

Thalassemia is the world’s most common form of inherited anemia, and in economically undeveloped countries still accounts for tens of thousands of premature deaths every year.1 The accumulation of free excess α-globin chains in red blood cells and their precursors, as a result of the decreased production of β-globin, is believed to be the main pathophysiological mechanism leading to hemolytic anemia and ineffective erythropoiesis in β-thalassemia.2 Clinical genetic data accumulated over the last 30 years indicate that a natural reduction in α-globin chain output by 25–50%, resulting from co-inherited α-thalassemia, ameliorates the disease phenotype in patients with β-thalassemia.3–5 Herein, we have developed and performed a targeted small molecule screen to identify compounds which downregulate α-globin expression. This identified IOX1, a pan-histone demethylase inhibitor, which selectively downregulates α-globin expression without perturbing erythroid differentiation or general gene expression, more specifically β-like globin expression. Our data show that selective silencing of α-globin expression in erythroid cells is pharmacologically feasible, and IOX1 is a lead compound to developing new therapy to treat β-thalassemia through the novel pathway of downregulating α-globin expression.
We first optimized a serum-free, miniature erythroid differentiation system starting from primary human CD34+ cells, the exact type of cells we would ultimately like to target in vivo (Figure 1). This culture system produced a sufficient number of viable, relatively pure, and synchronous populations of human erythroid cells in vitro to enable us to perform high throughput screens (Figure 1A,B). CD34+ cells were differentiated in 96-well plates over 21 days along the erythroid lineage, and the morphology and immunophenotypical characteristics of the resultant cells faithfully recapitulated normal erythropoiesis (Figure 1C,D). These cells demonstrated a gradual increase in expression of the globin genes (Figure 1E) and other erythroid-specific genes (Online Supplementary Figure S1), and the hemoglobin protein analysis confirmed the higher proportion of fetal hemoglobin (HbF) and adult hemoglobin (HbA) in cells differentiated from umbilical cord and adult CD34+ cells, respectively (Figure 1F).
Figure 1.
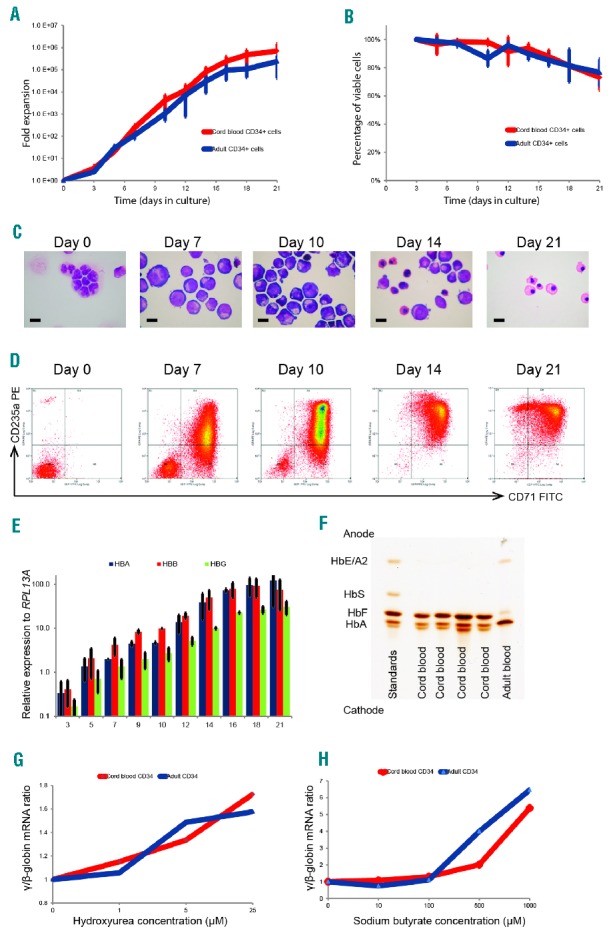
Characterization and validation of the small scale erythroid differentiation system used for small molecule screen. Human CD34+ hematopoietic stem and progenitor cells purified from umbilical cord or adult peripheral blood were cultured in a two-phase liquid culture system in a serum-free medium for 21 days. (A) Mean fold expansion during erythroid differentiation; error bars represent SD (n=3). (B) Mean percentage cell viability during erythroid differentiation; error bars represent SD (n=3). (C) Morphology of cells by cytospins stained using modified Wright’s stain at different time points (day 0–21), in culture representing different stages of erythroid differentiation, demonstrating progression through stages of pro-, basophilic and polychromatic to orthochromatic erythroblasts; scale bar – 10μm. (D) Representative flow cytometry plots of cells stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD71 and phycoerythrin (PE)-conjugated anti-CD235a antibodies, demonstrating sequential expression of CD71 followed by CD235a and subsequent loss of CD71. (E) Relative expression of α (HBA)-, β (HBB)- and γ (HBG)-globin mRNA levels quantified by qPCR and normalized to the housekeeping gene RPL13A at different time points in culture (adult blood CD34+ cells); error bars represent SD (n=3). (F) Hemoglobin subtypes of the erythroid cells differentiated from umbilical cord and adult CD34+ cells analyzed by isoelectric focusing. The samples were run against