

Abstract
Context
Partial lipodystrophy (PL) is associated with metabolic co-morbidities but may go undiagnosed as the disease spectrum is not fully described.
Objective
Define disease spectrum in PL using genetic, clinical (historical, morphometric) and laboratory characteristics.
Design
Cross-sectional evaluation.
Participants
23 patients (22 with familial, one acquired, 78.3% female, aged 12–64 years) with PL and non-alcoholic fatty liver disease (NAFLD).
Measurements
Genetic, clinical and laboratory characteristics, body composition indices, liver fat content by MRI, histopathological and immunofluorescence examinations of liver biopsies.
Results
7 patients displayed heterozygous pathogenic variants in LMNA. Two related patients had a heterozygous, likely pathogenic novel variant of POLD1 (NM002691.3: c.3199 G>A; p.E1067K). Most patients had high ratios (>1.5) of %fat trunk to %fat legs (FMR) when compared to reference normals. Liver fat quantified using MR Dixon method was high (11.3+6.3%) and correlated positively with hemoglobin A1c and triglycerides while leg fat by dual-energy X-ray absorptiometry (DEXA) correlated negatively with triglycerides. In addition to known metabolic comorbidities; chronic pain (78.3%), hypertension (56.5%) and mood disorders (52.2%) were highly prevalent. Mean NAFLD Activity Score (NAS) score was 5±1 and 78.3% had fibrosis. LMNA-immunofluorescence staining from select patients (including one with the novel POLD1 variant) showed a high degree of nuclear atypia and disorganization.
Conclusions
PL is a complex multi-system disorder. Metabolic parameters correlate negatively with extremity fat and positively with liver fat. DEXA-based FMR may prove useful as a diagnostic tool. Nuclear disorganization and atypia may be a common biomarker even in the absence of pathogenic variants in LMNA.
Keywords: Partial lipodystrophy, diabetes, hypertriglyceridemia
Introduction
Partial lipodystrophy (PL) is a cluster of rare heterogeneous diseases characterized by selective loss of adipose tissue, which places patients at risk for common conditions typically associated with metabolic syndrome(1–3). Commonly associated features are insulin resistance and diabetes, hypertriglyceridemia and nonalcoholic fatty liver disease (NAFLD)(4). PL may be acquired or inherited and pathogenic variants in multiple genes have been identified as causative for inherited PL including LMNA, PPARG, PLIN1, AKT2, CIDEC(2) and more recently LIPE(5) and ADRA2A(6).
One obstacle in development of novel treatment strategies for these disorders has been the lack of documentation of the differentiating features of these syndromes from more common forms of truncal obesity with metabolic syndrome and Type 2 diabetes. This was noted during the Advisory Meeting of the FDA for approval of Metreleptin for the treatment of lipodystrophy syndromes (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM379648.pdf). Another problem is the lack of objective description of natural history and disease burden. Stated differently, are patients with PL facing different clinical problems, comorbidity risk and mortality compared to patients with more common metabolic disorders such as Type 2 diabetes and truncal obesity? If the answers to these questions are yes, then the pursuit of specific treatments for these disorders will be of high value despite the small number of patients afflicted with PL.
Our study is one of the first in the field to specifically describe the baseline drug-naïve state of patients participating in a drug trial. Accumulation of data such as these presented here may allow more precise definition of the disease state, possibly helping the development of specific diagnostic criteria or disease severity scores. We also hope to increase recognition of similar patients by providing extensive clinical data to endocrinologists.
Methods
We have been recruiting patients with PL for an open-label study conducted at the University of Michigan funded by NIDDK (approved by University of Michigan IRBMED) to evaluate the molecular effects of an investigational drug on treating liver disease associated with PL (ClinicalTrials.gov identifier: NCT01679197, HUM00058708). Here, we describe baseline characteristics of 23 patients who completed screening and study initiation procedures but have not received any investigational drug. Diagnosis of PL was based on physician assessment with evidence of selective loss of body fat(3). Patients were included with both inherited and acquired forms of PL, excluding lipodystrophy associated with human immunodeficiency virus (HIV). Patients entering this study had a liver ultrasound showing presence of fatty liver disease or prior biopsy showing evidence of non-alcoholic steatohepatitis (NASH), no evidence of other forms of liver disease and alcohol consumption of less than 40 grams per week.
After informed consent was obtained from all patients or guardians, history and physical examinations were performed and previous medical records were examined where possible to verify historical details. Blood samples after overnight fast were analyzed for leptin levels using a commercial ELISA assay (EMDMilipore, Billerica, MA). Hemoglobin A1c, fasting lipids and hepatic function tests were determined in the Clinical Pathology Laboratory of University of Michigan using auto-analyzer equipment.
Clinical and research resources were used to determine the molecular basis of disease in patients (described in Supplemental Methods). Body composition was evaluated using anthropometric measurements, including skin thickness and waist and hip circumferences using standardized techniques. Fat and lean body mass were estimated using dual X-ray absorptiometry (DEXA) (GE Lunar Prodigy, model PA +41744, Madison, WI)(7). Ratio of percent fat mass of the trunk to percent fat mass of the legs (FMR) was calculated.
For determination of hepatic fat content, magnetic resonance (MR) imaging using quantitative multi-echo Dixon method and multi-echo MR-spectroscopy have been employed (method described in Supplemental Methods and Supplemental Figure 1). Transcutaneous liver biopsies were performed and total NASH scores were calculated from histopathological examination using the simplified clinical criteria as presented in Supplemental Table 1, which is a modified and clinically applied version of NIH NASH Clinical Network Criteria(8). NASH score equals the sum of scores for steatosis (0–3), lobular inflammation (0–3), hepatocellular ballooning (0–2) and fibrosis (0–4). NAFLD activity score (NAS) includes these parameters, but without fibrosis. In addition, select liver biopsy specimens were processed for lamin A/C immunofluorescence staining, then scored in a blinded fashion (methods described under Supplemental Methods).
