

Abstract
Evaluating cooperativity for cucurbit[8]uril (CB[8])‐mediated ternary complexation is required for understanding and advancing designs of such ternary self‐assembled systems. A key issue is to dissect the contributions of the binding steps of the first and second guest molecules to the overall ternary complex formation energy. This is addressed by performing concentration‐dependent titrations between CB[8] and guests by means of concentration‐dependent calorimetric and 1H‐NMR titrations. The sensitivity of the fitting of the cumulative heat of complexation of the calorimetric titrations is evaluated in terms of fitting error and enthalpy–entropy compensation and, together with the NMR spectroscopic analysis of the separate species, non‐cooperative binding is conceived to be the most probable binding scenario. The binding behavior of CB[8] homoternary complexes is similar to CB[8] heteroternary complexes, with an enthalpy‐driven tight fit of the guests in the CB[8] cavity overcoming the entropic penalty. Also for these types of complexes, a non‐cooperative binding is the most probable.
Keywords: complexation, cooperativity, cucurbit[n]uril, self-assembly, titration
Specific molecular recognition properties between ligands (guests) and receptors (hosts) allow non‐covalent synthesis of artificial receptor–ligand complexes to occur.1, 2, 3, 4 Cucurbit[n]urils (CB[n]) form a new class of macrocyclic hosts that show remarkable molecular recognition properties in water.5 The highest affinities between CB[n]s and their guests occur when high energy solvation water molecules are released from the cavity, which generates an enthalpic gain upon complexation.3 CB[8] is the first homologue large enough to promote binding of two equivalents of guest forming a ternary complex.6, 7 For example, a heteroternary complex forms through the well‐defined sequential binding of two different guests inside the CB[8] cavity and this can drive the self‐assembly of copolymers,8 hydrogels,9 particles,10, 11 and monolayers.12 Also homoternary complexes can be used for such purposes, in particular as demonstrated for the binding of N‐terminal aromatic amino acidic residues such as tryptophan (Trp) or phenylalanine (Phe) to CB[8].13 This type of CB[8]‐peptide complex extends the application of CB[8] assemblies into the biological arena.14, 15, 16, 17, 18
A ternary complex offers the opportunity for tuning the assembly properties by cooperativity. Cooperativity describes the relationship between the affinities of binding of the first and second equivalent of guest by the host.19 In comparison to the affinity of the first guest molecule, the binding of the second guest can either be favored, unfavored, or unaffected (i.e., positive, negative, or non‐cooperative, respectively). The principle of cooperative interactions is common in living systems and modulates the function of a receptor by the concentration of the ligands. For example, the binding of oxygen to the four pockets of hemoglobin is a positive cooperative process resulting in an increase of the binding affinity of hemoglobin for the substrate oxygen upon each molecule of oxygen bound.20 Proper design of the stability and dynamics of self‐assembled systems based on ternary interactions requires a thorough understanding of the, possibly cooperative, binding behavior of the ternary complex interaction motif. In a systematic study of the sequence‐specific recognition of peptides by CB[8], the homoternary complex between PheGly2 and CB[8] was proposed as a synthetic, positively cooperative receptor–ligand interaction.13 An overall ternary binding constant K ter of 1.5×1011 m −2 was reported for the complex CB[8]⋅(PheGly2)2.13 The positively cooperative nature of this complex was suggested on the basis of 1H‐NMR experiments, but the extent of cooperativity was not quantified.13 Here, we assess the degree of cooperativity for ternary complexes of CB[8] and two peptides both with an N‐terminal phenylalanine, followed by either two (PheGly2) or six glycine (PheGly6) residues. Isothermal titration calorimetry (ITC) and 1H‐NMR titrations were used to study the dependence of the affinity of CB[8] on the concentration of the guest. A key issue is to dissect the contributions of the bindings of the first and second guest molecules to the overall ternary complex formation. This is addressed by performing concentration‐dependent titrations, an evaluation of the error sensitivity in the ITC experiments, and by a spectroscopic analysis of the separate species by 1H NMR spectroscopy.
Figure 1 shows the first and the second binding events between the host CB[8] (H) and the peptide guest (G), leading to the formation of the 1:1 complex HG and the homoternary 1:2 complex HG2, respectively. The first equilibrium binding constant K 1 arises from the interaction of a single guest G with the host H. For the second binding step, the dissociation of a guest is associated with a pre‐factor 2 (2*k d,2) to account for the presence of two identical guest molecules in the cavity. Overall, the degree of cooperativity, defined by the ratio K 1/K 2, governs which of the three scenarios, positive, negative, and non‐cooperativity, applies, depending on whether K 2 is larger than, smaller than, or equal to K 1, respectively.