a commercial set of standards. (G and H) γ/β mRNA ratio after incubation of erythroid cells in a dose range of hydroxyurea and sodium butyrate. Compounds were added to the liquid culture medium on day 7 of erythroid cell differentiation (corresponding to the proerythroblast stage), and the cells were then incubated in a 5% CO2 atmosphere at 37°C for 72 hours. Data on erythroid cells differentiated from umbilical cord and adult CD34+ cells are presented in red and blue, respectively. mRNA: messenger ribonucleic acid; HbF: hemoglobin F; HbA: hemoglobin A; HbS: hemoglobin S; HbE; hemoglobin E; HBA: α-globin; HBB: β-globin; HBG: γ-globin; HbA2: hemoglobin A2.
We then validated the culture system using hydroxyurea and sodium butyrate, which were previously shown to alter globin gene expression. Erythroid cells incubated with these compounds demonstrated a dose dependent increase in the γ/β messenger ribonucleic acid (mRNA) ratio, consistent with previously reported data6 (Figure 1G,H). Next, we transfected erythroid cells with two validated small interfering RNAs targeting human α-globin RNA, which resulted in the expected knockdown of α-globin expression (Online Supplementary Figure S2). These observations confirm that the small-scale erythroid differentiation system which we have optimized is a valid tool to examine changes in globin gene expression in vitro.
Previous studies have revealed contrasting epigenetic environments containing the human α- and β-globin genes.7,8 The human α-globin gene cluster is located on chromosome 16, in a gene dense, early replicating, open chromatin environment and its promoter is associated with unmethylated CpG islands and, in non-erythroid cells, is enriched for H3K27me3 which signals transcriptional silencing.9,10 By contrast, in non-erythroid cells the β-globin gene is situated in a relatively gene sparse, late replicating, closed heterochromatic environment on chromosome 11, and the promoter of the β-globin gene is methylated rather than enriched for H3K27me3.11 Therefore, in the search for drugs which specifically alter expression of α-globin, we performed a selective screen, using a small molecule library of epigenetically active cell permeable compounds, potentially targeting these different epigenetic environments. This library contains a collection of 37 compounds that were designed to inhibit a wide range of epigenetic pathways (Online Supplementary Table S1). Erythroid cells were incubated for 72 hours with these compounds, and gene expression levels were obtained using Fluidigm high throughput quantitative polymerase chain reaction (qPCR) system. The primary screening criterion was downregulation of α-globin expression without altering β-globin expression, and an α/β globin mRNA ratio of less than 0.75 was considered as the cutoff for identifying high-scoring compounds. This screen identified four compounds that downregulate α-globin expression: histone demethylase (KDM) inhibitor, IOX1; histone deacetylase inhibitor, vorinostat; histone methyltransferase inhibitor, chaetocin and lysine-specific histone demethylase 1 inhibitor, tranylcypromine (Figure 2B and Online Supplementary Figures S3–S5). Of these compounds, the novel KDM inhibitor IOX1 provided the most promising results with the desired effects on globin gene expression. Chaetocin decreased the viability of erythroid cells at low concentrations and tranylcypromine markedly retarded erythroid differentiation, as evidenced by immature cell morphology and lack of expression of erythroid-specific cell surface proteins. Therefore these two compounds were not followed up further (Online Supplementary Figure S6). Vorinostat downregulated α-globin expression whilst inducing γ-globin expression (Online Supplementary Figure S7) and is currently under further investigation.
Figure 2.