Statistical analyses were performed using GraphPad Prism version 6.05 (LaJolla, CA) and SAS version 9.2 (Cary, NC). Correlations were investigated by calculating Pearson’s correlation coefficient. Two tailed p-value was calculated, and P < 0.05 was considered statistically significant. For quantitation of lamin immunofluorescence data, statistical significance was determined via one-way ANOVA followed by Tukey post-hoc test at a threshold of P < 0.05.
Results
Patient characteristics and genetic evaluation
Twenty-three patients with PL were enrolled who were 12–64 years old (median age 43), 18 female and 5 male. The majority, 20 patients were Caucasian, 2 Hispanic and one African-American. Summary characteristics are shown in Table 1, and detailed clinical data can be found in Supplemental Table 2–5. Of known genes, LMNA variants were quite common, identified in 7 patients. Of these 7 patients, 5 had a heterozygous pathogenic variant at position 482, the hot spot. Four of these of these patients harbored R482Q pathogenic variant (2 sisters, patients 13 and 20, and two unrelated patients, 17 and 18). One patient (patient 21) harbored the previously reported R482W pathogenic variant and was also diagnosed with muscular dystrophy(9). Patient 10 also harbored a previously reported R60G pathogenic variant (10) and had a history of dilated non-ischemic cardiomyopathy requiring heart transplant at age 25. Patient 22 was heterozygous for LMNA R349W and had unique clinical features including loss of fat from the face, hands and feet, a large dorsocervical fat pad, proteinuria, cardiomyopathy (believed to be both primary and ischemic), infertility, hyperandrogenism and alopecia (Figure 1, Supplemental Table 2–5). Another interesting laboratory abnormality in patient 22 is low alkaline phosphatase level, typically associated with progeria(11, 12). There have been two other published instances of this mutation, one in a male patient with similar fat distribution, but without the exaggerated dorsocervical fat pad, diabetes, or other features described here(13). In addition, a large kindred with focal segmental glomerulosclerosis and proteinuria in association with PL has been reported(14).
Table 1.
Demographics and characteristics
| Female/Male, n | 18/5 |
| Age (years), mean (range) | 43 (12–64) |
| Race, n (%) White Black Other |
20 (87.0) 1 (4.3) 2 (8.7) |
| Lipodystrophy subtype, n (%) Familial partial lipodystrophy Acquired partial lipodystrophy |
22 (95.7) 1 (4.3) |
| Mutation if known, n LMNA POLD1 |
7 2, subjects are related |
| Body weight (kg), mean (SD) | 77.2 (21.4) |
| Body mass index (kg/m2), mean (SD) | 27.3 (5.9) |
| Waist to hip ratio, mean (SD) | 0.99 (0.08) |
| Skin fold thickness, anterior mid-thigh* (mm), mean (SD), [range] | 12.1 (7.7) [4–29] |
| %Fat trunk / %Fat legs*, mean (SD) | 1.78 (0.53)) |
| MR Spectroscopy liver fat (%), mean (SD) | 13.8 (6.1) |
| Dixon method MR liver fat (%), mean (SD) | 11.9 (6.3) |
| Hemoglobin A1c (%), mean (SD) | 8.6 (1.9) |
| Leptin level (ng/ml), mean (SD) | 22.3 (16.6) |
| Fasting Triglycerides (mg/dl), mean (SD) | 1058 (1745) |
| AST (IU/L), mean (SD) | 42 (28) |
| ALT (IU/L), mean (SD) | 52 (37) |
| Hemoglobin (g/dL), mean (SD) | 13.8 (1.3) |
| Platelets (K/uL), mean (SD) | 265 (55) |
| Creatinine (mg/dL), mean (SD) | 0.67 (0.22) |
| CK (IU/L), mean (SD) | 145 (101) |
excludes patient with acquired partial lipodystrophy; Abbreviations: MR, magnetic resonance; AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase
Figure 1.
