et al., 2012; Brand et al., 2013). These compounds act as noncompetitive P-site inhibitors. The lipophilicity of compounds may require high concentrations of organic solvents in cell experiments so that nonspecific solvent effects become a problem (Brunskole Hummel et al., 2013), but this aspect has not been generally considered in cell studies.
Three compounds, 2-amino-7-(2-furanyl)-7,8-dihydro-5(6H)-quinazolinone (NKY80), 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ22,536), and vidarabine (also referred to as adenine 9-β-d-arabinofuranoside or 9-β-d-arabinofuranosyl-adenine), have been systematically studied at all nine mAC isoforms, several native membrane systems, and sAC (Brand et al., 2013; Bitterman et al., 2013; Braeunig et al., 2013; Braeunig et al., 2014). Table 7 lists the pIC50 values of these compounds for inhibition of ACs and pKi values for interaction with adenosine receptors. Some compounds have been studied with defined ACs by several independent research groups. Overall, there is reasonably good agreement in the obtained pIC50 values (Table 7). SQ22,536, NKY80, and vidarabine exhibit some degree of selectivity among AC isoforms, AC2 being relatively insensitive compared with ACs 5 and 6 (Bitterman et al., 2013). ACs 8 and 9 and sAC are very resistant to inhibition by SQ22,536, NKY80, and vidarabine. pIC50 values for sGC of the compounds listed in Table 7 are not available. A recent study reported on the identification of noncompetitive inhibitors with some selectivity for AC2 relative to ACs 1 and 5, compound 8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-ol hydrobromide (SKF-83566) being a representative (Conley et al., 2013). However, SKF-83566 is also a potent dopamine D1 receptor antagonist (O’Boyle and Waddington, 1984).
TABLE 7.
Interaction of prototypical membrane-permeable P-site inhibitors with mACs, sAC, sGC, and adenosine receptors
AC activity was determined in the presence of Mg2+ and in the presence of stimulators (GαS-GTPγS and/or FSK) to ensure maximum catalytic activity and, hence, highest inhibitor potency possible. For ACs, pIC50 values are listed. For adenosine receptors, pKi values are listed. Structures of P-site inhibitors are shown in Fig. 13. 2′,5′-Dideoxy-3′-ATP (Table 8 and Fig. 15) is mechanistically related to the P-site inhibitors shown here.
| AC | SQ22,536 | NKY80 | Vidarabine |
|---|---|---|---|
| AC1 | 4.27a, 3.95b, 3.92c | 4.04a, 3.48b | 4.31a, 3.86b |
| AC2 | 3.71a, 3.41b, 3.17c, 3.54d | 3.11a, >3.00b, >3.00e, >3.00d | 3.61a, 3.26b, 3.15e |
| AC3 | 3.96a, 4.00d | 4.03a, 3.64e, 3.89d | 4.07a |
| AC4 | 3.55a | 3.95a | 4.03a |
| AC5 | 5.46a, 5.02b, 5.66d | 5.15a, 4.09b, 4.82e, 5.08d | 5.67a, 4.70b, 5.00e |
| AC6 | 5.25a | 4.80a | 5.34a |
| AC7 | 3.38a | 3.09a | 3.64a |
| AC8 | >3.00a, 3.92c | >3.00a | >3.00a |
| AC9 | > 3.00a | >3.00a | >3.00a |
| 5C1:2C2 | N.D. | N.D. | N.D. |
| sAC | >>3.00f | >>3.00f | >>3.00f |
| sGC | N.D. | N.D. | N.D. |
| Mouse heart AC, wild type | 5.20b | 4.37b | 4.98b |
| Mouse heart AC, AC5 knockout | 5.30b | 4.41b | 5.00b |
| Rat heart AC, left ventricle | N.D. | 4.48g | 4.73g |
| Rat heart, right ventricle | N.D. | 4.62g | 5.12g |
| Adenosine A1 receptor | 4.86h | 4.62h | >4.00h |
| Adenosine A2A receptor | 4.76h | 5.52h | >4.00h |
| Adenosine A3 receptor | >4.22h | >4.00h | >4.00h |
N.D., not determined.
cis-N-(2-phenylcyclopentyl)-azacyclotridec-1-en-2-amine hydrochloride (MDL 12,330A) was one of the first AC inhibitors to be introduced into experimental pharmacology (Hunt and Evans, 1980) and is still widely used. The compound is membrane-permeable and has been reviewed critically (Seifert et al., 2012). MDL 12,330A cannot be recommended for intact-cell studies because of its low potency and pleiotropic effects. The compound has only been incompletely characterized so far at defined AC isoforms. MDL 12,330A inhibits a lysate of mAC with a pIC50 of 3.4 and purified sAC with a pIC50 of 3.6 (Bitterman et al., 2013). Thus, MDL 12,330A does not discriminate between mACs and sAC, and the potency of the compound is lower than the potency of many membrane-permeable P-site inhibitors (Table 7).
5. Adenylyl Cyclase 1 Inhibitors and Modulation of Calmodulin.
A nucleotidyl cyclase that has attracted considerable attention as drug target is AC1. It was proposed that AC1 inhibition could be used to treat various neuropsychiatric disorders such as neuropathic pain (Wang et al., 2011a). 5-[[2-(6-Amino-9H-purin-9-yl)ethyl]amino]-1-pentanol (NB001) was suggested to inhibit AC1 in intact cells selectively (Wang et al., 2011a), but this proposition was not confirmed with recombinant AC1 in membrane preparations (Brand et al., 2013). Thus, the precise mechanism of action of NB001 remains elusive.
AC1 forms a unique interface with its activator CaM compared with other mammalian CaM targets (Vorherr et al., 1993; Diel et al., 2008; Masada et al., 2012; Lübker and Seifert, 2015; Lübker et al., 2015a,b). Principally, such a unique protein-protein interaction interface could be exploited pharmacologically. In this context, it is interesting to note that calmidazolium chloride, which is very potent at inhibiting numerous CaM-regulated effectors, only very ineffectively inhibits AC1 (Lübker and Seifert, 2015). However, potent and selective inhibitors of the AC1-CaM interaction have not yet been identified in a small-scale screening study (Lübker and Seifert, 2015). Calmidazolium also inhibits AC9, probably by binding to the catalytic site (Haunso et al., 2003). Currently, there is no clinical use of AC1 inhibitors.
Ageing processes are related to the oxidation of methionine residues in CaM (Bigelow and Squier, 2005), and oxidized CaM is less efficient than native CaM at activating AC1 (Lübker et al., 2015a,b). Thus, one could envisage that reduction of oxidized methionine residues in CaM by increased expression of methionine sulfoxide reductase is therapeutically useful in CNS diseases associated with impaired learning and memory (Moskovitz and Oien, 2010; Lübker et al., 2015a,b).
6. Adenylyl Cyclase 5 Inhibitors.
Currently, there is great interest in AC5-selective inhibitors. Based on experiments with AC5 knockout mice (see section II.E.3.a), it has been proposed that selective AC5 inhibition may be beneficial in the treatment of heart failure and other age-related diseases (Vatner et al., 2013; Bravo et al., 2016). However, AC5 is broadly expressed in many organs, including kidney (Erdorf and Seifert, 2011) and brain (Kim and Han, 2009), so that substantial organ toxicity could emerge as a result of global AC5 inhibition (Table 3). Most notably, AC5 inhibition in the brain could result in Parkinson-like symptoms (Iwamoto et al., 2003; Seifert, 2014). In addition, the available competitive and noncompetitive AC inhibitors do not discriminate between AC5 and the closely related AC6 (Gille et al., 2004; Brand et al., 2013) (Table 7). This is supported by the observation that vidarabine shows very similar potency in heart membranes from wild-type mice and AC5 knockout mice (Table 7). The lack of discrimination between ACs 5 and 6 is problematic, because AC6 also plays an important functional role in the heart (Göttle et al., 2009; Tang et al., 2013).
Another complication constitutes the fact that several P-site inhibitors show significant affinity to adenosine receptors (Klotz and Kachler, 2016) (Table 7) and inhibit cellular phosphorylation cascades via an as yet unknown mechanism (Emery et al., 2013), so that interpretation of pharmacological data as AC5 inhibition is very problematic. In some instances, because of solubility issues and low compound potency, it has been impossible to obtain sufficiently high inhibitor concentrations in intact cells to observe AC inhibition (Brunskole Hummel et al., 2013). Similarly, it is questionable whether the required micromolar concentrations of P-site inhibitors can be obtained in the intact organism without off-target effects (Seifert, 2016). Overall, NKY80, SQ22,536, and vidarabine, which are very widely used as “selective mAC inhibitors” (or even more problematic as “selective AC5 inhibitors”) (e.g., Rangel-Barajas et al., 2011; Bravo et al., 2016; Liu and Steketee, 2016; Wada et al., 2016; Wei et al., 2016), should be used with great caution as pharmacological tools, and any observed effects should be carefully checked for off-target actions (Seifert, 2014, 2016). It is particularly important to confirm that the AC inhibitor used actually prevents a GPCR- or FSK-mediated cAMP increase (Brunskole Hummel et al., 2013). Because vidarabine constitutes an FDA-approved antiviral drug, the clinical use of this compound for AC5 inhibition has been proposed (Vatner et al., 2013; Bravo et al., 2016). Unfortunately, vidarabine possesses a high potential for toxic effects so that systemic clinical use would potentially be dangerous (Seifert, 2014, 2016).
A newly emerging potential clinical application of AC5 inhibitors is the treatment of extrapyramidal movement disorders such as FDFM, caused by gain-of-function mutations in the ADCY5 gene (Chen et al., 2012c, 2014b). Thus, reduction of the pathologically increased catalytic activity of the R418W AC5 mutant and A726T AC5 mutant may be beneficial. However, like with wild-type ACs, selectivity of inhibitors is a concern. As a first step toward the development of mutant-selective AC5 inhibitors, a comprehensive pharmacological characterization of AC5 mutants with the available tools (Figs. 12 and 13; Tables 6 and 7) is required.
7. Soluble Adenylyl Cyclase Inhibitors.
In general, MANT- and TNP-nucleotides inhibit purified sAC much less potently than ACs 1, 2, and 5 and purified 5C1:2C2 and sGC (Table 6), but data sets are incomplete. Along the same line, membrane-permeable P-site inhibitors, often used in intact cell studies, inhibit sAC considerably less potently than most mAC isoforms (Table 7). The membrane-impermeable P-site inhibitor 2′,5′-dideoxy-3′-ATP (Fig. 15) is less potent at inhibiting sAC than purified 5C1:2C2 and various mAC isoforms (Table 8), fitting to the data on membrane-permeable P-site inhibitors.
Fig. 15.

Structures of representative inhibitors targeting sAC. Pharmacological data for inhibitors are listed in Table 8. All compounds are noncompetitive sAC inhibitors. 2CE, KH7, and RU-0204277 are membrane-permeable. KH7 has been broadly used in intact cell studies, but cell toxicity is of concern. RU-0204277 is a recently developed compound with less toxic liability than KH7 and binds to the bicarbonate site of sAC. 2CE exhibits different potencies at purified sAC and in intact cells. 2′,5′-dideoxy-3′-ATP is a classic P-site inhibitor related to SQ 22536, NKY80, and vidarabine (for structures, see Fig. 13 and for pharmacological data see Table 7) that also potently inhibits mACs and cannot penetrate the plasma membrane.
TABLE 8.
Interaction of prototypical membrane-permeable sAC inhibitors and the membrane-impermeable P-site inhibitor 2’,5′-dd-3′-ATP with sAC and mACs
pIC50 values are listed. Structures of prototypical sAC inhibitors and 2′,5′-dideoxy-3′-ATP are shown in Figure 15. pIC50 values for ACs 3, 4 and 6 and sGC are not available. 2′,5′-dideoxy-3′-ATP is mechanistically related to the P-site inhibitors documented in Fig. 13 and Table 7.
| AC | 2CE | KH7 | RU-0204277 | 2’,5′-dideoxy-3′-ATP |
|---|---|---|---|---|
| Purified sAC | ∼5.7a | 5.52b | ∼5.0c | 6.16d |
| 5C1:2C2 | N.D. | N.D. | N.D. | 7.42d |
| mAC lysate | ∼4a | no inhibition at 500 μMb | N.D. | 7.40d |
| AC1 | ∼4a | N.D. | no inhibition at 50 μMc | 7.43d |
| AC2 | ∼4a | N.D. | no inhibition at 50 μMc | 6.66d |
| AC5 | ∼4a | N.D. | no inhibition at 50 μMc | 7.43d |
| AC7 | ∼5.7a | N.D. | N.D. | N.D. |
| AC8 | N.D. | N.D. | no inhibition at 50 μMc | N.D. |
| AC9 | N.D. | N.D. | no inhibition at 50 μMc | N.D. |
N.D., not determined. Specific experimental conditions for analysis of inhibitors varied in the different studies and are given in:
Considerable effort has been directed toward the development of sAC inhibitors to probe the role of sAC in cell physiology. Catechol estrogens are noncompetitive sAC inhibitors, binding to a hydrophobic region in the vicinity of the catalytic site (Steegborn et al., 2005). 2-Hydroxyestradiol (2CE) is a prototypical compound of this class (Fig. 15). 2CE exhibits selectivity toward AC1, 2, and 5, but not AC7 (Table 8). The potency of 2CE in intact cells and cell-free systems differs for as yet unknown reasons (Steegborn et al., 2005; Bitterman et al., 2013).
(±)-2-(1H-Benzimidazol-2-ylthio)propanoic acid 2-[(5-bromo-2-hydroxyphenyl)methylene]hydrazide (KH7) is the most widely used sAC inhibitor in intact-cell studies (Kumar et al., 2009; Schmid et al., 2010; Kolodecik et al., 2012; Bitterman et al., 2013; Dey et al., 2014; Hasan et al., 2014; Nickols et al., 2015; De Rasmo et al., 2015; Sosa et al., 2016). Caution must be exerted when interpreting intact-cell studies with KH7 because of toxic effects of this compound in mitochondria (Di Benedetto et al., 2013) and β cells (Tian et al., 2011). KH7 inhibits purified sAC with similar potency as 2CE and shows selectivity when examined against a lysate containing mACs (Table 8). However, data for individual mAC isoforms are not available. The exact binding site of KH7 on sAC is not known.
6-Chloro-N4-cyclopropyl-N4-[(thiophen-3-yl)methyl]pyrimidine-2,4-diamine (RU-0204277) has recently emerged as a novel sAC inhibitor from a high-throughput screening strategy (Ramos-Espiritu et al., 2016). RU-0204277 binds to the bicarbonate-binding site of sAC and exhibits potency comparable to the potencies of 2CE and KH7 (Ramos-Espiritu et al., 2016) (Table 8). At a concentration of 50 μM, the substance did not affect ACs 1, 2, 5, 8, and 9. Given the IC50 for sAC of ∼10 μM, it will be necessary to examine inhibitor concentrations of 100–500 μM to assess specificity more comprehensively. Initial studies indicate that RU-0204277 is also useful for intact-cell studies and devoid of toxicity (Ramos-Espiritu et al., 2016).
Inhibition of sGC is a problematic property when aiming at the clinical application of mAC and sAC inhibitors (Stasch et al., 2011, 2015; Dove et al., 2014). Unfortunately, pIC50 values for sGC of the compounds listed in Table 8 are not yet available.
V. Conclusions and Future Directions
Since the initial identification of cAMP in biologic systems almost 60 years ago by Rall and Sutherland (1958), substantial progress has been made with respect to structure, expression, regulation, and compartmentation of both mACs and sAC. This knowledge constitutes the basis for the development and clinical application of mAC activators and inhibitors as well as sAC inhibitors. Despite our large increase in knowledge, there are still important gaps. Most importantly, a systematic pharmacological characterization of ACs is, with one exception for a small group of membrane-permeable inhibitors (Brand et al., 2013), not available. The incomplete pharmacological mAC characterization has resulted in misconceptions regarding the supposed selectivity of certain compounds (Seifert, 2014, 2016; Bravo et al., 2016).
Most crystal structures were obtained with a 5C1:2C2 hybrid from different species, with parts of the cytosolic domains and the entire transmembrane domains missing. From the tremendous progress in GPCR crystallography (Venkatakrishnan et al., 2013; Piscitelli et al., 2015), it can be extrapolated that resolution of holo-mAC crystal structures with homologous cytosolic domains and transmembrane domains would substantially increase our knowledge on these enzymes, specifically with regard to hitherto unexplored ligand binding sites. This would require, as a first step, systematic testing and optimization of detergents that may stabilize holo-mACs (Cho et al., 2015). In case of GPCRs, this effort was ultimately successful but took almost 20 years (Rasmussen et al., 2007).
Although numerous experimental tools are available to the AC research community, lack of selectivity and potency of such compounds has often turned out to be a problem. Because of the structural differences between mACs (class IIIa) and sAC (class IIIb) (Linder and Schultz, 2003), the development of global mAC inhibitors and sAC inhibitors with selectivity against the other AC class may be feasible, but cross-inhibition of sGC, definitively an off-target effect (Stasch et al., 2011, 2015; Dove et al., 2014), has to be considered as well. From a medicinal chemistry point-of-view, the development of isoform-selective mAC inhibitors is particularly challenging because of the high degree of conservation of the catalytic core. One approach toward achieving this ambitious goal would be to pharmacologically address the poorly conserved transmembrane domains of mACs (Beltz et al., 2016), but this is not trivial for two reasons: First, it is difficult to conduct site-directed mutagenesis and chimeric studies with the transmembrane domains of mACs. But these studies constitute a first step toward delineation of potential ligand binding sites within transmembrane spans. Second, it is expected that ligands binding to the transmembrane domains would be highly hydrophobic, but this would also increase the risk of off-target effects considerably (Daniel, 2003).