Figure 1.
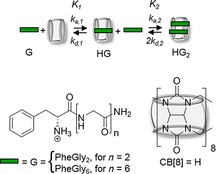
Equilibria of complexation of CB[8] (host, H) and peptide PheGlyn (guest, G).
An important aspect for the assessment of the degree of cooperativity is to work in an as wide as possible range of concentrations of H and G to make use of the different concentration dependencies of the binding constants for the formation of HG and HG2. For a given overall binding constant K ter, different degrees of cooperativity are expected to give different species distributions. This means that the distributions of the concentrations of H, HG, and HG2, while keeping the initial concentrations of host and guest constant, correspond to unique scenarios of K 1/K 2. To be able to accurately determine the ratio K 1/K 2, different distributions of H, HG, and HG2 can be measured starting from different initial concentrations of host and guest. A proper working range of concentrations was determined to be between 1 and 50 μm (see the Supporting Information for details). ITC studies were performed to determine the ratio between K 1 and K 2 for the ternary complexes of CB[8] with the peptides PheGly2 and PheGly6. The simultaneous fitting of the ITC data sets measured at three different host concentrations provided a restricted range of physically acceptable K 1/K 2 ratios. Specifically, consistent with the optimal range of concentrations, CB[8] was loaded in the cell at concentrations between 10 and 50 μm and titrated with a solution of the peptide guest. The enthalpograms obtained for each host–guest complex are given in Figure 2 a, e. A mathematical model was used to fit the experimental heats with a least‐squares minimization routine (see the Supporting Information for details). Briefly, the heat of complex formation was expressed as a function of the species concentrations, and the thermodynamic parameters K 1, K 2, ΔH 0 1, and ΔH 0 2 were used as fit parameters. Heats of dilution for each set of initial concentration were also included in the model, and calculated values were confirmed by reference experiments. The best fits provided the optimal four parameters ΔH 0 1, ΔH 0 2, K 1 and K 2, and thus the optimal K 1/K 2 ratio for each peptide guest. K 1/K 2 values of around 2 were found for both peptides (K 1/K 2=2.1±0.8 for PheGly2 and 1.8±0.4 for PheGly6, Table 1), which agrees with a non‐cooperative binding scenario.
Figure 2.
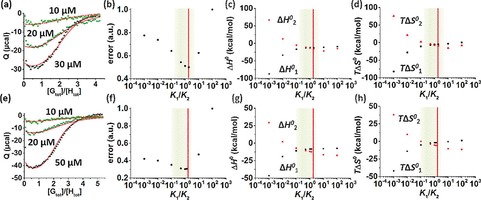
ITC data (markers) of binding CB[8] (H, three initial concentrations) with PheGly2 (G) (a) and PheGly6 (G) (e) in PBS (10 mm phosphate buffer, 2.7 mm KCl and 137 mm NaCl, pH 7.4). ITC data (see also Figures S1–S6 in the Supporting Information) were simultaneously fitted (solid lines) to a model with K 1, K 2, ΔH 0 1, and ΔH 0 2 as fit parameters. Representative plots of the normalized least‐squares fit error, ΔH 0 and −TΔS 0 calculated at fixed values of the K 1/K 2 ratio for PheGly2 (b–d) and PheGly6 (f–h). Red vertical lines indicate the non‐cooperative case (K 1/K 2=2), green areas represent the acceptable ranges of K 1/K 2 within 20 % of the minimum fit error and of enthalpy–entropy compensation.
Table 1.
Thermodynamic binding constants for complexes of CB[8][a] and PheGlyn.