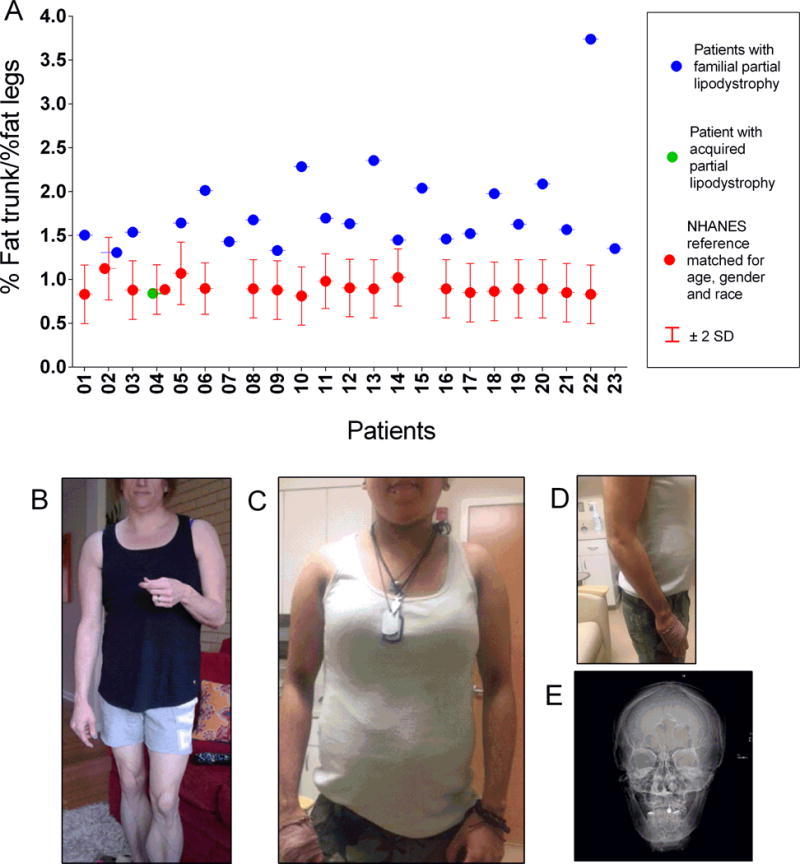
A. Comparison of %fat trunk / %fat legs (fat mass ratio, FMR) in 23 patients with partial lipodystrophy, one of whom has acquired partial lipodystrophy (patient 4) to age, gender and ethnic group matched reference values (median with 95% confidence interval) from NHANES data(15). Reference data was not available for pediatric patients (patients 7, 15, 23). B. Picture of patient 22 who presented with a very high fat mass ratio harboring a possibly pathogenic variant at position 349 (R349W). Patient displayed loss of fat from face, extremities, including loss of supportive fat from hands and feet, but with excessive fat deposition in the back displaying an exaggerated buffalo hump. Other clinical features included ischemic and non-ischemic cardiac disease, PCO-S with severe hyper-androgenism and infertility, alopecia, hematuria (due to thin basement membrane disease on kidney biopsy). Laboratory features included a high testosterone level and a very low alkaline phosphatase level typically associated with progeroid disorders. (C, D Pictures of patient 7 with a possibly pathogenic POLD1 variant. This patient and her mother displayed very severe insulin resistance, preservation of subcutaneous fat around the face and neck and even in the abdominal wall. There was a paucity of fat in the extremities. Patients also demonstrated stiffness in joints, elevated CK levels (mother displaying muscle weakness), proteinuria (massive in one leading to kidney biopsy showing membranoproliferative glomerulonephritis). The patient and her mother did not have deafness. E. X-ray of the narrow mandible for patient 6 with possibly pathogenic POLD1 variant.
Two related patients (mother and daughter, 6 and 7) had a variant in Exon 26 of POLD1 (NM002691.3: c.3199 G>A; p.E1067K, Figure 1, Supplemental Figures 2 and 3 and Supplemental Tables 2–5) which we classified as variant of unknown significance which is most likely pathogenic. Two patients (4 and 9) did not harbor mutations in the known 15 LD genes (see Supplemental Methods) interrogated with whole exome sequencing (WES). WES for the other 11 patients is currently in progress but they did not harbor LMNA p.R482 variants using specific sequencing for this locus as described in Supplemental Methods.
Body composition
All but one patient had a pattern of fat loss consistent with that seen in Familial Partial Lipodystrophy (FPLD) in which there is fat atrophy of the upper and lower extremities as well as the pelvis, with relative fat sparing or accumulation in the neck, trunk and abdomen. One patient had Acquired Partial Lipodystrophy with fat atrophy in the upper body and preservation of fat in the legs. FMR in this patient was 0.84, while in the other 22 patients, FMR was higher, 1.78 ± 0.53. Using the Third National Health and Nutrition Examination Survey (NHANES III) reference data(15) and the conversion to Lunar equipment previously reported(16), patients were matched for gender, race and age and the majority had FMR well above 95% confidence interval of reference median values (Figure 1). We also compared the FMR obtained from our adult female patients with a group of normal and overweight women with a BMI of 20–30 kg/m2 as well as a group of female diabetic patients with truncal obesity and fatty liver disease (Supplemental Figure 4) who were age-matched. A cut-off value of 1.5 would be well above the normal for adult female populations from NHANESIII as well as our two comparator groups with a sensitivity of 86.7% and specificity of 96.8%. There were distinctive patients with substantially higher values, most notably patient 22.
Leg fat mass was negatively correlated with log-transformed triglycerides (p=0.0498, Figure 2). No correlation was found between leg fat mass and hemoglobin A1c (R2=0.06, Figure 2), or between leg fat mass and liver fat (R2=0.01). Mid-thigh skin fold thickness, mean: 12.1 ± 7.7, range 4–29 mm, was low compared to age, gender and race matched data from NHANES data(17), mean: 26.3 ± 7.2, range 12.3–32.4 mm.
Figure 2.
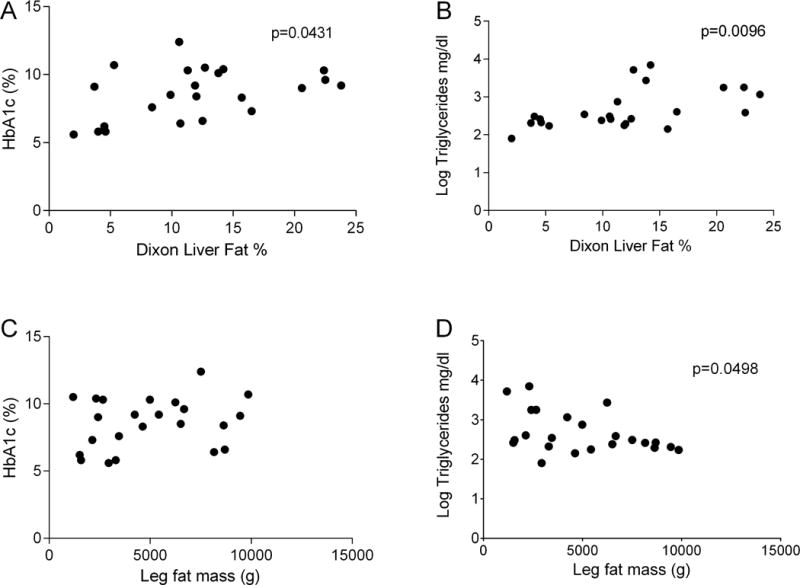
Correlation was noted between metabolic parameters of hemoglobin A1c and liver fat % (A) as well as log transformed triglycerides and liver fat % (B). Hemoglobin A1c was not significantly correlated with leg fat mass (C). A significant correlation was seen between log transformed triglycerides and leg fat mass (D). Leg fat mass and liver fat % did not correlate (data not shown).