Very few, if any, studies from the pharmaceutical industry on the development on mAC and sAC inhibitors have been published, in marked contrast to the situation regarding allosterically acting NO-dependent sGC stimulators and NO-independent sGC activators (Stasch et al., 2011, 2015; Friebe et al., 2015). One reason for these differences may be the intrinsic difficulties from the medicinal chemistry perspective. However, another reason for this situation is certainly the fact that the pathophysiological relevance of a given mAC isoform for specific human diseases is much less clear than for sGC. The complex and overlapping expression pattern of mACs makes it difficult to obtain clinically useful pharmacological modulation without toxicity. One solution to the problem could be approaches to apply mAC inhibitors and activators locally. Such a strategy would, however, limit possible clinical applications to locally accessible organs such as the eye, skin, and airways. sAC inhibition could be a strategy to achieve the elusive goal of a male contraceptive (Roth and Amory, 2016), but the proposed broad expression and function of sAC beyond the male reproductive system (Steegborn, 2014; Levin and Buck, 2015) would probably result in many side effects. It remains to be determined whether the recently introduced microRNA approach (Zhuang et al., 2014) constitutes a clinically useful approach to target mACs.
There are some similarities between ACs and PDEs as drug targets, covered in Pharmacological Reviews a number of years ago (Bender and Beavo, 2006). ACs and PDEs exist in multiple isoforms that have different biochemical properties, functions, and tissue distribution (Conti and Beavo, 2007). For PDE4 and PDE5 inhibitors, important clinical applications have already been established (Francis and Corbin, 2005; Garnock-Jones, 2015), and a number of novel therapeutic applications of selective PDE inhibitors are emerging (Heckman et al., 2015; Lee et al., 2015). Given the fact that any given AC and PDE possess a unique tissue distribution, one could envisage unique therapeutic applications of isoform-selective AC activators and inhibitors.
The role of defined AC isoforms in human diseases is just beginning to emerge. Proof-of-principle for a pathophysiological role of an AC isoform was recently provided by linking gain-of-function mutations in ADCY5 to a extrapyramidal movement disorders (Chen et al., 2012c, 2014b, 2015b). These data also raise the question whether development of mutant-specific AC isoform inhibitors is useful. The development of mutant-specific protein kinase inhibitors has been highly successful for targeted cancer therapy (Dhillon, 2016).
The recent advances in our knowledge on the pathophysiological roles of AC isoforms call for more studies on links between ADCY gene polymorphisms and human diseases. In addition, upregulation or downregulation in the expression and function of wild-type AC isoforms in human diseases must be studied in greater detail. At the mRNA/RT-PCR level, such studies are possible, but at the protein level, the paucity of specific antibodies constitutes a bottleneck. Once a role of AC isoforms in human diseases is better defined, we hope that the pharmaceutical industry is willing to take the risk for such AC inhibitor and activator development programs that, in principle, are feasible.
Acknowledgments
The authors thank Drs. Erich Schneider (Hannover Medical School) and Jochen Schultz (University of Tübingen) for critical reading of the manuscript and Annette Stanke for careful compilation of the reference list and formal edition of the manuscript. A special thanks to the external reviewers for thoughtful comments and suggestions.
Abbreviations
- AC
adenylyl cyclase
- AKAP
A-kinase-anchoring protein
- βAR
β-adrenoceptor
- 2C2
C2 catalytic domain of AC2
- 5C1
C1 catalytic domain of AC5
- CaM
calmodulin
- 2CE
2-hydroxyestradiol
- cNMP
nucleoside 3,5-monophosphate
- CNS
central nervous system
- FDFM
familial dyskinesia with facial myokymia
- FSK
forskolin
- Gαi
α-subunit of the inhibitory G-protein of adenylyl cyclase Gi
- Gαo
α-subunit of the G-protein Go that is widely expressed in the CNS and neuroendocrine cells and mediates inhibition of calcium channels
- Gαolf
α-subunit of the olfactory G-protein that stimulates adenylyl cyclase
- Gαq
α-subunit of a G-protein that stimulates phospholipase C
- Gαs
α-subunit of the stimulatory G-protein of adenylyl cyclase Gs
- Gβγ
βγ-subunits of G-proteins
- GPCR
G-protein-coupled receptor
- HEK
human embryonic kidney
- IBMX
isobutylmethylxanthine
- KH7
(±)-2-(1H-benzimidazol-2-ylthio)propanoic acid 2-[(5-bromo-2-hydroxyphenyl)methylene]hydrazide
- mAC
membrane adenylyl cyclase
- MANT
2′,(3′)-O-(N-methylanthraniloyl)
- MRP
multidrug resistance associated protein
- NB001
5-[[2-(6-amino-9H-purin-9-yl)ethyl]amino]-1-pentanol
- NKY80
2-amino-7-(2-furanyl)-7,8-dihydro-5(6H)-quinazolinone
- NO
nitric oxide
- PDE
phosphodiesterase
- PKA
cAMP-dependent protein kinase
- PKC
protein kinase C
- sAC
soluble adenylyl cyclase
- sGC
soluble guanylyl cyclase
- pGC
particulate guanylyl cyclase
- RGS proteins
regulator of G-protein signaling proteins
- RU-0204277
6-chloro-N4-cyclopropyl-N4-[(thiophen-3-yl)methyl]pyrimidine-2,4-diamine
- 6-chloro-N4-cyclopropyl-N4-[(thiophen-3-yl)methyl]pyrimidine-2
4-diamine
- SKF-83566
8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-ol hydrobromide
- SQ22,536
9-(tetrahydro-2-furanyl)-9H-purin-6-amine
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Dessauer, Watts, Ostrom, Conti, Dove, and Seifert.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM107094 (to R.S.O.), GM60419 (to C.W.D.)], National Institute of Neurological Disorders and Stroke [Grant NS091759 to C.W.D.], National Institute of Mental Health [Grants MH096927 and MH101673 to V.J.W.], the American Heart Association [AHA 005140Z, AHA 0450120Z, 14GRNT20380762], and the Deutsche Forschungsgemeinschaft [Se 529/5-1, Se 529/5-2].
We dedicate this review to the late Alfred G. Gilman, Nobel laureate for Physiology 1994, who made seminal contributions to the field of adenylyl cyclase research.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE. (1998) Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet 19:289–291. [DOI] [PubMed] [Google Scholar]
- Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. (2009) Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, MacDougall DA, Tyser R, Pugh SD, Calaghan SC, Harvey RD. (2011) Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J Mol Cell Cardiol 50:500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Yang PC, Rice M, Singer CA, Nikolaev VO, Lohse MJ, Clancy CE, Harvey RD. (2014) Role of membrane microdomains in compartmentation of cAMP signaling. PLoS One 9:e95835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alasbahi RH, Melzig MF. (2012) Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie 67:5–13. [PubMed] [Google Scholar]
- Aldehni F, Tang T, Madsen K, Plattner M, Schreiber A, Friis UG, Hammond HK, Han PL, Schweda F. (2011) Stimulation of renin secretion by catecholamines is dependent on adenylyl cyclases 5 and 6. Hypertension 57:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C, et al. CGTP Collaborators (2015) The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172:6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allerston CK, von Delft F, Gileadi O. (2013) Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS One 8:e57644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alousi AA, Jasper JR, Insel PA, Motulsky HJ. (1991) Stoichiometry of receptor-Gs-adenylate cyclase interactions. FASEB J 5:2300–2303. [DOI] [PubMed] [Google Scholar]
- Angel-Chavez LI, Acosta-Gómez EI, Morales-Avalos M, Castro E, Cruzblanca H. (2015) Forskolin suppresses delayed-rectifier K+ currents and enhances spike frequency-dependent adaptation of sympathetic neurons. PLoS One 10:e0126365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni FA, Palkovits M, Simpson J, Smith SM, Leitch AL, Rosie R, Fink G, Paterson JM. (1998) Ca2+/calcineurin-inhibited adenylyl cyclase, highly abundant in forebrain regions, is important for learning and memory. J Neurosci 18:9650–9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni FA, Wiegand UK, Black J, Simpson J. (2006) Cellular localisation of adenylyl cyclase: a post-genome perspective. Neurochem Res 31:287–295. [DOI] [PubMed] [Google Scholar]
- Appel NM, Robbins JD, De Souza EB, Seamon KB. (1992) [125I]-labeled forskolin analogs which discriminate adenylyl cyclase and a glucose transporter: pharmacological characterization and localization of binding sites in rat brain by in vitro receptor autoradiography. J Pharmacol Exp Ther 263:1415–1423. [PubMed] [Google Scholar]
- Artymiuk PJ, Poirrette AR, Rice DW, Willett P. (1997) A polymerase I palm in adenylyl cyclase? Nature 388:33–34. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähre H, Danker KY, Stasch JP, Kaever V, Seifert R. (2014) Nucleotidyl cyclase activity of soluble guanylyl cyclase in intact cells. Biochem Biophys Res Commun 443:1195–1199. [DOI] [PubMed] [Google Scholar]
- Barker A, Sharp SJ, Timpson NJ, Bouatia-Naji N, Warrington NM, Kanoni S, Beilin LJ, Brage S, Deloukas P, Evans DM, et al. (2011) Association of genetic Loci with glucose levels in childhood and adolescence: a meta-analysis of over 6,000 children. Diabetes 60:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudouin-Legros M, Hamdaoui N, Borot F, Fritsch J, Ollero M, Planelles G, Edelman A. (2008) Control of basal CFTR gene expression by bicarbonate-sensitive adenylyl cyclase in human pulmonary cells. Cell Physiol Biochem 21:75–86. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, et al. (2006) Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell 23:925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayewitch ML, Avidor-Reiss T, Levy R, Pfeuffer T, Nevo I, Simonds WF, Vogel Z. (1998) Inhibition of adenylyl cyclase isoforms V and VI by various Gbetagamma subunits. FASEB J 12:1019–1025. [DOI] [PubMed] [Google Scholar]
- Beazely MA, Watts VJ. (2006) Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol 535:1–12. [DOI] [PubMed] [Google Scholar]
- Beltz S, Bassler J, Schultz JE. (2016) Regulation by the quorum sensor from Vibrio indicates a receptor function for the membrane anchors of adenylate cyclases. eLife 5:e13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520. [DOI] [PubMed] [Google Scholar]
- Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, Justice AE, Monda KL, Croteau-Chonka DC, Day FR, et al. (2013) Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet 45:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste KY, Burhenne H, Kaever V, Stasch JP, Seifert R. (2012) Nucleotidyl cyclase activity of soluble guanylyl cyclase α1β1. Biochemistry 51:194–204. [DOI] [PubMed] [Google Scholar]
- Beste KY, Spangler CM, Burhenne H, Koch KW, Shen Y, Tang WJ, Kaever V, Seifert R. (2013) Nucleotidyl cyclase activity of particulate guanylyl cyclase A: comparison with particulate guanylyl cyclases E and F, soluble guanylyl cyclase and bacterial adenylyl cyclases CyaA and edema factor. PLoS One 8:e70223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow DJ, Squier TC. (2005) Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta 1703:121–134. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Abramowitz J, Brown AM. (1990) Receptor-effector coupling by G proteins. Biochim Biophys Acta 1031:163–224. [DOI] [PubMed] [Google Scholar]
- Bitterman JL, Ramos-Espiritu L, Diaz A, Levin LR, Buck J. (2013) Pharmacological distinction between soluble and transmembrane adenylyl cyclases. J Pharmacol Exp Ther 347:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogard AS, Adris P, Ostrom RS. (2012) Adenylyl cyclase 2 selectively couples to E prostanoid type 2 receptors, whereas adenylyl cyclase 3 is not receptor-regulated in airway smooth muscle. J Pharmacol Exp Ther 342:586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogard AS, Birg AV, Ostrom RS. (2014) Non-raft adenylyl cyclase 2 defines a cAMP signaling compartment that selectively regulates IL-6 expression in airway smooth muscle cells: differential regulation of gene expression by AC isoforms. Naunyn Schmiedebergs Arch Pharmacol 387:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogard AS, Xu C, Ostrom RS. (2011) Human bronchial smooth muscle cells express adenylyl cyclase isoforms 2, 4, and 6 in distinct membrane microdomains. J Pharmacol Exp Ther 337:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böl GF, Hülster A, Pfeuffer T. (1997) Adenylyl cyclase type II is stimulated by PKC via C-terminal phosphorylation. Biochim Biophys Acta 1358:307–313. [DOI] [PubMed] [Google Scholar]
- Braeunig JH, Albrecht-Küpper B, Seifert R. (2014) Adenylyl cyclase regulation in heart failure due to myocardial infarction in rats. Naunyn Schmiedebergs Arch Pharmacol 387:389–398. [DOI] [PubMed] [Google Scholar]
- Braeunig JH, Schweda F, Han PL, Seifert R. (2013) Similarly potent inhibition of adenylyl cyclase by P-site inhibitors in hearts from wild type and AC5 knockout mice. PLoS One 8:e68009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand CS, Hocker HJ, Gorfe AA, Cavasotto CN, Dessauer CW. (2013) Isoform selectivity of adenylyl cyclase inhibitors: characterization of known and novel compounds. J Pharmacol Exp Ther 347:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand CS, Sadana R, Malik S, Smrcka AV, Dessauer CW. (2015) Adenylyl cyclase 5 regulation by Gβγ involves isoform-specific use of multiple interaction sites. Mol Pharmacol 88:758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T. (1991) Purification of soluble form of adenylyl cyclase from testes. Methods Enzymol 195:130–136. [DOI] [PubMed] [Google Scholar]
- Braun T, Dods RF. (1975) Development of a Mn-2+-sensitive, “soluble” adenylate cyclase in rat testis. Proc Natl Acad Sci USA 72:1097–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo CA, Vatner DE, Pachon R, Zhang J, Vatner SF. (2016) A Food and Drug Administration-approved antiviral agent that inhibits adenylyl cyclase type 5 protects the ischemic heart even when administered after reperfusion. J Pharmacol Exp Ther 357:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskole Hummel I, Reinartz MT, Kälble S, Burhenne H, Schwede F, Buschauer A, Seifert R. (2013) Dissociations in the effects of β2-adrenergic receptor agonists on cAMP formation and superoxide production in human neutrophils: support for the concept of functional selectivity. PLoS One 8:e64556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust TF, Conley JM, Watts VJ. (2015) Gα(i/o)-coupled receptor-mediated sensitization of adenylyl cyclase: 40 years later. Eur J Pharmacol 763 (Pt B):223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. (1999) Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA 96:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali JJ, Zwaagstra JC, Mons N, Cooper DM, Krupinski J. (1994) Type VIII adenylyl cyclase. A Ca2+/calmodulin-stimulated enzyme expressed in discrete regions of rat brain. J Biol Chem 269:12190–12195. [PubMed] [Google Scholar]
- Carapito R, Paul N, Untrau M, Le Gentil M, Ott L, Alsaleh G, Jochem P, Radosavljevic M, Le Caignec C, David A, et al. (2015) A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov Disord 30:423–427. [DOI] [PubMed] [Google Scholar]
- Carvalho AF, Köhler CA, Brunoni AR, Miskowiak KW, Herrmann N, Lanctôt KL, Hyphantis TN, Quevedo J, Fernandes BS, Berk M. (2016) Bias in peripheral depression biomarkers. Psychother Psychosom 85:81–90. [DOI] [PubMed] [Google Scholar]
- Cassel D, Selinger Z. (1977) Mechanism of adenylate cyclase activation by cholera toxin: inhibition of GTP hydrolysis at the regulatory site. Proc Natl Acad Sci USA 74:3307–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Gabay M, Wright FA, Kan W, Oner SS, Lanier SM, Smrcka AV, Blumer JB, Tall GG. (2011) Purification of heterotrimeric G protein alpha subunits by GST-Ric-8 association: primary characterization of purified G alpha(olf). J Biol Chem 286:2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang FCF, Westenberger A, Dale RC, Smith M, Pall HS, Perez-Dueñas B, Grattan-Smith P, Ouvrier RA, Mahant N, Hanna BC, et al. (2016) Phenotypic insights into ADCY5-associated disease. Mov Disord 31:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Méneret A, Friedman JR, Korvatska O, Gad A, Bonkowski ES, Stessman HA, Doummar D, Mignot C, Anheim M, et al. (2015b) ADCY5-related dyskinesia. Broader spectrum and genotype-penotype correlations. Neurology 85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, DeVivo M, Dingus J, Harry A, Li J, Sui J, Carty DJ, Blank JL, Exton JH, Stoffel RH, et al. (1995) A region of adenylyl cyclase 2 critical for regulation by G protein β γ subunits. Science 268:1166–1169. [DOI] [PubMed] [Google Scholar]
- Chen J, Iyengar R. (1993) Inhibition of cloned adenylyl cyclases by mutant-activated Gi-α and specific suppression of type 2 adenylyl cyclase inhibition by phorbol ester treatment. J Biol Chem 268:12253–12256. [PubMed] [Google Scholar]