| ITC PheGly2 [b] | ITC PheGly2 [c] | ITC PheGly6 [b] | 1H NMR PheGly2 [d] | 1H NMR PheGly6 [d] | |
|---|---|---|---|---|---|
| K 1/K 2 | 2.1 (0.8) | – | 1.8 (0.4) | 0.5 | 1.2 |
| K 1 [M−1] | 2.2 (1.1)×105 | – | 8.7 (0.6)×104 | 3.8×105 | 9.2×104 |
| K 2 [M−1] | 1.0 (0.2)×105 | – | 5.1 (1.3)×104 | 7.8×105 | 7.7×104 |
| K ter [M‐2][e] | 2.3 (1.4)×1010 | 1.5 (0.2)×1011 | 4.4 (1.1)×109 | 3.0×1011 | 7.1×109 |
| ΔH 0 1 [kcal mol−1] | −11.6 (0.3) | −29.6 (0.2) | −8.3 (0.2) | – | – |
| ΔH 0 2 [kcal mol−1] | −13.7 (1.7) | −14.7 (2.5) | – | – | |
| ΔG 0 1 [kcal mol−1] | −7.2 (0.3) | −15.4 (0.1) | −6.7 (0.1) | −7.6 | −6.8 |
| ΔG 0 2 [kcal mol−1] | −6.8 (0.1) | −6.4 (0.2) | −8.0 | −6.7 | |
| TΔS 0 1 [kcal mol−1][f] | −4.3 (0.5) | −14.2 (0.3) | −1.5 (0.2) | – | – |
| TΔS 0 2 [kcal mol−1][f] | −6.9 (2.2) | −8.3 (3.3) | – | – |
Standard deviations are given in parentheses. [a] Concentration of CB[8] was spectrophotometrically determined.24 [b] See Figure 2 and text for details. Data obtained at 25 °C in PBS (10 mm phosphate buffer, 2.7 mm KCl and 137 mm NaCl, pH 7.4). [c] Data as reported13 for the overall ternary complex HG2. Data based on three ITC experiments titrating 2 mm of PheGly2 into 0.1 mm CB[8] in 10 mm sodium phosphate, pH 7.0 at 27 °C. [d] See Figure 3 and text for details. Data obtained at 25 °C in D2O [e] Product of K 1 and K 2 gives Kter. [f] Difference ΔG and ΔH gives TΔS 0.
To evaluate how sensitive the fit error is to variations of the K 1/K 2 ratio, the least‐squares error was calculated for different degrees of cooperativity. Thus, the parameters ΔH 0 1, ΔH 0 2, and K 2 (correlated to K 1) were optimized for chosen values of K 1/K 2. Figure 2 shows the dependence of the fit error (Figure 2 b, f) on the ratio K 1/K 2, and the correlated enthalpies (Figure 2 c, g) and entropies (Figure 2 d, h). Figure S4 in the Supporting Information shows the changes in fit of the ITC titrations at very high and very low K 1/K 2. The trends in fit show, in short, that: (a) a much higher K 1/K 2 should be visible by a plateau of Q at low [Gtot]/[Htot] combined with a clear inflection at [Gtot]/[Htot]=1, and (b) a much lower K 1/K 2 should lead to a rather shallow slope at around [Gtot]/[Htot]=1 (in Figure S4 a, most visible in the two higher concentrations, and in Figure S4 b, at the two lower concentrations), which clearly conflict with the observed data. An evaluation of all thermodynamic parameters presented in Figure 2 allowed for the determination of a range of possible degrees of cooperativity (indicated in green in Figure 2). Values of the fit error within 20 % from the minimum error were defined as acceptable. This 20 % cut‐off value was selected based on the variability of the minimum error observed in triplicate calorimetric experiments. Therefore, the upper boundary of the range of acceptable degrees of cooperativity was set at values of K 1/K 2 equal to 6 for PheGly2 and to 3.5 for PheGly6. For higher values of K 1/K 2 (strongly negative cooperativity), the fit errors became quickly unacceptably high (Figure 2 b, f). Regarding the thermodynamic parameters, such high K 1/K 2 ratios gave more exothermic enthalpies and less favorable entropies for the second step (Figure 2).
The lower limit of the range was determined considering that, even though the fit errors did not rise as quickly as at the upper limit, the binding enthalpies and entropies for the first and second binding events diverged more and more for values of K 1/K 2 lower than 0.5. Specifically, an inversion of the signs and order of ΔH 0 1 and ΔH 0 2, as well as of TΔS 0 1 and TΔS 0 2, was observed for values of K 1/K 2 below 0.2 for PheGly2 and below 0.1 for PheGly6 (Figure 2). Under these conditions, the second binding event became less enthalpically favored (and more entropically favored) than the first step. Both steps would thus be associated with large enthalpy–entropy compensation effects and opposite driving forces, that is, strongly enthalpy‐driven for the first step and strongly entropy‐driven for the second. In particular, the unfavorable positive enthalpy contribution (Figure 2 c, g) and the highly favorably entropy (Figure 2 d, h) for the second step are not realistic considering that CB[8] complexation is known to be enthalpically driven and entropically unfavorable.21, 22, 23 Overall, the considerations made in terms of fit error and of enthalpy–entropy compensation determined a range of acceptable K 1/K 2 ratios between 0.2 and 6 for PheGly2 and between 0.1 and 3.5 for PheGly6, which are highlighted in green in Figure 2. For both peptides, these ranges indicate either a non‐cooperative or a weakly, negative or positive cooperative system.