Leptin levels, mean: 22.3 ± 16.6, range 4.8–67.1 ng/ml, were variable and in general higher than noted in studies that enrolled only patients with leptin deficiency at the NIDDK(18), but similar to one of our previously published series(19). Leptin levels correlated with total fat mass (p=0.0071) and trunk fat (p=0.0044) (Supplemental Figure 5) and did not correlate with HbA1c (R2=0.00) or triglyceride levels (R2=0.08).
Comorbidities
Prevalence of comorbidities and medications used are shown in Figure 3. All patients had either diabetes (82.6%) or pre-diabetes (17.4%) with variable degree of control. Mean ± SD hemoglobin A1c was 8.6 ± 1.9%. One patient had Type 1 diabetes with undetectable C-peptide (patient 14) and another patient (patient 15) had positive anti-GAD antibody at baseline. Nearly half (11 patients) had very poor control with hemoglobin A1c above 9%. Nineteen patients (82.6%) used a glucose lowering agent. Ten patients (43.5%) used combination therapy with 2 or more agents. Insulin was used by 12 patients (52.2%) and 9 of these patients required over 100 units per day. Six patients (26.1%) used highly concentrated U-500 insulin.
Figure 3.
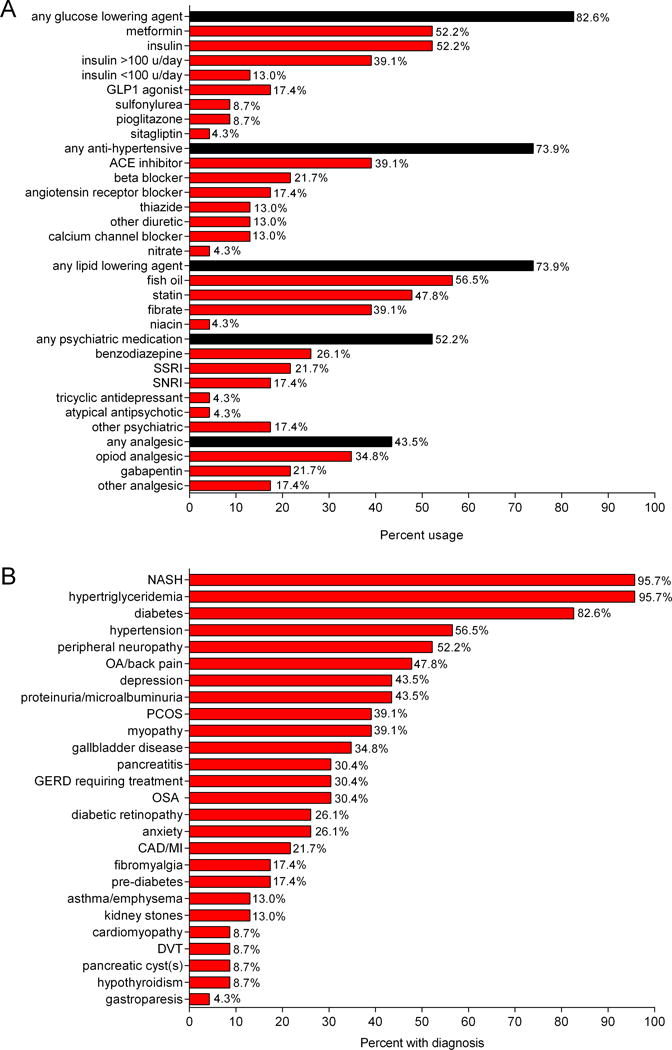
A. Frequency of medication use for diabetes, dyslipidemia, hypertension and mood disorders. B. Co-morbidity frequency among 23 patients with partial lipodystrophy.
Microvascular complications of diabetes were common. Twelve patients (52.2%) had peripheral neuropathy. One patient had a history of CIDP (Chronic Inflammatory Demyelinating Polyneuropathy). Six patients had known diabetic retinopathy based on patient report, though the prevalence may be higher as we did not have dilated examination results for all patients. Proteinuria or microalbuminuria was known in 10 patients (43.5%). Two patients had kidney biopsies for clinical reasons, one displaying membranoproliferative glomerulonephritis (patient 6, biopsied for massive proteinuria) and another (patient 22, biopsied for hematuria) displaying thin basement membrane disease. One patient had severe gastroparesis.
A total of 22 out of 23 patients had hypertriglyceridemia, mean 1058 ± 1745 mg/dl. Six had very high triglycerides over 1000 mg/dl. Most (17 patients) used a lipid lowering agent (Figure 3). Recurrent pancreatitis occurred in 7 of 23 patients (30.4%). At least three episodes of pancreatitis occurred while triglyceride levels were less than 800 mg/dL. Two patients who are sisters, with known LMNA gene mutations, were found to have pancreatic cystic lesions on MRI. One had a single 0.3 cm cystic lesion in the pancreatic neck and the other patient had multiple subcentimeter cystic lesions which may represent side branch intraductal mucinous neoplasms (Supplemental Figure 6).