Small molecule screen identified histone lysine demethylase inhibitor IOX1 as a potential compound to downregulate α-globin expression. (A) A schematic of the work flow of the small molecule screen. Small molecules were added to the liquid culture medium on day 7 of erythroid cell differentiation (corresponding to the proerythroblast stage), and the cells were then incubated in a 5% CO2 atmosphere at 37°C for 72 hours. Gene expression was analyzed using the Fluidigm high throughput qPCR system. (B) Representative heat map (one of 3 biological repeats) demonstrating fold differences of α- and β-globin mRNA levels in erythroid cells treated with small molecules. The expression levels were normalized to multiple housekeeping genes (RPL13A, RPL18, GAPDH and FTH1) and referenced to the vehicle (DMSO) control. Each row represents a single compound and each column represents one of three technical repeats performed for the α- and β-globin genes, each with colors ranging from dark red (downward expression) to blue (upward expression); HBA, α-globin; HBB, β-globin. (see Online Supplementary Figure S3 for full heat map). Four compounds that downregulate α-globin expression are marked using green rectangles. (C) α/β-globin mRNA ratios in erythroid cells (differentiated from cord blood CD34+ cells) treated with a dose range of IOX1 analyzed by qPCR. Error bars represent SD (n=3); *P<0.05, **P<0.01 relative to DMSO control. (D) Globin mRNA levels in erythroid cells (differentiated from cord blood CD34+ cells) treated with IOX1 (40μM) quantified by nCounter Digital Analyzer (NanoString Technologies). The NanoString count for each globin gene was normalized to the counts of multiple housekeeping genes (RPL13A, RPL18, GAPDH, PABPC1, CA2, FTH1, PAIP2 and LAPTM4A). Error bars represent SD (n=3); *P<0.05, **P<0.01 relative to DMSO control. DMSO: dimethyl sulfoxide; mRNA: messenger ribonucleic acid; RNA: ribonucleic acid.
To further examine the effect of IOX1 on globin gene expression, we titrated the concentration of IOX1 with the developing erythroid cells. This confirmed initial observations: IOX1 caused a dose-dependent decrease in α-globin expression, whereas the expression of β-globin was largely unaffected (Online Supplementary Figures S8 and S9). The decrease in α/β-globin mRNA ratios was statistically significant at all doses tested (Figure 2C). We then analyzed the mRNA levels of all globin genes in erythroid cells treated with IOX1 using the nCounter Digital Analyzer (NanoString Technologies), which found that IOX1 significantly downregulated α-, γ-, μ- and ζ-globin expression (Figure 2D). Interestingly, with the exception of γ-globin, IOX1 downregulated α- and other α-like globin genes (μ and ζ) situated in the α-globin locus, whereas the expression levels of β-like globin genes (β, δ and ε) were unaffected, suggesting that IOX1 acts selectively on the α-globin locus.
IOX1 reduced cell expansion by about 40% (fold expansion dropped from 18-fold to 11-fold) at 40μM concentration, but the proportion of viable cells remained unchanged over all dose levels (Figure 3A,B). This suggests that IOX1 has a mild inhibitory action on erythroid cell proliferation in vitro, although it does not adversely affect cellular viability. Morphologically, erythroid cells treated with a dose range of IOX1 differentiated in a similar way to untreated cells, suggesting that IOX1 does not alter erythroid differentiation (Figure 3C). This was further confirmed by immunophenotypic cell surface marker expression, which demonstrated no significant differences in the expression levels of CD34, CD71 and CD235a between IOX1 treated and control cells (Figure 3D–G). This is of particular importance, as some other compounds that are currently being tested for the treatment of β-thalassemia via the upregulation of γ-globin and fetal hemoglobin alter erythroid cell differentiation.13
Figure 3.