- Chen J, Levin LR, Buck J. (2012a) Role of soluble adenylyl cyclase in the heart. Am J Physiol Heart Circ Physiol 302:H538–H543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Martinez J, Milner TA, Buck J, Levin LR. (2013a) Neuronal expression of soluble adenylyl cyclase in the mammalian brain. Brain Res 1518:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Eltzschig HK, Fredholm BB. (2013b) Adenosine receptors as drug targets--what are the challenges? Nat Rev Drug Discov 12:265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Baumlin N, Buck J, Levin LR, Fregien N, Salathe M. (2014a) A soluble adenylyl cyclase form targets to axonemes and rescues beat regulation in soluble adenylyl cyclase knockout mice. Am J Respir Cell Mol Biol 51:750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cao H, Saraf A, Zweifel LS, Storm DR. (2015a) Overexpression of the type 1 adenylyl cyclase in the forebrain leads to deficits of behavioral inhibition. J Neurosci 35:339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Luo J, Leng Y, Yang Y, Zweifel LS, Palmiter RD, Storm DR. (2016) Ablation of type III adenylyl cyclase in mice causes reduced neuronal activity, altered sleep pattern, and depression-like phenotypes. Biol Psychiatry 80:836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xia Z, Storm DR. (2012b) Stimulation of electro-olfactogram responses in the main olfactory epithelia by airflow depends on the type 3 adenylyl cyclase. J Neurosci 32:15769–15778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. (2000) Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 289:625–628. [DOI] [PubMed] [Google Scholar]
- Chen Y, Harry A, Li J, Smit MJ, Bai X, Magnusson R, Pieroni JP, Weng G, Iyengar R. (1997) Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Gαs stimulation. Proc Natl Acad Sci USA 94:14100–14104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Friedman JR, Chen DH, Chan GCK, Bloss CS, Hisama FM, Topol SE, Carson AR, Pham PH, Bonkowski ES, et al. (2014b) Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, Lipe H, Eichler EE, Nickerson DA, Bird TD, et al. (2012c) Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CL, Lin MS, Lai HL, Wu YS, Chang CP, Chen HM, Chang C, Su CK, Chern Y. (2013) Lack of type VI adenylyl cyclase (AC6) leads to abnormal sympathetic tone in neonatal mice. Exp Neurol 248:10–15. [DOI] [PubMed] [Google Scholar]
- Chien CL, Wu YS, Lai HL, Chen YH, Jiang ST, Shih CM, Lin SS, Chang C, Chern Y. (2010) Impaired water reabsorption in mice deficient in the type VI adenylyl cyclase (AC6). FEBS Lett 584:2883–2890. [DOI] [PubMed] [Google Scholar]
- Cho KH, Du Y, Scull NJ, Hariharan P, Gotfryd K, Loland CJ, Guan L, Byrne B, Kobilka BK, Chae PS. (2015) Novel xylene-linked maltoside amphiphiles (XMAs) for membrane protein stabilisation. Chemistry 21:10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EJ, Xia Z, Storm DR. (1992) Stimulation of the type III olfactory adenylyl cyclase by calcium and calmodulin. Biochemistry 31:6492–6498. [DOI] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, et al. (2012) Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75:1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, Igarashi P. (2011) Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci USA 108:10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Col JA, Matsuo T, Storm DR, Rodriguez I. (2007) Adenylyl cyclase-dependent axonal targeting in the olfactory system. Development 134:2481–2489. [DOI] [PubMed] [Google Scholar]
- Colledge M, Scott JD. (1999) AKAPs: from structure to function. Trends Cell Biol 9:216–221. [DOI] [PubMed] [Google Scholar]
- Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. (1993) Substitution of three amino acids switches receptor specificity of Gqa to that of Gia. Nature 363:274–276. [DOI] [PubMed] [Google Scholar]
- Conley JM, Brand CS, Bogard AS, Pratt EP, Xu R, Hockerman GH, Ostrom RS, Dessauer CW, Watts VJ. (2013) Development of a high-throughput screening paradigm for the discovery of small-molecule modulators of adenylyl cyclase: identification of an adenylyl cyclase 2 inhibitor. J Pharmacol Exp Ther 347:276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M, Beavo J. (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511. [DOI] [PubMed] [Google Scholar]
- Cooper DMF. (1998) Adenylyl Cyclases. Advances in Second Messenger and Phosphoprotein Research, Lippincott-Raven, New York. [PubMed] [Google Scholar]
- Cooper DM. (2003) Regulation and organization of adenylyl cyclases and cAMP. Biochem J 375:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Crossthwaite AJ. (2006) Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci 27:426–431. [DOI] [PubMed] [Google Scholar]
- Cooper DM, Tabbasum VG. (2014) Adenylate cyclase-centred microdomains. Biochem J 462:199–213. [DOI] [PubMed] [Google Scholar]
- Cooper DMF, Mons N, Karpen JW. (1995) Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature 374:421–424. [DOI] [PubMed] [Google Scholar]
- Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG, Russel FG, Shayo C, Davio C. (2011) Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem 286:6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros C, Brette F. (2013) Functional subcellular distribution of β1- and β2-adrenergic receptors in rat ventricular cardiac myocytes. Physiol Rep 1:e00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossthwaite AJ, Seebacher T, Masada N, Ciruela A, Dufraux K, Schultz JE, Cooper DM. (2005) The cytosolic domains of Ca2+-sensitive adenylyl cyclases dictate their targeting to plasma membrane lipid rafts. J Biol Chem 280:6380–6391. [DOI] [PubMed] [Google Scholar]
- Cumbay MG, Watts VJ. (2001) Heterologous sensitization of recombinant adenylate cyclases by activation of D(2) dopamine receptors. J Pharmacol Exp Ther 297:1201–1209. [PubMed] [Google Scholar]
- Cumbay MG, Watts VJ. (2004) Novel regulatory properties of human type 9 adenylate cyclase. J Pharmacol Exp Ther 310:108–115. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390:88–91. [DOI] [PubMed] [Google Scholar]
- Daly JW. (1984) Forskolin, adenylate cyclase, and cell physiology: an overview. Adv Cyclic Nucleotide Protein Phosphorylation Res 17:81–89. [PubMed] [Google Scholar]
- Daniel WA. (2003) Mechanisms of cellular distribution of psychotropic drug action and interactions. Prog Neuropsychopharmacol Biol Psychiatry 27:65–73. [DOI] [PubMed] [Google Scholar]
- Dayan G, Jault JM, Baubichon-Cortay H, Baggetto LG, Renoir JM, Baulieu EE, Gros P, Di Pietro A. (1997) Binding of steroid modulators to recombinant cytosolic domain from mouse P-glycoprotein in close proximity to the ATP site. Biochemistry 36:15208–15215. [DOI] [PubMed] [Google Scholar]
- de Mooij-van Malsen AJ, van Lith HA, Oppelaar H, Hendriks J, de Wit M, Kostrzewa E, Breen G, Collier DA, Olivier B, Kas MJ. (2009) Interspecies trait genetics reveals association of Adcy8 with mouse avoidance behavior and a human mood disorder. Biol Psychiatry 66:1123–1130. [DOI] [PubMed] [Google Scholar]
- De Rasmo D, Signorile A, Santeramo A, Larizza M, Lattanzio P, Capitanio G, Papa S. (2015) Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim Biophys Acta 1853:183–191. [DOI] [PubMed] [Google Scholar]
- Defer N, Best-Belpomme M, Hanoune J. (2000) Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol Renal Physiol 279:F400–F416. [DOI] [PubMed] [Google Scholar]
- Delint-Ramirez I, Willoughby D, Hammond GR, Ayling LJ, Cooper DM. (2011) Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J Biol Chem 286:32962–32975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Désaubry L, Johnson RA. (1998) Adenine nucleoside 3′-tetraphosphates are novel and potent inhibitors of adenylyl cyclases. J Biol Chem 273:24972–24977. [DOI] [PubMed] [Google Scholar]
- Désaubry L, Shoshani I, Johnson RA. (1996) 2′,5′-Dideoxyadenosine 3′-polyphosphates are potent inhibitors of adenylyl cyclases. J Biol Chem 271:2380–2382. [DOI] [PubMed] [Google Scholar]
- Desrivières S, Pronko SP, Lourdusamy A, Ducci F, Hoffman PL, Wodarz N, Ridinger M, Rietschel M, Zelenika D, Lathrop M, et al. (2011) Sex-specific role for adenylyl cyclase type 7 in alcohol dependence. Biol Psychiatry 69:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessauer CW. (2009) Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol 76:935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessauer CW, Chen-Goodspeed M, Chen J. (2002) Mechanism of Gα i-mediated inhibition of type V adenylyl cyclase. J Biol Chem 277:28823–28829. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Gilman AG. (1997) The catalytic mechanism of mammalian adenylyl cyclase. Equilibrium binding and kinetic analysis of P-site inhibition. J Biol Chem 272:27787–27795. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Scully TT, Gilman AG. (1997) Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem 272:22272–22277. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. (1998) Identification of a Giα binding site on type V adenylyl cyclase. J Biol Chem 273:25831–25839. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. (1999) The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol Sci 20:205–210. [DOI] [PubMed] [Google Scholar]
- Dey S, Roy D, Majumder GC, Bhattacharyya D. (2014) Extracellular regulation of sperm transmembrane adenylyl cyclase by a forward motility stimulating protein. PLoS One 9:e110669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhande OS, Bhatt S, Anishchenko A, Elstrott J, Iwasato T, Swindell EC, Xu HP, Jamrich M, Itohara S, Feller MB, et al. (2012) Role of adenylate cyclase 1 in retinofugal map development. J Comp Neurol 520:1562–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. (2016) Dabrafenib plus trametinib: a review in advanced melanoma with BRAF (V600) mutation. Target Oncol 11:417–428. [DOI] [PubMed] [Google Scholar]
- Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. (2013) Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab 17:965–975. [DOI] [PubMed] [Google Scholar]
- Diel S, Beyermann M, Lloréns JM, Wittig B, Kleuss C. (2008) Two interaction sites on mammalian adenylyl cyclase type I and II: modulation by calmodulin and G(betagamma). Biochem J 411:449–456. [DOI] [PubMed] [Google Scholar]
- Diel S, Klass K, Wittig B, Kleuss C. (2006) Gbg activation site in adenylyl cyclase type II. Adenylyl cyclase type III is inhibited by Gbg. J Biol Chem 281:288–294. [DOI] [PubMed] [Google Scholar]
- Ding Q, Gros R, Gray ID, Taussig R, Ferguson SS, Feldman RD. (2004) Raf kinase activation of adenylyl cyclases: isoform-selective regulation. Mol Pharmacol 66:921–928. [PubMed] [Google Scholar]
- Ding X, Staudinger JL. (2005) Induction of drug metabolism by forskolin: the role of the pregnane X receptor and the protein kinase a signal transduction pathway. J Pharmacol Exp Ther 312:849–856. [DOI] [PubMed] [Google Scholar]
- DiRocco DP, Scheiner ZS, Sindreu CB, Chan GC, Storm DR. (2009) A role for calmodulin-stimulated adenylyl cyclases in cocaine sensitization. J Neurosci 29:2393–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokphrom U, Qualls-Creekmore E, Yoshimura M. (2011) Effects of alcohols on recombinant adenylyl cyclase type 7 expressed in bacteria. Alcohol Clin Exp Res 35:1915–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlea JM, Ramanan N, Shaw PJ. (2009) Use-dependent plasticity in clock neurons regulates sleep need in Drosophila. Science 324:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove S, Danker KY, Stasch JP, Kaever V, Seifert R. (2014) Structure/activity relationships of (M)ANT- and TNP-nucleotides for inhibition of rat soluble guanylyl cyclase α1β1. Mol Pharmacol 85:598–607. [DOI] [PubMed] [Google Scholar]
- Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D, Jiang LI. (2010) Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol 185:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Dessauer CW. (2011) A kinase-anchoring proteins and adenylyl cyclase in cardiovascular physiology and pathology. J Cardiovasc Pharmacol 58:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW. (2010) AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem 285:14450–14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich AT, Furuyashiki T, Kitaoka S, Kakizuka A, Narumiya S. (2013) Prostaglandin E receptor EP1 forms a complex with dopamine D1 receptor and directs D1-induced cAMP production to adenylyl cyclase 7 through mobilizing G(βγ) subunits in human embryonic kidney 293T cells. Mol Pharmacol 84:476–486. [DOI] [PubMed] [Google Scholar]
- El-Haroun H, Bradbury D, Clayton A, Knox AJ. (2004) Interleukin-1β, transforming growth factor-β1, and bradykinin attenuate cyclic AMP production by human pulmonary artery smooth muscle cells in response to prostacyclin analogues and prostaglandin E2 by cyclooxygenase-2 induction and downregulation of adenylyl cyclase isoforms 1, 2, and 4. Circ Res 94:353–361. [DOI] [PubMed] [Google Scholar]
- Emery AC, Eiden MV, Eiden LE. (2013) A new site and mechanism of action for the widely used adenylate cyclase inhibitor SQ22,536. Mol Pharmacol 83:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Liu XH, Xu W, Eiden MV, Eiden LE. (2015) Cyclic adenosine 3′,5′-monophosphate elevation and biological signaling through a secretin family Gs-coupled G protein-coupled receptor are restricted to a single adenylate cyclase isoform. Mol Pharmacol 87:928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdorf M, Mou TC, Seifert R. (2011) Impact of divalent metal ions on regulation of adenylyl cyclase isoforms by forskolin analogs. Biochem Pharmacol 82:1673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdorf M, Seifert R. (2011) Pharmacological characterization of adenylyl cyclase isoforms in rabbit kidney membranes. Naunyn Schmiedebergs Arch Pharmacol 383:357–372. [DOI] [PubMed] [Google Scholar]
- Espinasse I, Iourgenko V, Defer N, Samson F, Hanoune J, Mercadier JJ. (1995) Type V, but not type VI, adenylyl cyclase mRNA accumulates in the rat heart during ontogenic development. Correlation with increased global adenylyl cyclase activity. J Mol Cell Cardiol 27:1789–1795. [DOI] [PubMed] [Google Scholar]
- Espinasse I, Iourgenko V, Richer C, Heimburger M, Defer N, Bourin MC, Samson F, Pussard E, Giudicelli JF, Michel JB, et al. (1999) Decreased type VI adenylyl cyclase mRNA concentration and Mg2+-dependent adenylyl cyclase activities and unchanged type V adenylyl cyclase mRNA concentration and Mn(2+)-dependent adenylyl cyclase activities in the left ventricle of rats with myocardial infarction and longstanding heart failure. Cardiovasc Res 42:87–98. [DOI] [PubMed] [Google Scholar]
- Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, van Duin M, Conti M, et al. (2004) Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA 101:2993–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler CE, Jackson T, Elliott LE, De Castro LM, Jonassaint J, Ashley-Koch A, Telen MJ. (2008) β(2)-Adrenergic receptor and adenylate cyclase gene polymorphisms affect sickle red cell adhesion. Br J Haematol 141:105–108. [DOI] [PubMed] [Google Scholar]
- Fagan KA, Mons N, Cooper DMF. (1998) Dependence of the Ca2+-inhibitable adenylyl cyclase of C6-2B glioma cells on capacitative Ca2+ entry. J Biol Chem 273:9297–9305. [DOI] [PubMed] [Google Scholar]
- Fagan KA, Smith KE, Cooper DM. (2000) Regulation of the Ca2+-inhibitable adenylyl cyclase type VI by capacitative Ca2+ entry requires localization in cholesterol-rich domains. J Biol Chem 275:26530–26537. [DOI] [PubMed] [Google Scholar]
- Fan P, Jiang Z, Diamond I, Yao L. (2009) Up-regulation of AGS3 during morphine withdrawal promotes cAMP superactivation via adenylyl cyclase 5 and 7 in rat nucleus accumbens/striatal neurons. Mol Pharmacol 76:526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federman AD, Conklin BR, Schrader KA, Reed RR, Bourne HR. (1992) Hormonal stimulation of adenylyl cyclase through Gi-protein β γ subunits. Nature 356:159–161. [DOI] [PubMed] [Google Scholar]
- Ferguson GD, Storm DR. (2004) Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda) 19:271–276. [DOI] [PubMed] [Google Scholar]
- Francis SH, Corbin JD. (2005) Sildenafil: efficacy, safety, tolerability and mechanism of action in treating erectile dysfunction. Expert Opin Drug Metab Toxicol 1:283–293. [DOI] [PubMed] [Google Scholar]
- Francis SH, Sekhar KR, Ke H, Corbin JD. (2011) Inhibition of cyclic nucleotide phosphodiesterases by methylxanthines and related compounds. Handb Exp Pharmacol 200:93–133. [DOI] [PubMed] [Google Scholar]
- Franks-Skiba K, Cooke R. (1995) The conformation of the active site of myosin probed using mant-nucleotides. Biophys J 68(4, Suppl)142S–149S. [PMC free article] [PubMed] [Google Scholar]
- Freyaldenhoven AM, Gutierrez GE, Lifschitz MD, Katz MS. (1992) Protein kinase C differentially modulates PTH- and PGE2-sensitive adenylate cyclase in osteoblast-like cells. Am J Physiol 262:E87–E95. [DOI] [PubMed] [Google Scholar]
- Friebe A, Sandner P, Seifert R. (2015) From bedside to bench--meeting report of the 7th International Conference on cGMP “cGMP: generators, effectors and therapeutic implications” in Trier, Germany, from June 19th to 21st 2015. Naunyn Schmiedebergs Arch Pharmacol 388:1237–1246. [DOI] [PubMed] [Google Scholar]
- Friedman J, Babu B, Clark RB. (2002) β2-adrenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells: lack of evidence for Gs/Gi switching. Mol Pharmacol 62:1094–1102. [DOI] [PubMed] [Google Scholar]
- Fu R, Ceballos-Picot I, Torres RJ, Larovere LE, Yamada Y, Nguyen KV, Hegde M, Visser JE, Schretlen DJ, Nyhan WL, et al. Lesch-Nyhan Disease International Study Group (2014) Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 137:1282–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gancedo JM. (2013) Biological roles of cAMP: variations on a theme in the different kingdoms of life. Biol Rev Camb Philos Soc 88:645–668. [DOI] [PubMed] [Google Scholar]