Mood disorder requiring medication was identified in 12 patients (52.2%). Depression was the most common psychiatric condition, present in 10 patients (43.5%). Six had anxiety (26.1%), one patient had bipolar disorder and one had an unidentified psychiatric condition for which he previously used an antipsychotic agent.
Chronic pain was common among our patients. Eighteen patients (78.3%) reported some type of painful condition including neuropathy (52.2%), arthritis (47.8%), chronic back pain (13.0%), fibromyalgia (17.4%) and myopathy (39.1% had either suspected or confirmed myopathy). One patient had muscular dystrophy.
Other noted conditions include coronary artery disease or myocardial infarction in 5 patients (22.7%), one patient with ischemic cardiomyopathy and one patient with LMNA mutation and non-ischemic cardiomyopathy who required a heart transplant. Hypertension was present in 13 patients (56.6%). Seven patients (30.4%) had sleep apnea. Three (13%) had asthma or emphysema. Nine patients (39.1%) had polycystic ovarian syndrome.
Hepatic evaluation
Liver fat quantified using magnetic resonance (MR) spectroscopy and also using MR Dixon method showed fat percentage, mean ± SD of 13.8 ± 6.1% and 11.9 ± 6.3%, respectively (normal <5%)(20). Liver fat was correlated with metabolic parameters. A positive correlation was found between liver fat quantified by MR Dixon method and hemoglobin A1c (p=0.0431) as well as log-transformed triglycerides (p=0.0096) Figure 2.
Of the 23 patients who underwent liver biopsy, 22 met histopathological criteria for NASH. Total NASH scores of those 22 patients ranged from 3–9 (mean 6±2). NAS in patients who met criteria for NASH ranged from 3–8 (mean 5±1). Eighteen patients (78.3%) had some degree of fibrosis present with variable severity (stage 1a-4). One 12-year-old female patient (patient 15) had stage 4 fibrosis (Figure 4).
Figure 4.
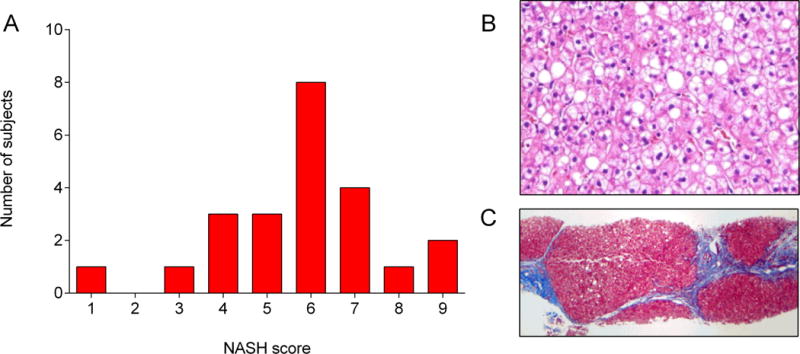
A. Frequency distribution of NASH scores of liver biopsy specimens. B. H&E and C. Trichrome staining of liver biopsy sample in patient 15 (a 12-year-old with atypical PL) showing hepatic injury and steatosis (B) stage 4 fibrosis (C).
Since LMNA mutations were common among the FPLD patients, and mice with hepatocyte-specific LMNA deletion develop spontaneous steatosis with nuclear alterations and susceptibility to diet-induced steatohepatitis (Kwan et al., manuscript submitted), we investigated nuclear envelope integrity from our liver biopsy samples. These early studies were performed on 3 patients with LMNA pathogenic variants and one patient with a POLD1 p.E1067K variant. Specimens from 2 patients with NASH (without lipodystrophy) were used as controls. Immunofluorescence staining of frozen liver sections showed dysmorphic nuclei and lamin disorganization in the patients with LMNA pathogenic variants compared to NASH controls (Figure 5). We also observed abnormal nuclear morphology in the patient with POLD1 variant, though not to the same extent as the patients with LMNA variants.
Figure 5.
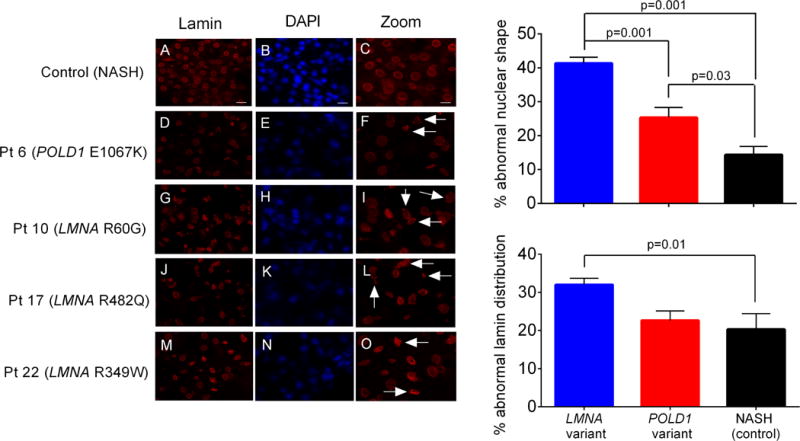
Abnormal nuclear morphology and lamin distribution in livers from patients with LMNA mutations. Lamin A/C distribution (red) and nuclei (blue) were visualized by immunofluorescence staining of frozen liver sections from patients with familial partial lipodystrophy (D-O). Liver biopsy specimens from two patients with nonalcoholic steatohepatitis but no history of PL and no pathogenic variants in LMNA were used as controls; representative images from one of these patients are shown (A-C). Dysmorphic nuclei and lamin disorganization are indicated by arrows in high magnification images (right column). Scale bars 20 μm (left and middle column), 10 μm (right column). Right panels show quantification of abnormal nuclear shape (upper right) and abnormal lamin distribution (lower right) in control NASH livers versus those from patients with LMNA and POLD1 variants. For each group, 3–4 randomly selected high-power fields were scored for each patient liver section (>150 nuclei per field, >700 nuclei per patient). Statistical significance was determined by one-way ANOVA followed by Tukey post-hoc test at a threshold of P < 0.05. Error bars represent the standard error of the mean.