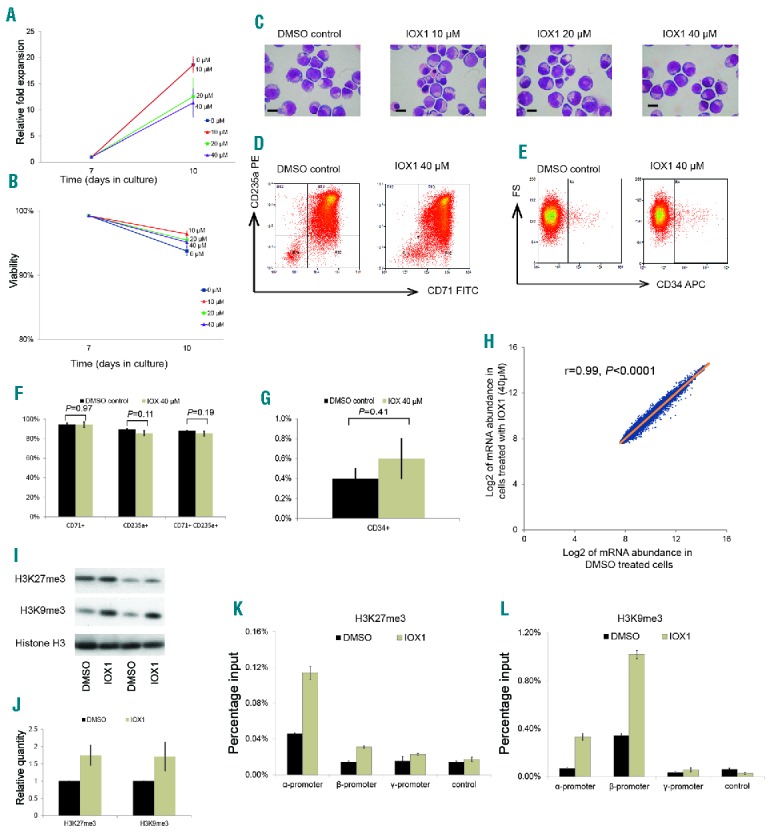
Effects of IOX1 treatment on erythroid cells. Erythroid cells were incubated with IOX1 (40μM concentration, unless specified otherwise) or DMSO (vehicle) control for 72 hours from day 7 of culture. (A) Mean cell proliferation shown as relative fold expansions of erythroid cells treated with a dose range of IOX1. Error bars represent SEM (n=3). (B) Mean percentage viability of erythroid cells treated with a dose range of IOX1. Error bars represent SEM (n=3). (C) Representative cytospins of cells on day 10 of erythroid cell differentiation (corresponding to basophilic erythroblasts stage), treated with a dose range of IOX1 and stained by modified Wright–s stain; scale bar – 10μm. (D) Representative flow cytometry plots of cells on day 10 of erythroid cell differentiation treated with IOX1, stained with FITC-conjugated anti-CD71 and PE-conjugated anti-CD235a antibodies. (E) Representative flow cytometry plots of the same cells shown in (D) stained with APC-conjugated anti-CD34. (F) Percentages of cells expressing CD71 and CD235a in IOX1 treated and control groups; error bars represent SD (n=3). (G) Percentage of cells expressing CD34 in IOX1 treated and control groups; error bars represent SD (n=3). (H) Microarray analysis comparing global gene expression of IOX1 treated and control cells (n=4). (I) Western blot of histone extracts from erythroid cells treated with IOX1 showing abundance of H3K27me3 and H3K9me3 histone modifications and histone H3 (internal control). Two technical replicates (different loading dilutions) of one of the two biologically independent experiments are shown. (J) Relative quantification of abundance of H3K27me3 and H3K9me3 histone modifications analyzed by western blot. (K&L) ChIP-PCR assay demonstrating abundance of H3K27me3 (K) and H3K9me3 (L) histone modifications at the α-, β- and γ-globin promoters in erythroid cells treated with IOX1 compared to a DMSO control. An intergenic region between the ε- and γ-globin genes was used as the negative control. Result of one of two biologically independent experiments is shown; error bars represent SD of technical repeats. DMSO: dimethyl sulfoxide; mRNA: messenger ribonucleic acid; FITC: fluorescein isothiocyanate; PE: phycoerythrin; APC: allophycocyanin.
We then conducted microarray analysis to examine the possible effects of IOX1 on global erythroid gene expression. Using this microarray, which assayed over 47 000 transcripts, mRNA abundance of most of the genes were similar in IOX1 treated cells when compared to the control, with a very high correlation coefficient (R=0.992) (Figure 3H). In total, only 162 genes were differentially expressed between the two groups (Online Supplementary Tables S2 and S3). Next, we analyzed the expression levels and fold differences of 52 genes which were reported as essential for erythroid physiology (adopted from the publicly available online database, Hembase). Expression levels in IOX1 treated and untreated cells were not significantly different in all but one of the 52 genes, further confirming the minimal effects of IOX1 on erythroid physiology (Online Supplementary Table S4).
Next we investigated the mechanism by which IOX1 exerts its effect on globin gene expression in erythroid cells. Gene ontology enrichment analysis performed on differentially regulated gene sets obtained by microarray analysis did not reveal a simple interpretation of how IOX1 specifically affects α-globin expression. However, previous reports on IOX1 demonstrate that it acts as a broad range inhibitor of histone demethylase enzymes.12,14 Therefore, we examined the changes in the pattern of histone methylation in erythroid cells treated with IOX1. Western blot of histone protein extracts from IOX1 treated erythroid cells showed an increase in two repressive chromatin modifications, H3K27me3 and H3K9me3 (Figure 3I,J).