- Gao BN, Gilman AG. (1991) Cloning and expression of a widely distributed (type IV) adenylyl cyclase. Proc Natl Acad Sci USA 88:10178–10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK. (1998) Increased expression of adenylylcyclase type VI proportionately increases β-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci USA 95:1038–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Sadana R, Dessauer CW, Patel TB. (2007) Conditional stimulation of type V and VI adenylyl cyclases by G protein betagamma subunits. J Biol Chem 282:294–302. [DOI] [PubMed] [Google Scholar]
- Garelick MG, Chan GC, DiRocco DP, Storm DR. (2009) Overexpression of type I adenylyl cyclase in the forebrain impairs spatial memory in aged but not young mice. J Neurosci 29:10835–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnock-Jones KP. (2015) Roflumilast: A review in COPD. Drugs 75:1645–1656. [DOI] [PubMed] [Google Scholar]
- Geduhn J, Dove S, Shen Y, Tang WJ, König B, Seifert R. (2011) Bis-halogen-anthraniloyl-substituted nucleoside 5′-triphosphates as potent and selective inhibitors of Bordetella pertussis adenylyl cyclase toxin. J Pharmacol Exp Ther 336:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW. (2005) Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol 288:C1305–C1316. [DOI] [PubMed] [Google Scholar]
- Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK. (1997) Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem 272:2587–2590. [DOI] [PubMed] [Google Scholar]
- Gille A, Seifert R. (2003) 2′(3′)-O-(N-methylanthraniloyl)-substituted GTP analogs: a novel class of potent competitive adenylyl cyclase inhibitors. J Biol Chem 278:12672–12679. [DOI] [PubMed] [Google Scholar]
- Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, Seifert R. (2004) Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem 279:19955–19969. [DOI] [PubMed] [Google Scholar]
- Gilman AG. (1987) G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56:615–649. [DOI] [PubMed] [Google Scholar]
- Godard MP, Johnson BA, Richmond SR. (2005) Body composition and hormonal adaptations associated with forskolin consumption in overweight and obese men. Obes Res 13:1335–1343. [DOI] [PubMed] [Google Scholar]
- Gong B, Wang H, Gu S, Heximer SP, Zhuo M. (2007) Genetic evidence for the requirement of adenylyl cyclase 1 in synaptic scaling of forebrain cortical neurons. Eur J Neurosci 26:275–288. [DOI] [PubMed] [Google Scholar]
- Gordeladze JO, Hansson V. (1981) Purification and kinetic properties of the soluble Mn2+-dependent adenylyl cyclase of the rat testis. Mol Cell Endocrinol 23:125–136. [DOI] [PubMed] [Google Scholar]
- Gordeladze JO, Abyholm T, Cusan L, Clausen OP, Hansson V. (1982) Cellular localization of the Mn2+-dependent adenylyl cyclase in the human testis. Arch Androl 8:199–204. [DOI] [PubMed] [Google Scholar]
- Göttle M, Geduhn J, König B, Gille A, Höcherl K, Seifert R. (2009) Characterization of mouse heart adenylyl cyclase. J Pharmacol Exp Ther 329:1156–1165. [DOI] [PubMed] [Google Scholar]
- Gros R, Van Uum S, Hutchinson-Jaffe A, Ding Q, Pickering JG, Hegele RA, Feldman RD. (2007) Increased enzyme activity and beta-adrenergic mediated vasodilation in subjects expressing a single-nucleotide variant of human adenylyl cyclase 6. Arterioscler Thromb Vasc Biol 27:2657–2663. [DOI] [PubMed] [Google Scholar]
- Guellich A, Gao S, Hong C, Yan L, Wagner TE, Dhar SK, Ghaleh B, Hittinger L, Iwatsubo K, Ishikawa Y, et al. (2010) Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am J Physiol Heart Circ Physiol 299:H707–H712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guellich A, Mehel H, Fischmeister R. (2014) Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch 466:1163–1175. [DOI] [PubMed] [Google Scholar]
- Guerreiro S, Marien M, Michel PP. (2011) Methylxanthines and ryanodine receptor channels. Handb Exp Pharmacol 200:135–150. [DOI] [PubMed] [Google Scholar]
- Hahn DK, Tusell JR, Sprang SR, Chu X. (2015) Catalytic mechanism of mammalian adenylyl cyclase: a computational investigation. Biochemistry 54:6252–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoune J, Defer N. (2001) Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41:145–174. [DOI] [PubMed] [Google Scholar]
- Hardin M, Zielinski J, Wan ES, Hersh CP, Castaldi PJ, Schwinder E, Hawrylkiewicz I, Sliwinski P, Cho MH, Silverman EK. (2012) CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am J Respir Cell Mol Biol 47:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman JG, Robison GA, Sutherland EW. (1971) Cyclic nucleotides. Annu Rev Physiol 33:311–336. [DOI] [PubMed] [Google Scholar]
- Harwood AJ, Agam G. (2003) Search for a common mechanism of mood stabilizers. Biochem Pharmacol 66:179–189. [DOI] [PubMed] [Google Scholar]
- Hasan A, Danker KY, Wolter S, Bähre H, Kaever V, Seifert R. (2014) Soluble adenylyl cyclase accounts for high basal cCMP and cUMP concentrations in HEK293 and B103 cells. Biochem Biophys Res Commun 448:236–240. [DOI] [PubMed] [Google Scholar]
- Haunsø A, Simpson J, Antoni FA. (2003) Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol Pharmacol 63:624–631. [DOI] [PubMed] [Google Scholar]
- Heckman PR, Wouters C, Prickaerts J. (2015) Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: a translational overview. Curr Pharm Des 21:317–331. [DOI] [PubMed] [Google Scholar]
- Heinick A, Husser X, Himmler K, Kirchhefer U, Nunes F, Schulte JS, Seidl MD, Rolfes C, Dedman JR, Kaetzel MA, et al. (2015) Annexin A4 is a novel direct regulator of adenylyl cyclase type 5. FASEB J 29:3773–3787. [DOI] [PubMed] [Google Scholar]
- Hellevuo K, Welborn R, Menninger JA, Tabakoff B. (1997) Human adenylyl cyclase type 7 contains polymorphic repeats in the 3′ untranslated region: investigations of association with alcoholism. Am J Med Genet 74:95–98. [DOI] [PubMed] [Google Scholar]
- Hellevuo K, Yoshimura M, Kao M, Hoffman PL, Cooper DMF, Tabakoff B. (1993) A novel adenylyl cyclase sequence cloned from the human erythroleukemia cell line. Biochem Biophys Res Commun 192:311–318. [DOI] [PubMed] [Google Scholar]
- Herrinton LJ, Curtis JR, Chen L, Liu L, Delzell E, Lewis JD, Solomon DH, Griffin MR, Ouellet-Hellstom R, Beukelman T, et al. (2011) Study design for a comprehensive assessment of biologic safety using multiple healthcare data systems. Pharmacoepidemiol Drug Saf 20:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt JD, Hanoune J, Birnbaumer L. (1982) Guanine nucleotide inhibition of cyc- S49 mouse lymphoma cell membrane adenylyl cyclase. J Biol Chem 257:14723–14725. [PubMed] [Google Scholar]
- Hill J, Howlett A, Klein C. (2000) Nitric oxide selectively inhibits adenylyl cyclase isoforms 5 and 6. Cell Signal 12:233–237. [DOI] [PubMed] [Google Scholar]
- Hines LM, Hoffman PL, Bhave S, Saba L, Kaiser A, Snell L, Goncharov I, LeGault L, Dongier M, Grant B, et al. World Health Organization/International Society for Biomedical Research on Alcoholism Study on State and Trait Markers of Alcohol Use and Dependence Investigators (2006) A sex-specific role of type VII adenylyl cyclase in depression. J Neurosci 26:12609–12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines LM and Tabakoff B (2005) Platelet: adenylyl cyclase activity: a biological maker for major depression and recent drug use. Biol Psychiatry 58:955–962. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T. (2003) Fluorescent and colored trinitrophenylated analogs of ATP and GTP. Eur J Biochem 270:3479–3485. [DOI] [PubMed] [Google Scholar]
- Ho D, Zhao X, Yan L, Yuan C, Zong H, Vatner DE, Pessin JE, Vatner SF. (2015) Adenylyl cyclase type 5 deficiency protects against diet-induced obesity and insulin resistance. Diabetes 64:2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson DJ, Mitchell RK, Marselli L, Pullen TJ, Gimeno Brias S, Semplici F, Everett KL, Cooper DM, Bugliani M, Marchetti P, et al. (2014) ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63:3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PL, Tabakoff B. (1982) Effects of ethanol on Arrhenius parameters and activity of mouse striatal adenylate cyclase. Biochem Pharmacol 31:3101–3106. [DOI] [PubMed] [Google Scholar]
- Holstein JD, Patzer O, Körner A, Stumvoll M, Kovacs P, Holstein A. (2013) Genetic variants in GCKR, GIPR, ADCY5 and VPS13C and the risk of severe sulfonylurea-induced hypoglycaemia in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes 121:54–57. [DOI] [PubMed] [Google Scholar]
- Horner K, Livera G, Hinckley M, Trinh K, Storm D, Conti M. (2003) Rodent oocytes express an active adenylyl cyclase required for meiotic arrest. Dev Biol 258:385–396. [DOI] [PubMed] [Google Scholar]
- Hosono M. (1999) [Cardiovascular effects of colforsin daropate hydrochloride, a novel drug for the treatment of acute heart failure]. Nihon Yakurigaku Zasshi 114:83–88. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Milligan G. (1997) Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem Sci 22:217–224. [DOI] [PubMed] [Google Scholar]
- Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, Depre C, Iwatsubo K, Ishikawa Y, Sadoshima J, et al. (2009) Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol 297:H1776–H1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, Zhang GM, Feng ZH. (2009) miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep 10:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner M, Dixit A, Mou TC, Lushington GH, Pinto C, Gille A, Geduhn J, König B, Sprang SR, Seifert R. (2011a) Structural basis for the high-affinity inhibition of mammalian membranous adenylyl cyclase by 2′,3′-o-(N-methylanthraniloyl)-inosine 5′-triphosphate. Mol Pharmacol 80:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner M, Dizayee S, Matthes J, Seifert R, Herzig S. (2011b) Effect of MANT-nucleotides on L-type calcium currents in murine cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol 383:573–583. [DOI] [PubMed] [Google Scholar]
- Hulur I, Gamazon ER, Skol AD, Xicola RM, Llor X, Onel K, Ellis NA, Kupfer SS. (2015) Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genomics 16:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt NH, Evans T. (1980) RMI 12330A, an inhibitor of cyclic nucleotide phosphodiesterases and adenylate cyclase in kidney preparations. Biochim Biophys Acta 613:499–506. [DOI] [PubMed] [Google Scholar]
- Ikoma E, Tsunematsu T, Nakazawa I, Shiwa T, Hibi K, Ebina T, Mochida Y, Toya Y, Hori H, Uchino K, et al. (2003) Polymorphism of the type 6 adenylyl cyclase gene and cardiac hypertrophy. J Cardiovasc Pharmacol 42 (Suppl 1):S27–S32. [DOI] [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. (2003) Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol 23:305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y. (1995) Regulation of adenylyl cyclase by protein kinase A. J Biol Chem 270:12481–12484. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Okumura S, Iwatsubo K, Kawabe J, Ohtsu K, Sakai I, Hashimoto Y, Izumitani A, Sango K, Ajiki K, et al. (2003) Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem 278:16936–16940. [DOI] [PubMed] [Google Scholar]
- Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, Umemura S, Scarborough RM, Levy DE, Ishikawa Y. (2004) Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem 279:40938–40945. [DOI] [PubMed] [Google Scholar]
- Iyengar R. (1993) Molecular and functional diversity of mammalian Gs-stimulated adenylyl cyclases. FASEB J 7:768–775. [DOI] [PubMed] [Google Scholar]
- Iyengar R, Hildebrandt JD. (2002) G protein pathways: Effector mechanisms, Part 3. Methods Enzymol 345, Acadamic Press, San Diego, CA. [Google Scholar]
- Jacobowitz O, Chen J, Iyengar R. (1994) Transient expression assays for mammalian adenylyl cyclases. Methods Enzymol 238:108–116. [DOI] [PubMed] [Google Scholar]
- Jacobowitz O, Chen J, Premont RT, Iyengar R. (1993) Stimulation of specific types of Gs-stimulated adenylyl cyclases by phorbol ester treatment. J Biol Chem 268:3829–3832. [PubMed] [Google Scholar]
- Jacobowitz O, Iyengar R. (1994) Phorbol ester-induced stimulation and phosphorylation of adenylyl cyclase 2. Proc Natl Acad Sci USA 91:10630–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs KH, Aktories K, Schultz G. (1983a) A nucleotide regulatory site for somatostatin inhibition of adenylate cyclase in S49 lymphoma cells. Nature 303:177–178. [DOI] [PubMed] [Google Scholar]
- Jakobs KH, Gehring U, Gaugler B, Pfeuffer T, Schultz G. (1983b) Occurrence of an inhibitory guanine nucleotide-binding regulatory component of the adenylate cyclase system in cyc- variants of S49 lymphoma cells. Eur J Biochem 130:605–611. [DOI] [PubMed] [Google Scholar]
- Jaiswal BS, Conti M. (2001) Identification and functional analysis of splice variants of the germ cell soluble adenylyl cyclase. J Biol Chem 276:31698–31708. [DOI] [PubMed] [Google Scholar]
- Jaiswal BS, Conti M. (2003) Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc Natl Acad Sci USA 100:10676–10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang LI, Sternweis PC, Wang JE. (2013) Zymosan activates protein kinase A via adenylyl cyclase VII to modulate innate immune responses during inflammation. Mol Immunol 54:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joeyen-Waldorf J, Nikolova YS, Edgar N, Walsh C, Kota R, Lewis DA, Ferrell R, Manuck SB, Hariri AR, Sibille E. (2012) Adenylate cyclase 7 is implicated in the biology of depression and modulation of affective neural circuitry. Biol Psychiatry 71:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Alvarez R, Salomon Y. (1994) Determination of adenylyl cyclase catalytic activity using single and double column procedures. Methods Enzymol 238:31–56. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Désaubry L, Bianchi G, Shoshani I, Lyons E, Jr, Taussig R, Watson PA, Cali JJ, Krupinski J, Pieroni JP, Iyengar R. (1997) Isozyme-dependent sensitivity of adenylyl cyclases to P-site-mediated inhibition by adenine nucleosides and nucleoside 3'-polyphosphates. J Biol Chem 272:8962–8966. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Sutherland EW. (1973) Detergent-dispersed adenylate cyclase from rat brain. Effects of fluoride, cations, and chelators. J Biol Chem 248:5114–5121. [PubMed] [Google Scholar]
- Johnson RA, Yeung SM, Stübner D, Bushfield M, Shoshani I. (1989) Cation and structural requirements for P site-mediated inhibition of adenylate cyclase. Mol Pharmacol 35:681–688. [PubMed] [Google Scholar]
- Johnston CA, Beazely MA, Bilodeau ML, Andrisani O, Watts VJ. (2004) Differentiation-induced alterations in cyclic AMP signaling in the Cath.a differentiated (CAD) neuronal cell line. J Neurochem 88:1497–1508. [DOI] [PubMed] [Google Scholar]
- Jones DT, Masters SB, Bourne HR, Reed RR. (1990) Biochemical characterization of three stimulatory GTP-binding proteins. The large and small forms of Gs and the olfactory-specific G-protein, Golf. J Biol Chem 265:2671–2676. [PubMed] [Google Scholar]
- Joost HG, Habberfield AD, Simpson IA, Laurenza A, Seamon KB. (1988) Activation of adenylate cyclase and inhibition of glucose transport in rat adipocytes by forskolin analogues: structural determinants for distinct sites of action. Mol Pharmacol 33:449–453. [PubMed] [Google Scholar]
- Jourdan KB, Mason NA, Long L, Philips PG, Wilkins MR, Morrell NW. (2001) Characterization of adenylyl cyclase isoforms in rat peripheral pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 280:L1359–L1369. [DOI] [PubMed] [Google Scholar]
- Kannt A, Wieland T. (2016) Managing risks in drug discovery: reproducibility of published findings. Naunyn Schmiedebergs Arch Pharmacol 389:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. (2009) An adenylyl cyclase-mAKAPβ signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem 284:23540–23546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada T, Amano T, Ui M. (1982) Modulation by islet-activating protein of adenylate cyclase activity in C6 glioma cells. J Biol Chem 257:3739–3746. [PubMed] [Google Scholar]
- Katada T, Northup JK, Bokoch GM, Ui M, Gilman AG. (1984) The inhibitory guanine nucleotide-binding regulatory component of adenylate cyclase. Subunit dissociation and guanine nucleotide-dependent hormonal inhibition. J Biol Chem 259:3578–3585. [PubMed] [Google Scholar]
- Katsushika S, Chen L, Kawabe J, Nilakantan R, Halnon NJ, Homcy CJ, Ishikawa Y. (1992) Cloning and characterization of a sixth adenylyl cyclase isoform: types V and VI constitute a subgroup within the mammalian adenylyl cyclase family. Proc Natl Acad Sci USA 89:8774–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe J, Iwami G, Ebina T, Ohno S, Katada T, Ueda Y, Homcy CJ, Ishikawa Y. (1994) Differential activation of adenylyl cyclase by protein kinase C isoenzymes. J Biol Chem 269:16554–16558. [PubMed] [Google Scholar]