Discussion
The goal of our study is to provide a complete description of the disease state in a cohort of 23 patients with PL. Lack of clear documentation of clinical manifestations and full appreciation of disease burden has become an obstacle in therapeutic development for rare metabolic diseases given the significant heterogeneity in presentation(21). With this report, we are reintroducing the idea of careful morphometric description using DEXA to document the presence of PL. This approach may be adopted widely as a clinical test following further validation. Also, we are reporting a number of important clinical lessons such as the presence of severe disease burden not only due to metabolic disease but also due to neuropsychiatric symptoms, chronic pain and multi-system involvement. It would be useful to collect similar data from a larger number of patients to develop disease severity scores taking into account the entire spectrum of clinical findings.
Our genetic investigations have recapitulated the genetic heterogeneity of PL with multiple patients having different molecular basis for their disease. Several genes have been implicated in various forms of FPLD including LMNA, PPARG, PLIN1, AKT2 CIDEC, LIPE and ADR2A(2, 5, 6, 22, 23). However, the molecular cause of disease remains unexplained in up to 40% of patients with the known single gene mutations. Cambridge group recently reported enrichment of multiple common variants in a cohort of genes in patients formerly classified as FLPD1, suggesting the possibility of polygeneic inheritance (24).
LMNA pathogenic variants are most commonly identified in FPLD and also the most common in our cohort(22). As novel findings, we have described two unique phenotypes, one of which is associated with an uncommon LMNA variant (R349W), characterized by specific body composition and clinical features. We are able to add previously unrecognized clinical characteristics (dorsocervical fat pad, ischemic cardiac disease, low alkaline phosphatase) to the description of this genotype(13). As another novel finding, two related patients (mother-daughter) were found to have a heterozygous variant of uncertain significance in POLD1 that is possibly pathogenic and related to dysmorphic nuclei in the liver. POLD1 has been previously implicated in lipodystrophy syndromes. An in-frame deletion mutation in POLD1 was discovered in two patients with mandibular hypoplasia, deafness and progeroid features (MDP) syndrome with lipodystrophy(25). Our patients did not have clinical evidence for deafness or progeroid features, but had small mandibles and fluctuating CK levels. Our findings need to be complemented with further functional characterization of the observed variant. However, glutamine in position 1067 is highly conserved, within the second zinc module of the zinc finger domain and projected to be pathogenic using in silico algorithms (Supplemental Figure 3).
Another area of interest for us was to determine if unique morphometric characteristics could be objectively used as diagnostic criteria. Previously, the use of mid-thigh skin thickness has been suggested as a field tool to screen for FPLD (3, 26), and this criterion has been adopted into the consensus statement developed by AACE(3) as well as the most recent multi-society effort (Brown RJ, et.al, currently in print). In fact, recently, researchers from Spain have described skin-fold characteristics of patients with the Koberling varierty of PL, attributing a Koberling Index, the ratio of subscapular/calf skinfolds (KöB index), with a cut-off value of 3.477 to be good predictor of this subtype of PL (sensitivity:89%; specificity:84%)(27). However, accurate measurement of skin thickness may be tricky in some patients in our experience and we have observed high inter-observer variability while collecting data. Body composition in our patients as analyzed by DEXA showed a high FMR when compared to normative data obtained from NHANES reference data(15), highlighting the loss of fat in extremities seen in all of our patients who were classified as having FPLD. Others have proposed this approach previously in both female patients harboring LMNA pathogenic variants and in patients with HIV-associated lipodystrophy(26, 28). In our hands, an FMR above 1.5 (obtained using GE Lunar DEXA systems) in adult females may indicate an abnormal ratio compared to a reference population taking multi-ethnic or age-related variation into account. We are not able to suggest a cut-off value for male patients or children in our cohort. Further analysis of fat related parameters in larger groups of lipodystrophy patients and comparison to patients with common metabolic syndrome and abdominal obesity may help establish objective diagnostic criteria for PL.
We also present novel data showing that low leg fat was correlated with higher triglycerides in our unique population, suggesting a pathogenic link between higher triglycerides and low leg fat. Other researchers have reported a similar association from other cohorts and our patients possibly display the most extreme version of this association(29, 30). Further strengthening a pathogenic role for absence of femoral fat, Van Pelt and colleagues have observed worsening lipemia after femoral lipectomy(31). In contrast, higher fat in the liver in our patients is associated with worse glucose control and higher triglycerides (shown for the first time in a PL cohort), similar to studies in Type 2 diabetic patients(32). These findings suggest that the development of hypertriglyceridemia may be related to absence of peripheral fat depots (and possibly to related changes in their adipokines), but also due to accumulation of ectopic fat, allowing both mechanisms to be exploited for therapeutic development.