We then looked at the changes of these chromatin modifications at α- and β-globin loci using chromatin immunoprecipitation (ChIP) assays (Figure 3K,L). In the untreated cells, H3K27me3 abundance at the β-globin promoter was similar to the level observed at the negative control region, whereas the level at the α-globin promoter was higher, which is consistent with our previous findings in which we showed that human α-globin expression may be reduced by polycomb-mediated repression.9 Furthermore, treatment with IOX1 increased H3K27me3 abundance at both the α- and β-globin promoters, with a more pronounced change at the α-promoter. In contrast, H3K9me3 was more abundant at the β-globin promoter compared to the α-globin promoter in untreated cells, which further increased after IOX1 treatment. These observations suggest that the α-globin silencing effect of IOX1 is likely to be mediated via the inhibition of the KDM enzymes responsible for the removal of H3K27 methylation marks at the α-globin locus. KDM enzymes known to act at this site are KDM6A and KDM6B, and IOX1 inhibits these enzymes at various IC50 values in vitro.14,15 During the initial compound screen, GSK-J4, a specific inhibitor of KDM6A/B, downregulated both α- and β-globin. However, a recent report suggests that GSK-J4 also inhibits KDM5 enzymes that demethylase H3K4me3, which might explain why it downregulated both α- and β-globin. Our attempts to phenocopy the effect of IOX1 by knocking down individual enzymes were not successful, suggesting the presence of additional KDM enzymes acting at the H3K27 locus or an alternative pathway of its action. However, this should not preclude the use of IOX1 as a lead compound for reducing α-globin expression.
In conclusion, we have demonstrated that selective silencing of α-globin expression, without affecting the β-like globin expression or erythroid differentiation, is pharmacologically feasible. The histone demethylase inhibitor, IOX1, exerts the desired changes in erythroid cells and has potential as a lead compound to develop a novel therapy for β-thalassemia, which is still a life-limiting disease without a definitive cure.
Supplementary Material
Acknowledgments
The authors would like to thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070) for the generation of the Gene Expression data. We also acknowledge Jennifer Eglington of Oxford University Hospital NHS Trust for helping with the isoelectric focusing of hemoglobin.
Footnotes
Funding: this work was supported by grants to DRH by the UK Medical Research Council [grant number MC_UU_12025/unit programme MC_UU_12009/4] and the NIHR Oxford Biomedical Research Centre. SM is a Commonwealth Scholar, funded by the UK government. We also acknowledge funding from the Helmut Horten Foundation. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust [106169/ZZ14/Z].
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379(9813):373–383. [DOI] [PubMed] [Google Scholar]
- 2.Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4 ed. Oxford: Blackwell Science, 2001. [Google Scholar]
- 3.Mettananda S, Gibbons RJ, Higgs DR. alpha-Globin as a molecular target in the treatment of beta-thalassemia. Blood. 2015; 125(24):3694–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galanello R, Sanna S, Perseu L, et al. Amelioration of Sardinian β0 thalassemia by genetic modifiers. Blood. 2009;114(18):3935–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Premawardhena A, Fisher CA, Olivieri NF, et al. Haemoglobin E beta thalassaemia in Sri Lanka. Lancet. 2005;366(9495):1467–1470. [DOI] [PubMed] [Google Scholar]
- 6.Bradner JE, Mak R, Tanguturi SK, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci USA. 2010; 107(28):12617–12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mettananda S, Gibbons RJ, Higgs DR. Understanding alpha-globin gene regulation and implications for the treatment of beta-thalassemia. Ann N Y Acad Sci. 2016;1368(1):16–24. [DOI] [PubMed] [Google Scholar]
- 8.Brown KE, Amoils S, Horn JM, et al. Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat Cell Biol. 2001;3(6):602–606. [DOI] [PubMed] [Google Scholar]
- 9.Garrick D, De Gobbi M, Samara V, et al. The role of the polycomb complex in silencing alpha-globin gene expression in nonerythroid cells. Blood. 2008;112(9):3889–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Gobbi M, Garrick D, Lynch M, et al. Generation of bivalent chromatin domains during cell fate decisions. Epigenetics Chromatin. 2011;4(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higgs DR, Garrick D, Anguita E, et al. Understanding alpha-globin gene regulation: Aiming to improve the management of thalassemia. Ann N Y Acad Sci. 2005;1054:92–102. [DOI] [PubMed] [Google Scholar]
- 12.King ONF, Li XS, Sakurai M, et al. Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PloS one. 2010;5(11):e15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi L, Cui S, Engel JD, Tanabe O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med. 2013; 19(3):291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkinson RJ, Tumber A, Yapp C, et al. 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chem Sci. 2013;4(8):3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Gen. 2007;8(11):829–833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.