- Keppler D. (2011) Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol 201:299–323. [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X. (2009) Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci 29:12115–12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Han PL. (2009) Mice lacking adenylyl cyclase-5 cope badly with repeated restraint stress. J Neurosci Res 87:2983–2993. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim H, Baek IS, Lee KW, Han PL. (2011a) Mice lacking adenylyl cyclase type 5 (AC5) show increased ethanol consumption and reduced ethanol sensitivity. Psychopharmacology (Berl) 215:391–398. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim H, Park SK, Han PL. (2012) The dorsal striatum expressing adenylyl cyclase-5 controls behavioral sensitivity of the righting reflex to high-dose ethanol. Brain Res 1489:27–36. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ye YM, Lee HY, Sin HJ, Park HS. (2011b) Combined pharmacogenetic effect of ADCY9 and ADRB2 gene polymorphisms on the bronchodilator response to inhaled combination therapy. J Clin Pharm Ther 36:399–405. [DOI] [PubMed] [Google Scholar]
- Kinast L, von der Ohe J, Burhenne H, Seifert R. (2012) Impairment of adenylyl cyclase 2 function and expression in hypoxanthine phosphoribosyltransferase-deficient rat B103 neuroblastoma cells as model for Lesch-Nyhan disease: BODIPY-forskolin as pharmacological tool. Naunyn Schmiedebergs Arch Pharmacol 385:671–683. [DOI] [PubMed] [Google Scholar]
- Kittikulsuth W, Stuart D, Kohan DE. (2014a) Adenylyl cyclase 4 does not regulate collecting duct water and sodium handling. Physiol Rep 2:e00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittikulsuth W, Stuart D, Van Hoek AN, Stockand JD, Bugaj V, Mironova E, Blount MA, Kohan DE. (2014b) Lack of an effect of collecting duct-specific deletion of adenylyl cyclase 3 on renal Na+ and water excretion or arterial pressure. Am J Physiol Renal Physiol 306:F597–F607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinboelting S, Diaz A, Moniot S, van den Heuvel J, Weyand M, Levin LR, Buck J, Steegborn C. (2014a) Crystal structures of human soluble adenylyl cyclase reveal mechanisms of catalysis and of its activation through bicarbonate. Proc Natl Acad Sci USA 111:3727–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinboelting S, Ramos-Espiritu L, Buck H, Colis L, van den Heuvel J, Glickman JF, Levin LR, Buck J, Steegborn C. (2016) Bithionol potently inhibits human soluble adenylyl cyclase through binding to the allosteric activator site. J Biol Chem 291:9776–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinboelting S, van den Heuvel J, Steegborn C. (2014b) Structural analysis of human soluble adenylyl cyclase and crystal structures of its nucleotide complexes-implications for cyclase catalysis and evolution. FEBS J 281:4151–4164. [DOI] [PubMed] [Google Scholar]
- Klotz KN, Kachler S. (2016) Inhibitors of membranous adenylyl cyclases with affinity for adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 389:349–352. [DOI] [PubMed] [Google Scholar]
- Knigge A, Klöting N, Schön MR, Dietrich A, Fasshauer M, Gärtner D, Lohmann T, Dreßler M, Stumvoll M, Kovacs P, et al. (2015) ADCY5 gene expression in adipose tissue is related to obesity in men and mice. PLoS One 10:e0120742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. (1988) Chimeric α 2-,β 2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240:1310–1316. [DOI] [PubMed] [Google Scholar]
- Kolodecik TR, Shugrue CA, Thrower EC, Levin LR, Buck J, Gorelick FS. (2012) Activation of soluble adenylyl cyclase protects against secretagogue stimulated zymogen activation in rat pancreaic acinar cells. PLoS One 7:e41320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou J, Yoshimura M. (2007) Isoform-specific enhancement of adenylyl cyclase activity by n-alkanols. Alcohol Clin Exp Res 31:1467–1472. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Coussen F, Bakalyar HA, Tang WJ, Feinstein PG, Orth K, Slaughter C, Reed RR, Gilman AG. (1989) Adenylyl cyclase amino acid sequence: possible channel- or transporter-like structure. Science 244:1558–1564. [DOI] [PubMed] [Google Scholar]
- Kudlacek O, Mitterauer T, Nanoff C, Hohenegger M, Tang WJ, Freissmuth M, Kleuss C. (2001) Inhibition of adenylyl and guanylyl cyclase isoforms by the antiviral drug foscarnet. J Biol Chem 276:3010–3016. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kostin S, Flacke JP, Reusch HP, Ladilov Y. (2009) Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J Biol Chem 284:14760–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HL, Yang TH, Messing RO, Ching YH, Lin SC, Chern Y. (1997) Protein kinase C inhibits adenylyl cyclase type VI activity during desensitization of the A2a-adenosine receptor-mediated cAMP response. J Biol Chem 272:4970–4977. [DOI] [PubMed] [Google Scholar]
- Lai NC, Roth DM, Gao MH, Fine S, Head BP, Zhu J, McKirnan MD, Kwong C, Dalton N, Urasawa K, et al. (2000) Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP-generating capacity. Circulation 102:2396–2401. [DOI] [PubMed] [Google Scholar]
- Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, Spellman M, Clopton P, Hammond HK. (2004) Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation 110:330–336. [DOI] [PubMed] [Google Scholar]
- Laurenza A, Khandelwal Y, De Souza NJ, Rupp RH, Metzger H, Seamon KB. (1987) Stimulation of adenylate cyclase by water-soluble analogues of forskolin. Mol Pharmacol 32:133–139. [PubMed] [Google Scholar]
- Laurenza A, Sutkowski EM, Seamon KB. (1989) Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol Sci 10:442–447. [DOI] [PubMed] [Google Scholar]
- Laux WH, Pande P, Shoshani I, Gao J, Boudou-Vivet V, Gosselin G, Johnson RA. (2004) Pro-nucleotide inhibitors of adenylyl cyclases in intact cells. J Biol Chem 279:13317–13332. [DOI] [PubMed] [Google Scholar]
- Lee D. (2015) Global and local missions of cAMP signaling in neural plasticity, learning, and memory. Front Pharmacol 6:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, et al. (2015) Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KL, Hoey DA, Spasic M, Tang T, Hammond HK, Jacobs CR. (2014) Adenylyl cyclase 6 mediates loading-induced bone adaptation in vivo. FASEB J 28:1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Hong JH, Choi IY, Che Y, Lee JK, Yang SD, Song CW, Kang HS, Lee JH, Noh JS, et al. (2002) Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 22:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Gyu Song G. (2015) Genome-wide pathway analysis in pancreatic cancer. J BUON 20:1565–1575. [PubMed] [Google Scholar]
- Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. (2009) Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol 11:433–442. [DOI] [PubMed] [Google Scholar]
- Levin LR, Buck J. (2015) Physiological roles of acid-base sensors. Annu Rev Physiol 77:347–362. [DOI] [PubMed] [Google Scholar]
- Levin LR, Han PL, Hwang PM, Feinstein PG, Davis RL, Reed RR. (1992) The Drosophila learning and memory gene rutabaga encodes a Ca2+/Calmodulin-responsive adenylyl cyclase. Cell 68:479–489. [DOI] [PubMed] [Google Scholar]
- Levin LR, Reed RR. (1995) Identification of functional domains of adenylyl cyclase using in vivo chimeras. J Biol Chem 270:7573–7579. [DOI] [PubMed] [Google Scholar]
- Li S, Lee ML, Bruchas MR, Chan GC, Storm DR, Chavkin C. (2006) Calmodulin-stimulated adenylyl cyclase gene deletion affects morphine responses. Mol Pharmacol 70:1742–1749. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen L, Kass RS, Dessauer CW. (2012) The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem 287:29815–29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, Li J, Zhao SD, Bradfield JP, Mentch FD, Maggadottir SM, Hou C, Abrams DJ, Chang D, Gao F, et al. (2015) Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat Med 21:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Chen BC. (1997) Involvement of protein kinase C in the UTP-mediated potentiation of cyclic AMP accumulation in mouse J774 macrophages. Br J Pharmacol 121:1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder JU, Schultz JE. (2003) The class III adenylyl cyclases: multi-purpose signalling modules. Cell Signal 15:1081–1089. [DOI] [PubMed] [Google Scholar]
- Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. (2003) Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem 278:15922–15926. [DOI] [PubMed] [Google Scholar]
- Liu K, Steketee JD. (2016) The role of adenylyl cyclase in the medial prefrontal cortex in cocaine-induced behavioral sensitization in rats. Neuropharmacology 111:70–77. [DOI] [PubMed] [Google Scholar]
- Liu L, Das S, Losert W, Parent CA. (2010) mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell 19:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ostrom RS, Insel PA. (2004) cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol 286:C1089–C1099. [DOI] [PubMed] [Google Scholar]
- Liu X, Thangavel M, Sun SQ, Kaminsky J, Mahautmr P, Stitham J, Hwa J, Ostrom RS. (2008) Adenylyl cyclase type 6 overexpression selectively enhances beta-adrenergic and prostacyclin receptor-mediated inhibition of cardiac fibroblast function because of colocalization in lipid rafts. Naunyn Schmiedebergs Arch Pharmacol 377:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livera G, Xie F, Garcia MA, Jaiswal B, Chen J, Law E, Storm DR, Conti M. (2005) Inactivation of the mouse adenylyl cyclase 3 gene disrupts male fertility and spermatozoon function. Mol Endocrinol 19:1277–1290. [DOI] [PubMed] [Google Scholar]
- Londos C, Wolff J. (1977) Two distinct adenosine-sensitive sites on adenylate cyclase. Proc Natl Acad Sci USA 74:5482–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübker C, Dove S, Tang WJ, Urbauer RJ, Moskovitz J, Urbauer JL, Seifert R. (2015a) Different roles of N-terminal and C-terminal domains in calmodulin for activation of Bacillus anthracis edema factor. Toxins (Basel) 7:2598–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübker C, Seifert R. (2015) Effects of 39 compounds on calmodulin-regulated adenylyl cyclases AC1 and Bacillus anthracis edema factor. PLoS One 10:e0124017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübker C, Urbauer RJ, Moskovitz J, Dove S, Weisemann J, Fedorova M, Urbauer JL, Seifert R. (2015b) Membranous adenylyl cyclase 1 activation is regulated by oxidation of N- and C-terminal methionine residues in calmodulin. Biochem Pharmacol 93:196–209. [DOI] [PubMed] [Google Scholar]
- Ludwig MG, Seuwen K. (2002) Characterization of the human adenylyl cyclase gene family: cDNA, gene structure, and tissue distribution of the nine isoforms. J Recept Signal Transduct Res 22:79–110. [DOI] [PubMed] [Google Scholar]
- Maas JW, Jr, Indacochea RA, Muglia LM, Tran TT, Vogt SK, West T, Benz A, Shute AA, Holtzman DM, Mennerick S, et al. (2005a) Calcium-stimulated adenylyl cyclases modulate ethanol-induced neurodegeneration in the neonatal brain. J Neurosci 25:2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas JW, Jr, Vogt SK, Chan GC, Pineda VV, Storm DR, Muglia LJ. (2005b) Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. J Neurosci 25:4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadeo DC, Janka-Junttila M, Smoot RL, Roselova P, Parent CA. (2007) A chemoattractant-mediated Gi-coupled pathway activates adenylyl cyclase in human neutrophils. Mol Biol Cell 18:512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Güell M, Serrano L. (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett 583:3966–3973. [DOI] [PubMed] [Google Scholar]
- Maiga B, Dolo A, Touré O, Dara V, Tapily A, Campino S, Sepulveda N, Risley P, Silva N, Corran P, et al. MalariaGEN Consortium (2013) Human candidate polymorphisms in sympatric ethnic groups differing in malaria susceptibility in Mali. PLoS One 8:e75675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed M, Nagabhushanam K, Natarajan S, Vaidyanathan P, Karri SK, Jose JA. (2015) Efficacy and safety of 1% forskolin eye drops in open angle glaucoma - An open label study. Saudi J Ophthalmol 29:197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin L, Gluenz E. (2015) cAMP signalling in trypanosomatids: role in pathogenesis and as a drug target. Trends Parasitol 31:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjurano A, Clark TG, Nadjm B, Mtove G, Wangai H, Sepulveda N, Campino SG, Maxwell C, Olomi R, Rockett KR, et al. MalariaGen Consortium (2012) Candidate human genetic polymorphisms and severe malaria in a Tanzanian population. PLoS One 7:e47463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann L, Heldman E, Bersudsky Y, Vatner SF, Ishikawa Y, Almog O, Belmaker RH, Agam G. (2009) Inhibition of specific adenylyl cyclase isoforms by lithium and carbamazepine, but not valproate, may be related to their antidepressant effect. Bipolar Disord 11:885–896. [DOI] [PubMed] [Google Scholar]
- Manolopoulos VG, Liu J, Unsworth BR, Lelkes PI. (1995) Adenylyl cyclase isoforms are differentially expressed in primary cultures of endothelial cells and whole tissue homogenates from various rat tissues. Biochem Biophys Res Commun 208:323–331. [DOI] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. (2002) Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295:496–499. [DOI] [PubMed] [Google Scholar]
- Masada N, Schaks S, Jackson SE, Sinz A, Cooper DM. (2012) Distinct mechanisms of calmodulin binding and regulation of adenylyl cyclases 1 and 8. Biochemistry 51:7917–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka I, Suzuki Y, Defer N, Nakanishi H, Hanoune J. (1997) Differential expression of type I, II, and V adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem 68:498–506. [DOI] [PubMed] [Google Scholar]
- Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. (2007) Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J Physiol 582:1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Hill J, Howlett A, Klein C. (1999) Adenylyl cyclase, a coincidence detector for nitric oxide. J Biol Chem 274:18887–18892. [DOI] [PubMed] [Google Scholar]
- Mencacci NE, Erro R, Wiethoff S, Hersheson J, Ryten M, Balint B, Ganos C, Stamelou M, Quinn N, Houlden H, et al. (2015) ADCY5 mutations are another cause of benign hereditary chorea. Neurology 85:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menninger JA, Barón AE, Conigrave KM, Whitfield JB, Saunders JB, Helander A, Eriksson CJ, Grant B, Hoffman PL, Tabakoff B, WHO/ISBRA Collaborative Study Investigators. International Society for Biomedical Research on Alcoholism (2000) Platelet adenylyl cyclase activity as a trait marker of alcohol dependence. Alcohol Clin Exp Res 24:810–821. [PubMed] [Google Scholar]
- Michel MC, Seifert R. (2015) Selectivity of pharmacological tools: implications for use in cell physiology. A review in the theme: Cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol 308:C505–C520. [DOI] [PubMed] [Google Scholar]
- Middelhaufe S, Leipelt M, Levin LR, Buck J, Steegborn C. (2012) Identification of a haem domain in human soluble adenylate cyclase. Biosci Rep 32:491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika D, Conti M. (2016) PDE4D phosphorylation: A coincidence detector integrating multiple signaling pathways. Cell Signal 28:719–724. [DOI] [PubMed] [Google Scholar]
- Morris DI, Speicher LA, Ruoho AE, Tew KD, Seamon KB. (1991) Interaction of forskolin with the P-glycoprotein multidrug transporter. Biochemistry 30:8371–8379. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, Oien DB. (2010) Protein carbonyl and the methionine sulfoxide reductase system. Antioxid Redox Signal 12:405–415. [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. (1979) Activation of adenylate cyclase by choleragen. Annu Rev Biochem 48:581–600. [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. (1989) Guanine nucleotide-binding proteins (G proteins) in activation of adenylyl cyclase: lessons learned from cholera and “travelers’ diarrhea”. J Lab Clin Med 113:258–268. [PubMed] [Google Scholar]
- Mou TC, Gille A, Fancy DA, Seifert R, Sprang SR. (2005) Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2 ‘(3’)-O-(N-Methylanthraniloyl)-guanosine 5 '-triphosphate. J Biol Chem 280:7253–7261. [DOI] [PubMed] [Google Scholar]
- Mou TC, Gille A, Suryanarayana S, Richter M, Seifert R, Sprang SR. (2006) Broad specificity of mammalian adenylyl cyclase for interaction with 2′,3′-substituted purine- and pyrimidine nucleotide inhibitors. Mol Pharmacol 70:878–886. [DOI] [PubMed] [Google Scholar]
- Mou TC, Masada N, Cooper DM, Sprang SR. (2009) Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry 48:3387–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühleisen TW, Leber M, Schulze TG, Strohmaier J, Degenhardt F, Treutlein J, Mattheisen M, Forstner AJ, Schumacher J, Breuer R, et al. (2014) Genome-wide association study reveals two new risk loci for bipolar disorder. Nat Commun 5:3339. [DOI] [PubMed] [Google Scholar]
- Neer EJ. (1978) Multiple forms of adenylate cyclase. Adv Cyclic Nucleotide Res 9:69–83. [PubMed] [Google Scholar]
- Ng MC, Saxena R, Li J, Palmer ND, Dimitrov L, Xu J, Rasmussen-Torvik LJ, Zmuda JM, Siscovick DS, Patel SR, et al. (2013) Transferability and fine mapping of type 2 diabetes loci in African Americans: the Candidate Gene Association Resource Plus Study. Diabetes 62:965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols J, Obiako B, Ramila KC, Putinta K, Schilling S, Sayner SL. (2015) Lipopolysaccharide-induced pulmonary endothelial barrier disruption and lung edema: critical role for bicarbonate stimulation of AC10. Am J Physiol Lung Cell Mol Physiol 309:L1430–L1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol X, Bennis M, Ishikawa Y, Chan GC, Repérant J, Storm DR, Gaspar P. (2006) Role of the calcium modulated cyclases in the development of the retinal projections. Eur J Neurosci 24:3401–3414. [DOI] [PubMed] [Google Scholar]
- Nicol X, Gaspar P. (2014) Routes to cAMP: shaping neuronal connectivity with distinct adenylate cyclases. Eur J Neurosci 39:1742–1751. [DOI] [PubMed] [Google Scholar]