As expected in patients with lipodystrophy, our patients had a high prevalence of diabetes, often associated with extreme insulin resistance. The incidence of diabetes was higher in our population (82.6%), compared to a previously reported cohort (50%)(33). This discrepancy may be due to recruitment of patients suspected to have NAFLD and these patients may have more severe metabolic co-morbidities. Microvascular complications of diabetes, particularly peripheral neuropathy and nephropathy were also common. Hypertriglyceridemia and NAFLD or NASH are known associations with lipodystrophy and most of our patients also demonstrated this(2). Other frequent conditions included recurrent pancreatitis, hypertension, PCOS, GERD, sleep apnea and coronary artery disease or myocardial infarction. Diabetes commonly co-exists with hypertension, hyperlipidemia, kidney disease and cardiovascular disease(34). However, we were surprised by the prevalence of chronic pain and mood disorder in this group. Overall, chronic pain was present in 78.3% of patients and was reported to be due to a combination of arthritis, back pain, fibromyalgia and myopathy. Depression has been shown to be prevalent in adults with diabetes to a varying degree (11–32%)(35), but appears higher in our patients (43.5%). It is not clear whether these under-appreciated manifestations are due to primary pathophysiological pathways, a manifestation of ectopic lipid (or other metabolite) deposition in muscles and nerves (or elsewhere) or due to some other adoptive (neurobehavioral, possibly functional) processes.
We further explored the extent of liver disease and NASH by histopathological criteria was present in nearly all patients (22 of 23). Since our trial specifically sought patients with fatty liver disease at baseline, the presence of NASH may be over-representative of the prevalence of NASH in the entire population of PL. It is noteworthy that significant fibrosis may be present even in young patients without diabetes as was shown in a 12-year-old female patient. Therefore, it would be important to consider a liver biopsy or a careful investigation for clinical fibrosis even in very young patients with PL.
Lastly, we present data that reflects relative nuclear integrity in the liver tissue of PL. Lamin A and C are integral components of the nuclear envelope and are expressed ubiquitously throughout the body. Alterations in the nuclear envelope may lead to premature cell apoptosis. LMNA pathogenic variants may increase risk of arrhythmia or cardiomyopathy, as occurred in two of our patients with non-ischemic cardiomyopathy (one requiring cardiac transplantation)(36). Given that patients had multi-system involvement, we examined the degree of nuclear disorganization in liver biopsy tissues of select patients. Patients with LMNA pathogenic variants had apparent nuclear disorganization in their liver tissue. We also noted a similar pattern, though not to the same degree, in a patient harboring a POLD1 variant of uncertain significance that is likely pathogenic. We posit nuclear atypia and disorganization may be a unifying cellular phenomenon in patients with PL depending on the mutation and the involved gene. This phenomenon may contribute to the multisystem involvement in PL. Further work is warranted in this arena.
Overall, PL was associated with involvement of at least 10 organ systems (with manifestations present in all 10 in nearly 40% of patients). The involvement of multiple organs in PL bears some resemblance to other heterogeneous rare metabolic diseases such as glycogen storage disease. Researchers studying Gaucher disease have developed a disease severity score tool(37), making use of the number of organ systems affected, severity of signs and symptoms and functional limitations. The diversity of comorbid conditions in our patients has inspired us to propose adoption of a similar approach to define the disease burden more objectively. If such a tool can be developed and validated, the impact of disease modifying treatments can be better measured.
Supplementary Material
Acknowledgments
We are indebted to the patients for volunteering in the study and the Lipodystrophy United Patient Foundation for the help in recruitment. We also thank the following sources of philanthropic gifts for Lipodystrophy Research at the University of Michigan: Mr. and Mrs. James Sopha, the White Point Foundation of Turkey (Istanbul, Turkey) and Ionis Pharmaceuticals (Carlsbad, CA).
Funding sources and disclosures:
This study is supported by National Institutes of Health (NIH) grant R01 DK088114 (EAO and HC) and R01 DK52951 (MBO). Infrastructure and data management support has been provided by the NIH Clinical and Translational Science Awards grant UL1TR000433, the Nutrition Obesity Research Centers grant P30 DK089503, and NIH institutional grant DK034933. JWI is supported by the Morton S and Henrietta K. Sellner Professorship in Human Genetics. GFB is supported by NIH training grant T32 DK094775. EAO received grant support from and served as an advisor to Amylin Pharmaceuticals LLC, Bristol-Myers-Squibb, AstraZeneca, Aegerion Pharmaceuticals, and Ionis Pharmaceuticals.
Footnotes
Clinical trial registration number: ClinicalTrials.gov identifier: NCT01679197 (UM HUM00058708)
References
- 1.Chan JL, Oral EA. Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr Pract. 2010;16(2):310–23. doi: 10.4158/EP09154.RA. [DOI] [PubMed] [Google Scholar]
- 2.Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–25. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract. 2013;19(1):107–16. doi: 10.4158/endp.19.1.v767575m65p5mr06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vigouroux C, Caron-Debarle M, Le Dour C, et al. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43(6):862–76. doi: 10.1016/j.biocel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Farhan SM, Robinson JF, McIntyre AD, et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30(12):1649–54. doi: 10.1016/j.cjca.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Garg A, Sankella S, Xing C, et al. Whole-exome sequencing identifies ADRA2A mutation in atypical familial partial lipodystrophy. JCI Insight. 2016;1(9) doi: 10.1172/jci.insight.86870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haarbo J, Gotfredsen A, Hassager C, et al. Validation of body composition by dual energy X-ray absorptiometry (DEXA) Clin Physiol. 1991;11(4):331–41. doi: 10.1111/j.1475-097x.1991.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 8.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 9.Speckman RA, Garg A, Du F, et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet. 2000;66(4):1192–8. doi: 10.1086/302836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanyam L, Simha V, Garg A. Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations. Clin Genet. 2010;78(1):66–73. doi: 10.1111/j.1399-0004.2009.01350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalil KA, Fargalley HS. Hypoparathyroidism in an Egyptian child with Hutchinson-Gilford progeria syndrome: a case report. J Med Case Rep. 2012;6:17. doi: 10.1186/1752-1947-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rauner M, Sipos W, Goettsch C, et al. Inhibition of lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J Bone Miner Res. 2009;24(1):78–86. doi: 10.1359/jbmr.080902. [DOI] [PubMed] [Google Scholar]
- 13.Mory PB, Crispim F, Freire MB, et al. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. Eur J Endocrinol. 2012;167(3):423–31. doi: 10.1530/EJE-12-0268. [DOI] [PubMed] [Google Scholar]
- 14.Thong KM, Xu Y, Cook J, et al. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron Clin Pract. 2013;124(1–2):31–7. doi: 10.1159/000354716. [DOI] [PubMed] [Google Scholar]
- 15.Kelly TL, Wilson KE, Heymsfield SB. Dual energy X-Ray absorptiometry body composition reference values from NHANES. PLoS One. 2009;4(9):e7038. doi: 10.1371/journal.pone.0007038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan B, Shepherd JA, Levine MA, et al. National Health and Nutrition Examination Survey whole-body dual-energy X-ray absorptiometry reference data for GE Lunar systems. J Clin Densitom. 2014;17(3):344–77. doi: 10.1016/j.jocd.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 17.McDowell MA, Fryar CD, Ogden CL. Anthropometric reference data for children and adults: United States, 1988–1994. Vital Health Stat. 2009;11(249):1–68. [PubMed] [Google Scholar]
- 18.Diker-Cohen T, Cochran E, Gorden P, et al. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. 2015;100(5):1802–10. doi: 10.1210/jc.2014-4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajluni N, Dar M, Xu J, Neidert AH, Oral EA. Efficacy and Safety of Metreleptin in Patients with Partial Lipodystrophy: Lessons from an Expanded Access Program. J Diabetes & Metab. 2016;7(3) doi: 10.4172/2155-6156.1000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288(2):E462–8. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 21.Bashaw ED. A clinical pharmacology-regulatory perspective on the approval of drugs for rare diseases. Clin Pharmacol Ther. 2016;100(4):327–9. doi: 10.1002/cpt.427. [DOI] [PubMed] [Google Scholar]
- 22.Jeru I, Vatier C, Araujo-Vilar D, et al. Clinical Utility Gene Card for: Familial partial lipodystrophy. Eur J Hum Genet. 2016 doi: 10.1038/ejhg.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeru I, Vatier C, Araujo-Vilar D, et al. Clinical Utility Gene Card for: Congenital Generalized Lipodystrophy. Eur J Hum Genet. 2016 doi: 10.1038/ejhg.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. doi: 10.1038/ng.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947–50. doi: 10.1038/ng.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83(1):18–34. doi: 10.1097/01.md.0000111061.69212.59. [DOI] [PubMed] [Google Scholar]
- 27.Guillin-Amarelle C, Sanchez-Iglesias S, Castro-Pais A, et al. Type 1 familial partial lipodystrophy: understanding the Kobberling syndrome. Endocrine. 2016 doi: 10.1007/s12020-016-1002-x. [DOI] [PubMed] [Google Scholar]
- 28.Beraldo RA, Vassimon HS, Aragon DC, et al. Proposed ratios and cutoffs for the assessment of lipodystrophy in HIV-seropositive individuals. Eur J Clin Nutr. 2015;69(2):274–8. doi: 10.1038/ejcn.2014.149. [DOI] [PubMed] [Google Scholar]
- 29.Van Pelt RE, Evans EM, Schechtman KB, et al. Contributions of total and regional fat mass to risk for cardiovascular disease in older women. Am J Physiol Endocrinol Metab. 2002;282(5):E1023–8. doi: 10.1152/ajpendo.00467.2001. [DOI] [PubMed] [Google Scholar]
- 30.Van Pelt RE, Jankowski CM, Gozansky WS, et al. Lower-body adiposity and metabolic protection in postmenopausal women. J Clin Endocrinol Metab. 2005;90(8):4573–8. doi: 10.1210/jc.2004-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez TL, Bessesen DH, Cox-York KA, et al. Femoral lipectomy increases postprandial lipemia in women. Am J Physiol Endocrinol Metab. 2015;309(1):E63–71. doi: 10.1152/ajpendo.00080.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Portillo-Sanchez P, Bril F, Maximos M, et al. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J Clin Endocrinol Metab. 2015;100(6):2231–8. doi: 10.1210/jc.2015-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2012;97(3):785–92. doi: 10.1210/jc.2011-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iglay K, Hannachi H, Joseph Howie P, et al. Prevalence and co-prevalence of comorbidities among patients with type 2 diabetes mellitus. Curr Med Res Opin. 2016;32(7):1243–52. doi: 10.1185/03007995.2016.1168291. [DOI] [PubMed] [Google Scholar]
- 35.Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069–78. doi: 10.2337/diacare.24.6.1069. [DOI] [PubMed] [Google Scholar]
- 36.Guenantin AC, Briand N, Bidault G, et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol. 2014;29:148–57. doi: 10.1016/j.semcdb.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 37.Weinreb NJ, Cappellini MD, Cox TM, et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12(1):44–51. doi: 10.1097/GIM.0b013e3181c39194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.