- Nicol X, Muzerelle A, Bachy I, Ravary A, Gaspar P. (2005) Spatiotemporal localization of the calcium-stimulated adenylate cyclases, AC1 and AC8, during mouse brain development. J Comp Neurol 486:281–294. [DOI] [PubMed] [Google Scholar]
- Nordman S, Abulaiti A, Hilding A, Långberg EC, Humphreys K, Ostenson CG, Efendic S, Gu HF. (2008) Genetic variation of the adenylyl cyclase 3 (AC3) locus and its influence on type 2 diabetes and obesity susceptibility in Swedish men. Int J Obes 32:407–412. [DOI] [PubMed] [Google Scholar]
- O’Boyle KM, Waddington JL. (1984) Identification of the enantiomers of SK&F 83566 as specific and stereoselective antagonists at the striatal D-1 dopamine receptor: comparisons with the D-2 enantioselectivity of Ro 22-1319. Eur J Pharmacol 106:219–220. [DOI] [PubMed] [Google Scholar]
- Oishi H, Takano K, Tomita K, Takebe M, Yokoo H, Yamazaki M, Hattori Y. (2012) Olprinone and colforsin daropate alleviate septic lung inflammation and apoptosis through CREB-independent activation of the Akt pathway. Am J Physiol Lung Cell Mol Physiol 303:L130–L140. [DOI] [PubMed] [Google Scholar]
- Okumura S, Kawabe J, Yatani A, Takagi G, Lee MC, Hong C, Liu J, Takagi I, Sadoshima J, Vatner DE, et al. (2003a) Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res 93:364–371. [DOI] [PubMed] [Google Scholar]
- Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, et al. (2003b) Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 100:9986–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura S, Vatner DE, Kurotani R, Bai Y, Gao S, Yuan Z, Iwatsubo K, Ulucan C, Kawabe J, Ghosh K, et al. (2007) Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation 116:1776–1783. [DOI] [PubMed] [Google Scholar]
- Oldenburger A, Maarsingh H, Schmidt M. (2012) Multiple facets of cAMP signalling and physiological impact: cAMP compartmentalization in the lung. Pharmaceuticals (Basel) 5:1291–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olianas MC, Onali P. (1986) Phorbol esters increase GTP-dependent adenylate cyclase activity in rat brain striatal membranes. J Neurochem 47:890–897. [DOI] [PubMed] [Google Scholar]
- Onda T, Hashimoto Y, Nagai M, Kuramochi H, Saito S, Yamazaki H, Toya Y, Sakai I, Homcy CJ, Nishikawa K, et al. (2001) Type-specific regulation of adenylyl cyclase. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J Biol Chem 276:47785–47793. [DOI] [PubMed] [Google Scholar]
- Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH. (2007) Adenylyl cyclase isoform v mediates renin release from juxtaglomerular cells. Hypertension 49:618–624. [DOI] [PubMed] [Google Scholar]
- Ostlund G, Sonnhammer EL. (2012) Quality criteria for finding genes with high mRNA-protein expression correlation and coexpression correlation. Gene 497:228–236. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Bogard AS, Gros R, Feldman RD. (2012) Choreographing the adenylyl cyclase signalosome: sorting out the partners and the steps. Naunyn Schmiedebergs Arch Pharmacol 385:5–12. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Insel PA. (2004) The evolving role of lipid rafts and caveolae in G proteincoupled receptor signaling: Implications for molecular pharmacology. Br J Pharmacol 143:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. (2002) Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol Pharmacol 62:983–992. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Naugle JE, Hase M, Gregorian C, Swaney JS, Insel PA, Brunton LL, Meszaros JG. (2003) Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J Biol Chem 278:24461–24468. [DOI] [PubMed] [Google Scholar]
- Ostrom RS, Violin JD, Coleman S, Insel PA. (2000) Selective enhancement of β-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes. Mol Pharmacol 57:1075–1079. [PubMed] [Google Scholar]
- Pagano M, Clynes MA, Masada N, Ciruela A, Ayling LJ, Wachten S, Cooper DM. (2009) Insights into the residence in lipid rafts of adenylyl cyclase AC8 and its regulation by capacitative calcium entry. Am J Physiol Cell Physiol 296:C607–C619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasevich S, Melén E, Hallberg J, Bergström A, Svartengren M, Pershagen G, Nyberg F. (2013) Investigation of novel genes for lung function in children and their interaction with tobacco smoke exposure: a preliminary report. Acta Paediatr 102:498–503. [DOI] [PubMed] [Google Scholar]
- Park HY, Kang YM, Kang Y, Park TS, Ryu YK, Hwang JH, Kim YH, Chung BH, Nam KH, Kim MR, et al. (2014) Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson’s disease. J Neurosci 34:11744–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TB, Du Z, Pierre S, Cartin L, Scholich K. (2001) Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene 269:13–25. [DOI] [PubMed] [Google Scholar]
- Pavan B, Biondi C, Dalpiaz A. (2009) Adenylyl cyclases as innovative therapeutic goals. Drug Discov Today 14:982–991. [DOI] [PubMed] [Google Scholar]
- Pavlikova N, Kortner TM, Arukwe A. (2010) Modulation of acute steroidogenesis, peroxisome proliferator-activated receptors and CYP3A/PXR in salmon interrenal tissues by tributyltin and the second messenger activator, forskolin. Chem Biol Interact 185:119–127. [DOI] [PubMed] [Google Scholar]
- Pfeuffer E, Dreher RM, Metzger H, Pfeuffer T. (1985) Catalytic unit of adenlyate cyclase: purification and identification by affinity crosslinking. Proc Natl Acad Sci USA 82:3086–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeuffer E, Mollner S, Lancet D, Pfeuffer T. (1989) Olfactory adenylyl cyclase. Identification and purification of a novel enzyme form. J Biol Chem 264:18803–18807. [PubMed] [Google Scholar]
- Pfeuffer E, Pfeuffer T. (1989) Affinity labeling of forskolin-binding proteins. Comparison between glucose carrier and adenylate cyclase. FEBS Lett 248:13–17. [DOI] [PubMed] [Google Scholar]
- Pfeuffer T. (1977) GTP-binding proteins in membranes and the control of adenylate cyclase activity. J Biol Chem 252:7224–7234. [PubMed] [Google Scholar]
- Pfeuffer T. (1991) Synthesis of forskolin-agarose affinity matrices. Methods Enzymol 195:44–51. [DOI] [PubMed] [Google Scholar]
- Pfeuffer T, Helmreich EJ. (1975) Activation of pigeon erythrocyte membrane adenylate cyclase by guanylnucleotide analogues and separation of a nucleotide binding protein. J Biol Chem 250:867–876. [PubMed] [Google Scholar]
- Pieroni JP, Harry A, Chen J, Jacobowitz O, Magnusson RP, Iyengar R. (1995) Distinct characteristics of the basal activities of adenylyl cyclases 2 and 6. J Biol Chem 270:21368–21373. [DOI] [PubMed] [Google Scholar]
- Pierre S, Eschenhagen T, Geisslinger G, Scholich K. (2009) Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov 8:321–335. [DOI] [PubMed] [Google Scholar]
- Piggott LA, Bauman AL, Scott JD, Dessauer CW. (2008) The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci USA 105:13835–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Hübner M, Gille A, Richter M, Mou TC, Sprang SR, Seifert R. (2009) Differential interactions of the catalytic subunits of adenylyl cyclase with forskolin analogs. Biochem Pharmacol 78:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Lushington GH, Richter M, Gille A, Geduhn J, König B, Mou TC, Sprang SR, Seifert R. (2011) Structure-activity relationships for the interactions of 2′- and 3′-(O)-(N-methyl)anthraniloyl-substituted purine and pyrimidine nucleotides with mammalian adenylyl cyclases. Biochem Pharmacol 82:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto C, Papa D, Hübner M, Mou TC, Lushington GH, Seifert R. (2008) Activation and inhibition of adenylyl cyclase isoforms by forskolin analogs. J Pharmacol Exp Ther 325:27–36. [DOI] [PubMed] [Google Scholar]
- Pinto CS, Reif GA, Nivens E, White C, Wallace DP. (2012) Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am J Physiol Renal Physiol 303:F1412–F1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli CL, Kean J, de Graaf C, Deupi X. (2015) A molecular pharmacologist’s guide to G protein-coupled receptor crystallography. Mol Pharmacol 88:536–551. [DOI] [PubMed] [Google Scholar]
- Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, Wells E, Greer CA, Wang T, Firestein S, et al. (2009) Functional expression of the olfactory signaling system in the kidney. Proc Natl Acad Sci USA 106:2059–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnam D, Shilpi S, Srinivas KV, Suiab L, Alam S, Amtul Z, Arigari NK, Jonnala KK, Siddiqui L, Dubey V, et al. (2014) Synthesis of cyclic 1,9-acetal derivatives of forskolin and their bioactivity evaluation. Eur J Med Chem 87:735–744. [DOI] [PubMed] [Google Scholar]
- Post SR, Ostrom RS, Insel PA. (2000) Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol Biol 126:363–374. [DOI] [PubMed] [Google Scholar]
- Premont RT. (1994) Identification of adenylyl cyclases by amplification using degenerate primers. Methods Enzymol 238:116–127. [DOI] [PubMed] [Google Scholar]
- Premont RT, Matsuoka I, Mattei MG, Pouille Y, Defer N, Hanoune J. (1996) Identification and characterization of a widely expressed form of adenylyl cyclase. J Biol Chem 271:13900–13907. [DOI] [PubMed] [Google Scholar]
- Price SA, Bernal AL. (2001) Uterine quiescence: the role of cyclic AMP. Exp Physiol 86:265–272. [DOI] [PubMed] [Google Scholar]
- Procopio DO, Saba LM, Walter H, Lesch O, Skala K, Schlaff G, Vanderlinden L, Clapp P, Hoffman PL, Tabakoff B. (2013) Genetic markers of comorbid depression and alcoholism in women. Alcohol Clin Exp Res 37:896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronko SP, Saba LM, Hoffman PL, Tabakoff B. (2010) Type 7 adenylyl cyclase-mediated hypothalamic-pituitary-adrenal axis responsiveness: influence of ethanol and sex. J Pharmacol Exp Ther 334:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam WC, Swenson SM, Reif GA, Wallace DP, Helmkamp GM, Jr, Grantham JJ. (2007) Identification of a forskolin-like molecule in human renal cysts. J Am Soc Nephrol 18:934–943. [DOI] [PubMed] [Google Scholar]
- Rall TW, Sutherland EW. (1958) Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem 232:1065–1076. [PubMed] [Google Scholar]
- Ramos-Espiritu L, Kleinboelting S, Navarrete FA, Alvau A, Visconti PE, Valsecchi F, Starkov A, Manfredi G, Buck H, Adura C, et al. (2016) Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat Chem Biol 12:838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Barajas C, Silva I, Lopéz-Santiago LM, Aceves J, Erlij D, Florán B. (2011) L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis 41:51–61. [DOI] [PubMed] [Google Scholar]
- Raoux M, Vacher P, Papin J, Picard A, Kostrzewa E, Devin A, Gaitan J, Limon I, Kas MJ, Magnan C, et al. (2015) Multilevel control of glucose homeostasis by adenylyl cyclase 8. Diabetologia 58:749–757. [DOI] [PubMed] [Google Scholar]
- Raslan Z, Naseem KM. (2015) Compartmentalisation of cAMP-dependent signalling in blood platelets: The role of lipid rafts and actin polymerisation. Platelets 26:349–357. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450:383–387. [DOI] [PubMed] [Google Scholar]
- Razzoli M, Andreoli M, Maraia G, Di Francesco C, Arban R. (2010) Functional role of Calcium-stimulated adenylyl cyclase 8 in adaptations to psychological stressors in the mouse: implications for mood disorders. Neuroscience 170:429–440. [DOI] [PubMed] [Google Scholar]
- Reddy R, Smith D, Wayman G, Wu Z, Villacres EC, Storm DR. (1995) Voltage-sensitive adenylyl cyclase activity in cultured neurons. A calcium-independent phenomenon. J Biol Chem 270:14340–14346. [DOI] [PubMed] [Google Scholar]
- Reed BY, Gitomer WL, Heller HJ, Hsu MC, Lemke M, Padalino P, Pak CY. (2002) Identification and characterization of a gene with base substitutions associated with the absorptive hypercalciuria phenotype and low spinal bone density. J Clin Endocrinol Metab 87:1476–1485. [DOI] [PubMed] [Google Scholar]
- Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. (2014) Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 25:232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees SD, Hydrie MZ, O’Hare JP, Kumar S, Shera AS, Basit A, Barnett AH, Kelly MA. (2011) Effects of 16 genetic variants on fasting glucose and type 2 diabetes in South Asians: ADCY5 and GLIS3 variants may predispose to type 2 diabetes. PLoS One 6:e24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers AE, Posner R, Neubig RR. (1994) Fluorescent guanine nucleotide analogs and G protein activation. J Biol Chem 269:13771–13778. [PubMed] [Google Scholar]
- Rieg T, Kohan DE. (2014) Regulation of nephron water and electrolyte transport by adenylyl cyclases. Am J Physiol Renal Physiol 306:F701–F709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieg T, Tang T, Murray F, Schroth J, Insel PA, Fenton RA, Hammond HK, Vallon V. (2010) Adenylate cyclase 6 determines cAMP formation and aquaporin-2 phosphorylation and trafficking in inner medulla. J Am Soc Nephrol 21:2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieg T, Tang T, Uchida S, Hammond HK, Fenton RA, Vallon V. (2013) Adenylyl cyclase 6 enhances NKCC2 expression and mediates vasopressin-induced phosphorylation of NKCC2 and NCC. Am J Pathol 182:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos-Hoyo A, Gutiérrez-Salmeán G. (2016) New dietary supplements for obesity: What we currently know. Curr Obes Rep 5:262–270. [DOI] [PubMed] [Google Scholar]
- Risøe PK, Rutkovskiy A, Ågren J, Kolseth IB, Kjeldsen SF, Valen G, Vaage J, Dahle MK. (2015) Higher TNFα responses in young males compared to females are associated with attenuation of monocyte adenylyl cyclase expression. Hum Immunol 76:427–430. [DOI] [PubMed] [Google Scholar]
- Risøe PK, Ryg U, Wang YY, Rutkovskiy A, Smedsrød B, Valen G, Dahle MK. (2011) Cecal ligation and puncture sepsis is associated with attenuated expression of adenylyl cyclase 9 and increased miR142-3p. Shock 36:390–395. [DOI] [PubMed] [Google Scholar]
- Rodbell M. (1984) Structure-function problems with the adenylate cyclase system. Adv Cyclic Nucleotide Protein Phosphorylation Res 17:207–214. [PubMed] [Google Scholar]
- Roos KP, Bugaj V, Mironova E, Stockand JD, Ramkumar N, Rees S, Kohan DE. (2013) Adenylyl cyclase VI mediates vasopressin-stimulated ENaC activity. J Am Soc Nephrol 24:218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos KP, Strait KA, Raphael KL, Blount MA, Kohan DE. (2012) Collecting duct-specific knockout of adenylyl cyclase type VI causes a urinary concentration defect in mice. Am J Physiol Renal Physiol 302:F78–F84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM. (1981) Physical separation of the catalytic and regulatory proteins of hepatic adenylate cyclase. J Biol Chem 256:1949–1953. [PubMed] [Google Scholar]
- Ross EM, Howlett AC, Ferguson KM, Gilman AG. (1978) Reconstitution of hormone-sensitive adenylate cyclase activity with resolved components of the enzyme. J Biol Chem 253:6401–6412. [PubMed] [Google Scholar]
- Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. (2002) Adenylyl cyclase increases survival in cardiomyopathy. Circulation 105:1989–1994. [DOI] [PubMed] [Google Scholar]
- Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. (1999) Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99:3099–3102. [DOI] [PubMed] [Google Scholar]
- Roth MY, Amory JK. (2016) Beyond the condom: Frontiers in male contraception. Semin Reprod Med 34:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottlaender D, Matthes J, Vatner SF, Seifert R, Herzig S. (2007) Functional adenylyl cyclase inhibition in murine cardiomyocytes by 2'(3')-O-(N-methylanthraniloyl)-guanosine 5'-[g-thio]triphosphate. J Pharmacol Exp Ther 321:608–615. [DOI] [PubMed] [Google Scholar]
- Sabbatini ME, D’Alecy L, Lentz SI, Tang T, Williams JA. (2013) Adenylyl cyclase 6 mediates the action of cyclic AMP-dependent secretagogues in mouse pancreatic exocrine cells via protein kinase A pathway activation. J Physiol 591:3693–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini ME, Gorelick F, Glaser S. (2014) Adenylyl cyclases in the digestive system. Cell Signal 26:1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana R, Dessauer CW. (2009) Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals 17:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalau-Bethell SM, Berdini V, Cleasby A, Congreve M, Coyle JE, Lock V, Murray CW, O’Brien MA, Rich SJ, Sambrook T, et al. (2014) Crystal structure of human soluble adenylate cyclase reveals a distinct, highly flexible allosteric bicarbonate binding pocket. ChemMedChem 9:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim S, Dessauer CW. (2004) Analysis of the interaction between RGS2 and adenylyl cyclase. Methods Enzymol 390:83–99. [DOI] [PubMed] [Google Scholar]
- Salim S, Sinnarajah S, Kehrl JH, Dessauer CW. (2003) Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem 278:15842–15849. [DOI] [PubMed] [Google Scholar]
- Sanabra C, Mengod G. (2011) Neuroanatomical distribution and neurochemical characterization of cells expressing adenylyl cyclase isoforms in mouse and rat brain. J Chem Neuroanat 41:43–54. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N. (1996) Myocardial adenylyl cyclase type V and VI MRNA: Differential regulation with age. J Cardiovasc Pharmacol 27:86–90. [DOI] [PubMed] [Google Scholar]
- Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, Nardi A, Hartman RE, Vogt SK, Luedke CE, et al. (2000) Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J Neurosci 20:4809–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidig AJ, Franken SM, Corrie JE, Reid GP, Wittinghofer A, Pai EF, Goody RS. (1995) X-ray crystal structure analysis of the catalytic domain of the oncogene product p21H-ras complexed with caged GTP and mant dGppNHp. J Mol Biol 253:132–150. [DOI] [PubMed] [Google Scholar]
- Schmid A, Meili D, Salathe M. (2014) Soluble adenylyl cyclase in health and disease. Biochim Biophys Acta 1842 (12 Pt B):2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Sutto Z, Nlend MC, Horvath G, Schmid N, Buck J, Levin LR, Conner GE, Fregien N, Salathe M. (2007) Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J Gen Physiol 130:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Sutto Z, Schmid N, Novak L, Ivonnet P, Horvath G, Conner G, Fregien N, Salathe M. (2010) Decreased soluble adenylyl cyclase activity in cystic fibrosis is related to defective apical bicarbonate exchange and affects ciliary beat frequency regulation. J Biol Chem 285:29998–30007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholich K, Barbier AJ, Mullenix JB, Patel TB. (1997) Characterization of soluble forms of nonchimeric type V adenylyl cyclases. Proc Natl Acad Sci USA 94:2915–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwencke C, Yamamoto M, Okumura S, Toya Y, Kim SJ, Ishikawa Y. (1999) Compartmentation of cyclic adenosine 3',5'-monophosphate signaling in caveolae. Mol Endocrinol 13:1061–1070. [DOI] [PubMed] [Google Scholar]
- Scott TL, Christian PA, Kesler MV, Donohue KM, Shelton B, Wakamatsu K, Ito S, D’Orazio J. (2012) Pigment-independent cAMP-mediated epidermal thickening protects against cutaneous UV injury by keratinocyte proliferation. Exp Dermatol 21:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamon KB, Padgett W, Daly JW. (1981) Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA 78:3363–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamon KB, Vaillancourt R, Edwards M, Daly JW. (1984) Binding of [3H]forskolin to rat brain membranes. Proc Natl Acad Sci USA 81:5081–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebacher T, Linder JU, Schultz JE. (2001) An isoform-specific interaction of the membrane anchors affects mammalian adenylyl cyclase type V activity. Eur J Biochem 268:105–110. [DOI] [PubMed] [Google Scholar]
- Seebeck T, Schaub R, Johner A. (2004) cAMP signalling in the kinetoplastid protozoa. Curr Mol Med 4:585–599. [DOI] [PubMed] [Google Scholar]
- Seeger F, Quintyn R, Tanimoto A, Williams GJ, Tainer JA, Wysocki VH, Garcin ED. (2014) Interfacial residues promote an optimal alignment of the catalytic center in human soluble guanylate cyclase: heterodimerization is required but not sufficient for activity. Biochemistry 53:2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R. (2014) Vidarabine is neither a potent nor a selective AC5 inhibitor. Biochem Pharmacol 87:543–546. [DOI] [PubMed] [Google Scholar]
- Seifert R. (2015) cCMP and cUMP: emerging second messengers. Trends Biochem Sci 40:8–15. [DOI] [PubMed] [Google Scholar]
- Seifert R. (2016) Does vidarabine mediate cardioprotection via inhibition of AC5? J Pharmacol Exp Ther 358:242–243. [DOI] [PubMed] [Google Scholar]
- Seifert R, Beste KY. (2012) Allosteric regulation of nucleotidyl cyclases: an emerging pharmacological target. Sci Signal 5:pe37. [DOI] [PubMed] [Google Scholar]
- Seifert R, Dove S. (2012) Towards selective inhibitors of adenylyl cyclase toxin from Bordetella pertussis. Trends Microbiol 20:343–351. [DOI] [PubMed] [Google Scholar]
- Seifert R, Dove S. (2013) Inhibitors of Bacillus anthracis edema factor. Pharmacol Ther 140:200–212. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. (2002) Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366:381–416. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E, Kobilka BK. (1998) Different effects of Gsα splice variants on β2-adrenoceptor-mediated signaling. The β2-adrenoceptor coupled to the long splice variant of Gsα has the properties of a constitutively active receptor. J Biol Chem 273:5109–5116. [PubMed] [Google Scholar]
- Seifert R, Lushington GH, Mou TC, Gille A, Sprang SR. (2012) Inhibitors of membranous adenylyl cyclases. Trends Pharmacol Sci 33:64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Schneider EH, Bähre H. (2015) From canonical to non-canonical cyclic nucleotides as second messengers: pharmacological implications. Pharmacol Ther 148:154–184. [DOI] [PubMed] [Google Scholar]
- Serazin-Leroy V, Morot M, de Mazancourt P, Giudicelli Y. (2001) Differences in type II, IV, V and VI adenylyl cyclase isoform expression between rat preadipocytes and adipocytes. Biochim Biophys Acta 1550:37–51. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Klee WA, Nirenberg M. (1975b) Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA 72:3092–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Nirenberg M, Klee WA. (1975a) Morphine receptors as regulators of adenylate cyclase activity. Proc Natl Acad Sci USA 72:590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shear M, Insel PA, Melmon KL, Coffino P. (1976) Agonist-specific refractoriness induced by isoproterenol. Studies with mutant cells. J Biol Chem 251:7572–7576. [PubMed] [Google Scholar]
- Shen JX, Cooper DM. (2013) AKAP79, PKC, PKA and PDE4 participate in a Gq-linked muscarinic receptor and adenylate cyclase 2 cAMP signalling complex. Biochem J 455:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Suzuki Y, Poyard M, Best-Belpomme M, Defer N, Hanoune J. (1997) Localization and differential expression of adenylyl cyclase messenger ribonucleic acids in rat adrenal gland determined by in situ hybridization. Endocrinology 138:4591–4598. [DOI] [PubMed] [Google Scholar]
- Shoshani I, Laux WH, Périgaud C, Gosselin G, Johnson RA. (1999) Inhibition of adenylyl cyclase by acyclic nucleoside phosphonate antiviral agents. J Biol Chem 274:34742–34744. [DOI] [PubMed] [Google Scholar]
- Sikora E, Llinas M, Garcia-Celma MJ, Escribano E, Solans C. (2015) Transdermal delivery of forskolin from emulsions differing in droplet size. Colloids Surf B Biointerfaces 126:541–545. [DOI] [PubMed] [Google Scholar]
- Simon NC, Aktories K, Barbieri JT. (2014) Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol 12:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. (2007) Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron 53:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha SC, Sprang SR. (2006) Structures, mechanism, regulation and evolution of class III nucleotidyl cyclases. Rev Physiol Biochem Pharmacol 157:105–140. [DOI] [PubMed] [Google Scholar]
- Sinnarajah S, Dessauer CW, Srikumar D, Chen J, Yuen J, Yilma S, Dennis JC, Morrison EE, Vodyanoy V, Kehrl JH. (2001) RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature 409:1051–1055. [DOI] [PubMed] [Google Scholar]
- Small KM, Brown KM, Theiss CT, Seman CA, Weiss ST, Liggett SB. (2003) An Ile to Met polymorphism in the catalytic domain of adenylyl cyclase type 9 confers reduced β2-adrenergic receptor stimulation. Pharmacogenetics 13:535–541. [DOI] [PubMed] [Google Scholar]
- Smigel MD. (1986) Purification of the catalyst of adenylate cyclase. J Biol Chem 261:1976–1982. [PubMed] [Google Scholar]
- Smith KE, Gu C, Fagan KA, Hu B, Cooper DM. (2002) Residence of adenylyl cyclase type 8 in caveolae is necessary but not sufficient for regulation by capacitative Ca2+ entry. J Biol Chem 277:6025–6031. [DOI] [PubMed] [Google Scholar]
- Sohma H, Hashimoto E, Shirasaka T, Tsunematsu R, Ozawa H, Boissl KW, Böning J, Riederer P, Saito T. (1999) Quantitative reduction of type I adenylyl cyclase in human alcoholics. Biochim Biophys Acta 1454:11–18. [DOI] [PubMed] [Google Scholar]
- Sosa CM, Zanetti MN, Pocognoni CA, Mayorga LS. (2016) Acrosomal swelling is triggered by cAMP downstream of the opening of store-operated calcium channels during acrosomal exocytosis in human sperm. Biol Reprod 94:57. [DOI] [PubMed] [Google Scholar]
- Spry ML, Vanover JC, Scott T, Abona-Ama O, Wakamatsu K, Ito S, D’Orazio JA. (2009) Prolonged treatment of fair-skinned mice with topical forskolin causes persistent tanning and UV protection. Pigment Cell Melanoma Res 22:219–229. [DOI] [PubMed] [Google Scholar]
- Stansfield DA, Franks DJ. (1970) Adenylate cyclase and ‘adenosine triphosphatase’ in corpus luteum. Biochem J 120:5P–7P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Pacher P, Evgenov OV. (2011) Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123:2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Schlossmann J, Hocher B. (2015) Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr Opin Pharmacol 21:95–104. [DOI] [PubMed] [Google Scholar]
- Steegborn C. (2014) Structure, mechanism, and regulation of soluble adenylyl cyclases - similarities and differences to transmembrane adenylyl cyclases. Biochim Biophys Acta 1842 (12 Pt B):2535–2547. [DOI] [PubMed] [Google Scholar]
- Steegborn C, Litvin TN, Hess KC, Capper AB, Taussig R, Buck J, Levin LR, Wu H. (2005) A novel mechanism for adenylyl cyclase inhibition from the crystal structure of its complex with catechol estrogen. J Biol Chem 280:31754–31759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel D, Hanoune J. (1984) The sperm adenylate cyclase. Ann N Y Acad Sci 438:18–28. [DOI] [PubMed] [Google Scholar]
- Stergiakouli E, Gaillard R, Tavaré JM, Balthasar N, Loos RJ, Taal HR, Evans DM, Rivadeneira F, St Pourcain B, Uitterlinden AG, et al. (2014) Genome-wide association study of height-adjusted BMI in childhood identifies functional variant in ADCY3. Obesity (Silver Spring) 22:2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazzabosco M, Fiorotto R, Melero S, Glaser S, Francis H, Spirli C, Alpini G. (2009) Differentially expressed adenylyl cyclase isoforms mediate secretory functions in cholangiocyte subpopulation. Hepatology 50:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Rama JJ, Arrojo M, Sobrino B, Amigo J, Brenlla J, Agra S, Paz E, Brión M, Carracedo Á, Páramo M, et al. (2015) Resequencing and association analysis of coding regions at twenty candidate genes suggest a role for rare risk variation at AKAP9 and protective variation at NRXN1 in schizophrenia susceptibility. J Psychiatr Res 66-67:38–44. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Beuve A, Tesmer JJ, Sprang SR, Garbers DL, Gilman AG. (1998) Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J Biol Chem 273:16332–16338. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG. (1996) Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol 36:461–480. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Whisnant RE, Kleuss C, Gilman AG. (1997) Interaction of Gsα with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem 272:22265–22271. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Taussig R. (2002) Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv 2:168–184. [DOI] [PubMed] [Google Scholar]
- Suryanarayana S, Göttle M, Hübner M, Gille A, Mou TC, Sprang SR, Richter M, Seifert R. (2009a) Differential inhibition of various adenylyl cyclase isoforms and soluble guanylyl cyclase by 2′,3′-O-(2,4,6-trinitrophenyl)-substituted nucleoside 5′-triphosphates. J Pharmacol Exp Ther 330:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryanarayana S, Pinto C, Mou TC, Richter M, Lushington GH, Seifert R. (2009b) The C1 homodimer of adenylyl cyclase binds nucleotides with high affinity but possesses exceedingly low catalytic activity. Neurosci Lett 467:1–5. [DOI] [PubMed] [Google Scholar]
- Sutherland EW, Robison GA. (1966) The role of cyclic-3′,5′-AMP in responses to catecholamines and other hormones. Pharmacol Rev 18:145–161. [PubMed] [Google Scholar]
- Sutherland EW, Rall TW, Menon T. (1962) Adenyl cylase. I. Distribution, preparation, and properties. J Biol Chem 237:1220–1227. [PubMed] [Google Scholar]
- Sutkowski EM, Tang WJ, Broome CW, Robbins JD, Seamon KB. (1994) Regulation of forskolin interactions with type I, II, V, and VI adenylyl cyclases by Gsα. Biochemistry 33:12852–12859. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Lee LJ, Hayashi Y, Muglia L, Itohara S, Erzurumlu RS, Iwasato T. (2015) Thalamic adenylyl cyclase 1 is required for barrel formation in the somatosensory cortex. Neuroscience 290:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Sato M, Ota S, Fukushima T, Ota A, Ota T, Goto K. (2012) Intraarterial colforsin may improve the outcome of patients with aneurysmal subarachnoid hemorrhage: a retrospective study. World Neurosurg 78:295–299. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Shen T, Poyard M, Best-Belpomme M, Hanoune J, Defer N. (1998) Expression of adenylyl cyclase mRNAs in the denervated and in the developing mouse skeletal muscle. Am J Physiol 274:C1674–C1685. [DOI] [PubMed] [Google Scholar]
- Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, Yuan JX, Roth DM, Hammond HK. (2008) Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 117:61–69. [DOI] [PubMed] [Google Scholar]
- Tang T, Lai NC, Roth DM, Drumm J, Guo T, Lee KW, Han PL, Dalton N, Gao MH. (2006) Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to β-adrenergic stimulation. Basic Res Cardiol 101:117–126. [DOI] [PubMed] [Google Scholar]
- Tang T, Lai NC, Wright AT, Gao MH, Lee P, Guo T, Tang R, McCulloch AD, Hammond HK. (2013) Adenylyl cyclase 6 deletion increases mortality during sustained β-adrenergic receptor stimulation. J Mol Cell Cardiol 60:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WJ, Gilman AG. (1991) Type-specific regulation of adenylyl cyclase by G protein β γ subunits. Science 254:1500–1503. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Gilman AG. (1992) Adenylyl cyclases. Cell 70:869–872. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Gilman AG. (1995) Construction of a soluble adenylyl cyclase activated by Gs α and forskolin. Science 268:1769–1772. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Hurley JH. (1998) Catalytic mechanism and regulation of mammalian adenylyl cyclases. Mol Pharmacol 54:231–240. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Krupinski J, Gilman AG. (1991) Expression and characterization of calmodulin-activated (type I) adenylylcyclase. J Biol Chem 266:8595–8603. [PubMed] [Google Scholar]
- Tardif JC, Rhéaume E, Lemieux Perreault LP, Grégoire JC, Feroz Zada Y, Asselin G, Provost S, Barhdadi A, Rhainds D, L’Allier PL, et al. (2015) Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet 8:372–382. [DOI] [PubMed] [Google Scholar]
- Taussig R, Iñiguez-Lluhi JA, Gilman AG. (1993a) Inhibition of adenylyl cyclase by Gi α. Science 261:218–221. [DOI] [PubMed] [Google Scholar]
- Taussig R, Quarmby LM, Gilman AG. (1993b) Regulation of purified type I and type II adenylylcyclases by G protein β γ subunits. J Biol Chem 268:9–12. [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Hepler JR, Gilman AG. (1994a) Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J Biol Chem 269:6093–6100. [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Gilman AG. (1994b) Expression and purification of recombinant adenylyl cyclases in Sf9 cells. Methods Enzymol 238:95–108. [DOI] [PubMed] [Google Scholar]
- Tempel BL, Bonini N, Dawson DR, Quinn WG. (1983) Reward learning in normal and mutant Drosophila. Proc Natl Acad Sci USA 80:1482–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepe NM, Lorenz JN, Yatani A, Dash R, Kranias EG, Dorn GW, 2nd, Liggett SB. (1999) Altering the receptor-effector ratio by transgenic overexpression of type V adenylyl cyclase: enhanced basal catalytic activity and function without increased cardiomyocyte β-adrenergic signalling. Biochemistry 38:16706–16713. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sprang SR. (1998) The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr Opin Struct Biol 8:713–719. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Dessauer CW, Sunahara RK, Murray LD, Johnson RA, Gilman AG, Sprang SR. (2000) Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 39:14464–14471. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα⋅GTPγS. Science 278:1907–1916. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, Sprang SR. (1999) Two-metal-Ion catalysis in adenylyl cyclase. Science 285:756–760. [DOI] [PubMed] [Google Scholar]
- Thangavel M, Liu X, Sun SQ, Kaminsky J, Ostrom RS. (2009) The C1 and C2 domains target human type 6 adenylyl cyclase to lipid rafts and caveolae. Cell Signal 21:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Hoffman BB. (1996) Isoform-specific sensitization of adenylyl cyclase activity by prior activation of inhibitory receptors: role of beta gamma subunits in transducing enhanced activity of the type VI isoform. Mol Pharmacol 49:907–914. [PubMed] [Google Scholar]
- Tian G, Sandler S, Gylfe E, Tengholm A. (2011) Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 60:1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov-Yarovoy V, et al. (2013) Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ Res 112:1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeyev V, Porter CA, Tuteja D, Qiu H, Li N, Tang T, Singapuri A, Han PL, Lopez JE, Hammond HK, et al. (2010) Disruption of adenylyl cyclase type V does not rescue the phenotype of cardiac-specific overexpression of Gαq protein-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 299:H1459–H1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobise K, Ishikawa Y, Holmer SR, Im MJ, Newell JB, Yoshie H, Fujita M, Susannie EE, Homcy CJ. (1994) Changes in type VI adenylyl cyclase isoform expression correlate with a decreased capacity for cAMP generation in the aging ventricle. Circ Res 74:596–603. [DOI] [PubMed] [Google Scholar]
- Toya Y, Schwencke C, Ishikawa Y. (1998) Forskolin derivatives with increased selectivity for cardiac adenylyl cyclase. J Mol Cell Cardiol 30:97–108. [DOI] [PubMed] [Google Scholar]
- Toyota T, Hattori E, Meerabux J, Yamada K, Saito K, Shibuya H, Nankai M, Yoshikawa T. (2002) Molecular analysis, mutation screening, and association study of adenylate cyclase type 9 gene (ADCY9) in mood disorders. Am J Med Genet 114:84–92. [DOI] [PubMed] [Google Scholar]
- Tsunematsu T, Okumura S, Mototani Y, Ohnuki Y, Jin H, Cai W, Suita K, Sato I, Umemura M, Yokoyama U, et al. (2015) Coupling of β1-adrenergic receptor to type 5 adenylyl cyclase and its physiological relevance in cardiac myocytes. Biochem Biophys Res Commun 458:531–535. [DOI] [PubMed] [Google Scholar]
- Tucker CL, Hurley JH, Miller TR, Hurley JB. (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc Natl Acad Sci USA 95:5993–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadakkan KI, Wang H, Ko SW, Zastepa E, Petrovic MJ, Sluka KA, Zhuo M. (2006) Genetic reduction of chronic muscle pain in mice lacking calcium/calmodulin-stimulated adenylyl cyclases. Mol Pain 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga EV, Stropova D, Rubenzik M, Wang M, Landsman RS, Roeske WR, Yamamura HI. (1998) Identification of adenylyl cyclase isoenzymes in CHO and B82 cells. Eur J Pharmacol 348:R1–R2. [DOI] [PubMed] [Google Scholar]
- Vatner SF, Park M, Yan L, Lee GJ, Lai L, Iwatsubo K, Ishikawa Y, Pessin J, Vatner DE. (2013) Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am J Physiol Heart Circ Physiol 305:H1–H8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. [DOI] [PubMed] [Google Scholar]
- Visel A, Alvarez-Bolado G, Thaller C, Eichele G. (2006) Comprehensive analysis of the expression patterns of the adenylate cyclase gene family in the developing and adult mouse brain. J Comp Neurol 496:684–697. [DOI] [PubMed] [Google Scholar]
- Vorherr T, Knöpfel L, Hofmann F, Mollner S, Pfeuffer T, Carafoli E. (1993) The calmodulin binding domain of nitric oxide synthase and adenylyl cyclase. Biochemistry 32:6081–6088. [DOI] [PubMed] [Google Scholar]
- Vortherms TA, Nguyen CH, Bastepe M, Jüppner H, Watts VJ. (2006) D2 dopamine receptor-induced sensitization of adenylyl cyclase type 1 is Gαs independent. Neuropharmacology 50:576–584. [DOI] [PubMed] [Google Scholar]
- Wada T, Nakamura Y, Cao X, Ohara H, Izumi-Nakaseko H, Ando K, Nakazato Y, Sugiyama A. (2016) Antiviral drug vidarabine possessing cardiac type 5 adenylyl cyclase inhibitory property did not affect cardiohemodynamic or electrophysiological variables in the halothane-anesthetized dogs. J Toxicol Sci 41:115–122. [DOI] [PubMed] [Google Scholar]
- Wagner R, Dudziak K, Herzberg-Schäfer SA, Machicao F, Stefan N, Staiger H, Häring HU, Fritsche A. (2011) Glucose-raising genetic variants in MADD and ADCY5 impair conversion of proinsulin to insulin. PLoS One 6:e23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walseth TF, Graff G, Krick TP, Goldberg ND. (1981) The fate of 18O in guanosine monophosphate during enzymic transformations leading to guanosine 3′,5′-monophosphate generation. J Biol Chem 256:2176–2179. [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR. (2004) Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7:635–642. [DOI] [PubMed] [Google Scholar]
- Wang H, Pineda VV, Chan GC, Wong ST, Muglia LJ, Storm DR. (2003) Type 8 adenylyl cyclase is targeted to excitatory synapses and required for mossy fiber long-term potentiation. J Neurosci 23:9710–9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. (2003) Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol 63:463–468. [DOI] [PubMed] [Google Scholar]
- Wang H, Wu M, Zhu W, Shen J, Shi X, Yang J, Zhao Q, Ni C, Xu Y, Shen H, et al. (2010) Evaluation of the association between the AC3 genetic polymorphisms and obesity in a Chinese Han population. PLoS One 5:e13851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xu H, Wu LJ, Kim SS, Chen T, Koga K, Descalzi G, Gong B, Vadakkan KI, Zhang X, et al. (2011a) Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med 3:65ra3. [DOI] [PubMed] [Google Scholar]
- Wang T, Brown MJ. (2004) Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol Cell Endocrinol 223:55–62. [DOI] [PubMed] [Google Scholar]
- Wang Z, Storm DR. (2011) Maternal behavior is impaired in female mice lacking type 3 adenylyl cyclase. Neuropsychopharmacology 36:772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Balet Sindreu C, Li V, Nudelman A, Chan GC, Storm DR. (2006) Pheromone detection in male mice depends on signaling through the type 3 adenylyl cyclase in the main olfactory epithelium. J Neurosci 26:7375–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li V, Chan GC, Phan T, Nudelman AS, Xia Z, Storm DR. (2009) Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One 4:e6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Nudelman A, Storm DR. (2007) Are pheromones detected through the main olfactory epithelium? Mol Neurobiol 35:317–323. [DOI] [PubMed] [Google Scholar]
- Wang Z, Phan T, Storm DR. (2011b) The type 3 adenylyl cyclase is required for novel object learning and extinction of contextual memory: role of cAMP signaling in primary cilia. J Neurosci 31:5557–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington NM, Howe LD, Paternoster L, Kaakinen M, Herrala S, Huikari V, Wu YY, Kemp JP, Timpson NJ, St Pourcain B, et al. (2015) A genome-wide association study of body mass index across early life and childhood. Int J Epidemiol 44:700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Krupinski J, Kempinski AM, Frankenfield CD. (1994) Molecular cloning and characterization of the type VII isoform of mammalian adenylyl cyclase expressed widely in mouse tissues and in S49 mouse lymphoma cells. J Biol Chem 269:28893–28898. [PubMed] [Google Scholar]
- Watts VJ, Neve KA. (2005) Sensitization of adenylate cyclase by Galpha i/o-coupled receptors. Pharmacol Ther 106:405–421. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Wei J, Wong S, Storm DR. (1996) Regulation of type I adenylyl cyclase by calmodulin kinase IV in vivo. Mol Cell Biol 16:6075–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wayman G, Storm DR. (1996) Phosphorylation and inhibition of type III adenylyl cyclase by calmodulin-dependent protein kinase II in vivo. J Biol Chem 271:24231–24235. [DOI] [PubMed] [Google Scholar]
- Wei R, Ma S, Wang C, Ke J, Yang J, Li W, Liu Y, Hou W, Feng X, Wang G, et al. (2016) Exenatide exerts direct protective effects on endothelial cells through the AMPK/Akt/eNOS pathway in a GLP-1 receptor-dependent manner. Am J Physiol Endocrinol Metab 310:E947–E957. [DOI] [PubMed] [Google Scholar]
- Wen W, Cho YS, Zheng W, Dorajoo R, Kato N, Qi L, Chen CH, Delahanty RJ, Okada Y, Tabara Y, et al. Genetic Investigation of ANthropometric Traits (GIANT) Consortium (2012) Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet 44:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner K, Neumann D, Seifert R. (2014) Analysis of the histamine H2-receptor in human monocytes. Biochem Pharmacol 92:369–379. [DOI] [PubMed] [Google Scholar]
- Werner K, Neumann D, Seifert R. (2016) High constitutive Akt2 activity in U937 promonocytes: effective reduction of Akt2 phosphorylation by the histamine H2-receptor and the β2-adrenergic receptor. Naunyn Schmiedebergs Arch Pharmacol 389:87–101. [DOI] [PubMed] [Google Scholar]
- Whisnant RE, Gilman AG, Dessauer CW. (1996) Interaction of the two cytosolic domains of mammalian adenylyl cyclase. Proc Natl Acad Sci USA 93:6621–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek L, Maas JW, Jr, Muglia LM, Vogt SK, Muglia LJ. (2010) Temporal and regional regulation of gene expression by calcium-stimulated adenylyl cyclase activity during fear memory. PLoS One 5:e13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland T, Ronzani M, Jakobs KH. (1992) Stimulation and inhibition of human platelet adenylylcyclase by thiophosphorylated transducin β γ-subunits. J Biol Chem 267:20791–20797. [PubMed] [Google Scholar]
- Williams C. (2004) cAMP detection methods in HTS: selecting the best from the rest. Nat Rev Drug Discov 3:125–135. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Cooper DM. (2007) Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev 87:965–1010. [DOI] [PubMed] [Google Scholar]
- Willoughby D, Halls ML, Everett KL, Ciruela A, Skroblin P, Klussmann E, Cooper DM. (2012) A key phosphorylation site in AC8 mediates regulation of Ca2+-dependent cAMP dynamics by an AC8-AKAP79-PKA signalling complex. J Cell Sci 125:5850–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, Ciruela A, Cooper DM. (2010) AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem 285:20328–20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Ong HL, De Souza LB, Wachten S, Ambudkar IS, Cooper DM. (2014) TRPC1 contributes to the Ca2+-dependent regulation of adenylate cyclases. Biochem J 464:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windholz J, Kovacs P, Tönjes A, Dittrich K, Blüher S, Kiess W, Stumvoll M, Körner A. (2011) Effects of genetic variants in ADCY5, GIPR, GCKR and VPS13C on early impairment of glucose and insulin metabolism in children. PLoS One 6:e22101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Rasmusson AM, Mitchell KS, Logue MW, Baldwin CT, Miller MW. (2014) A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depress Anxiety 31:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Storm DR. (2002) Generation of adenylyl cyclase knockout mice. Methods Enzymol 345:206–231. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. (1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23:787–798. [DOI] [PubMed] [Google Scholar]
- Wong ST, Trinh K, Hacker B, Chan GC, Lowe G, Gaggar A, Xia Z, Gold GH, Storm DR. (2000) Disruption of the type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron 27:487–497. [DOI] [PubMed] [Google Scholar]
- Woon CW, Soparkar S, Heasley L, Johnson GL. (1989) Expression of a Gαs/Gαi chimera that constitutively activates cyclic AMP synthesis. J Biol Chem 264:5687–5693. [PubMed] [Google Scholar]
- Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR, Ripke S, MacIntyre DJ, McGhee KA, Maclean AW, et al. (2012) Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 17:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. (1995) Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci USA 92:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Storm DR. (2012) Role of signal transduction crosstalk between adenylyl cyclase and MAP kinase in hippocampus-dependent memory. Learn Mem 19:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, He L, Takemoto M, Jalanko H, Chan GC, Storm DR, Betsholtz C, Tryggvason K, Patrakka J. (2011) Glomerular podocytes express type 1 adenylate cyclase: inactivation results in susceptibility to proteinuria. Nephron, Exp Nephrol 118:e39–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Garcia MA, Carlson AE, Schuh SM, Babcock DF, Jaiswal BS, Gossen JA, Esposito G, van Duin M, Conti M. (2006) Soluble adenylyl cyclase (sAC) is indispensable for sperm function and fertilization. Dev Biol 296:353–362. [DOI] [PubMed] [Google Scholar]
- Xie K, Masuho I, Brand C, Dessauer CW, Martemyanov KA. (2012) The complex of G protein regulator RGS9-2 and Gβ(5) controls sensitization and signaling kinetics of type 5 adenylyl cyclase in the striatum. Sci Signal 5:ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Rich TC, Scheitrum C, Conti M, Richter W. (2011) Inactivation of multidrug resistance proteins disrupts both cellular extrusion and intracellular degradation of cAMP. Mol Pharmacol 80:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Isaacs C, Hall IP, Emala CW. (2001) Human airway smooth muscle expresses 7 isoforms of adenylyl cyclase: a dominant role for isoform V. Am J Physiol Lung Cell Mol Physiol 281:L832–L843. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Götz ME, Ozawa H, Luckhaus C, Saito T, Rösler M, Riederer P. (2000) Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim Biophys Acta 1535:60–68. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Pohli S, Durany N, Ozawa H, Saito T, Boissl KW, Zöchling R, Riederer P, Böning J, Götz ME. (2001) Increased levels of calcium-sensitive adenylyl cyclase subtypes in the limbic system of alcoholics: evidence for a specific role of cAMP signaling in the human addictive brain. Brain Res 895:233–237. [DOI] [PubMed] [Google Scholar]
- Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. (2007) Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130:247–258. [DOI] [PubMed] [Google Scholar]
- Yan SZ, Beeler JA, Chen Y, Shelton RK, Tang WJ. (2001) The regulation of type 7 adenylyl cyclase by its C1b region and Escherichia coli peptidylprolyl isomerase, SlyD. J Biol Chem 276:8500–8506. [DOI] [PubMed] [Google Scholar]
- Yan SZ, Hahn D, Huang ZH, Tang WJ. (1996) Two cytoplasmic domains of mammalian adenylyl cyclase form a Gs α- and forskolin-activated enzyme in vitro. J Biol Chem 271:10941–10945. [DOI] [PubMed] [Google Scholar]
- Yan SZ, Huang ZH, Andrews RK, Tang WJ. (1998) Conversion of forskolin-insensitive to forskolin-sensitive (mouse-type IX) adenylyl cyclase. Mol Pharmacol 53:182–187. [DOI] [PubMed] [Google Scholar]
- Yan SZ, Huang ZH, Shaw RS, Tang WJ. (1997) The conserved asparagine and arginine are essential for catalysis of mammalian adenylyl cyclase. J Biol Chem 272:12342–12349. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang YL, Liu S, Zhang PP, Chen Z, Liu M, Tang H. (2014) miR-181b promotes cell proliferation and reduces apoptosis by repressing the expression of adenylyl cyclase 9 (AC9) in cervical cancer cells. FEBS Lett 588:124–130. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Cooper DM. (1992) Cloning and expression of a Ca(2+)-inhibitable adenylyl cyclase from NCB-20 cells. Proc Natl Acad Sci USA 89:6716–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Tabakoff B. (1999) Ethanol’s actions on cAMP-mediated signaling in cells transfected with type VII adenylyl cyclase. Alcohol Clin Exp Res 23:1457–1461. [PubMed] [Google Scholar]
- Yoshimura M, Pearson S, Kadota Y, Gonzalez CE. (2006) Identification of ethanol responsive domains of adenylyl cyclase. Alcohol Clin Exp Res 30:1824–1832. [DOI] [PubMed] [Google Scholar]
- Yu HJ, Unnerstall JR, Green RD. (1995) Determination and cellular localization of adenylyl cyclase isozymes expressed in embryonic chick heart. FEBS Lett 374:89–94. [DOI] [PubMed] [Google Scholar]
- Yu P, Sun M, Villar VA, Zhang Y, Weinman EJ, Felder RA, Jose PA. (2014) Differential dopamine receptor subtype regulation of adenylyl cyclases in lipid rafts in human embryonic kidney and renal proximal tubule cells. Cell Signal 26:2521–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Liu R, LaPlant Q, Xiao G, Renthal W, Chan GC, Storm DR, Aghajanian G, Nestler EJ. (2008) Distinct roles of adenylyl cyclases 1 and 8 in opiate dependence: behavioral, electrophysiological, and molecular studies. Biol Psychiatry 63:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu Y, Ruoho AE, Hurley JH. (1997) Structure of the adenylyl cyclase catalytic core. Nature 386:247–253. [DOI] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, Hell JW. (2013) Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. J Biol Chem 288:17918–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Storm DR, Wang H. (2011) Bidirectional synaptic plasticity and spatial memory flexibility require Ca2+-stimulated adenylyl cyclases. J Neurosci 31:10174–10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Xiang N, Chen Y, Sliwerska E, McInnis MG, Burmeister M, Zöllner S. (2010) Family-based association analysis to finemap bipolar linkage peak on chromosome 8q24 using 2,500 genotyped SNPs and 15,000 imputed SNPs. Bipolar Disord 12:786–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XB, Lutz S, Steffens F, Korth M, Wieland T. (2007) Oxytocin receptors differentially signal via Gq and Gi proteins in pregnant and nonpregnant rat uterine myocytes: implications for myometrial contractility. Mol Endocrinol 21:740–752. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Wang GX, Huneke B, Wieland T, Korth M. (2000) Pregnancy switches adrenergic signal transduction in rat and human uterine myocytes as probed by BKCa channel activity. J Physiol 524:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Oxford GS. (2011) Differential gene expression of neonatal and adult DRG neurons correlates with the differential sensitization of TRPV1 responses to nerve growth factor. Neurosci Lett 500:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang LK, Xu GP, Pan XR, Lou YJ, Zou QP, Xia D, Yan WW, Zhang YT, Jia PM, Tong JH. (2014) MicroRNA-181a-mediated downregulation of AC9 protein decreases intracellular cAMP level and inhibits ATRA-induced APL cell differentiation. Cell Death Dis 5:e1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke RA, Wierzbicki IH, Baarda BI, Sikora AE. (2015) The Neisseria gonorrhoeae Obg protein is an essential ribosome-associated GTPase and a potential drug target. BMC Microbiol 15:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. (2003) Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J 17:82–84. [DOI] [PubMed] [Google Scholar]