For both PheGly2 and PheGly6, the second binding event has a larger enthalpic gain than the first, as well as a larger entropy loss (Table 1). This indicates a tighter fit for the second guest in the CB[8] cavity, which is logical as it involves interaction with an already partially filled cavity. It is also in agreement with studies performed by Biedermann and co‐workers22 that show, in the case of heteroternary complexes, a more favorable enthalpy for the second aromatic guest correlates with a less favorable entropy contribution. Similar to what was shown for the heteroternary complexes, this can be expected also in the case of the homoternary complexes studied here; the first guest reduces the cavity volume of CB[8] in such a way that the potential energy of the residual cavity water molecules is increased, thus leading to a stronger enthalpic response upon release of these water molecules upon the binding of the second guest. In constrast, the tightly packed ternary complex reduces the degrees of freedom of both guests and therefore brings an additional unfavorable entropy contribution.22
Another observation from our calorimetric results is that when comparing the thermodynamic data for the two peptides, a stronger binding affinity was found for PheGly2 with respect to PheGly6, arising from differences for both the first and second guest binding steps. In particular, the first PheGly6 seems to have a weaker interaction with the host (less favorable ΔH 0 1).
Moreover, our results reveal a slightly weaker overall binding than the one reported in the literature13 for the overall ternary complexation of the peptide PheGly2 with CB[8] (see K ter in Table 1), which can be explained by a higher concentration of cations competing with the guest for the binding to the host in our buffer.25 The crystal structure of the complex13 shows that the shorter PheGly2 can assume a circular conformation to maximize its dipole–dipole interactions of the amidic protons with the carbonyl on the CB[8] rims. This cannot be achieved for a longer chain in the case of PheGly6, which may explain the observed difference in affinity. Unfortunately, the X‐ray structure of the complex CB[8]⋅(PheGly6)2 is not available to confirm this hypothesis. Our observations are in agreement with calorimetric experiments on heteroternary complexes of CB[8], paraquat and TrpGly2 or TrpGly5 that have shown a tighter binding for the short peptide compared to the long one.22 Taken together, the calorimetric data indicate that the most realistic scenario is the non‐cooperative binding of the peptides.
However, further narrowing the range of possible K 1/K 2 values could not be achieved by ITC alone, due to both the restricted operative concentration range (see above) and the convolution of the heat effects arising from the first and the second binding events. To overcome the latter limitation, 1H‐NMR was used to provide direct spectroscopic insight into the (relative) concentrations of all participating species separately. This technique has a relatively low sensitivity, so fairly high concentrations are preferred; however, to prevent precipitation of CB[8], experiments were performed at 50 μm, which contrasts an earlier study that used CB[8] at a concentration that exceeded the solubility limit.13 A titration experiment was performed at a constant total CB[8] concentration (in D2O) of 50 μm, while titrating from 0.5–4 equivalents of the peptides (Figure 3 a, d and see full spectra in Figure S3 of the Supporting Information). The three species G, HG, and HG2 were distinguished based on the signals of the aryl protons of the guests.13 Upon the first complexation, the upfield shifts of the phenyl protons of the Phe residue verified the shielding of the surrounding CB[8] host molecule. With the second complexation, the interaction among the two guests in the cavity of the CB[8] caused an additional upfield shift.26 Under non‐saturation conditions for CB[8], the HG complex is well visible at low concentrations for both peptides, thus excluding a strongly positive cooperative system, in contrast to what has been described in an earlier study.13 By monitoring the signals of the aromatic protons (Figure 3 a, d), the distributions of all species G, HG, and HG2 were determined for each titration step (Figure 3 b, e). These distributions were fitted to a model expressing the calculated distributions of species as a function of the fitting parameters K 2 and K 1 (see the Supporting Information for details). The calculated data are shown as lines in Figure 3 b, e for the peptides PheGly2 and PheGly6, respectively. Table 1 summarizes the values found for the optimized parameters K 1, K 2, the corresponding free energies ΔG 0 1, ΔG 0 2 (see also Figure S7), and the overall binding constant K ter. Higher overall binding affinities (Kter in Table 1) were found as expected because the cations in the PBS solutions used for ITC can compete with the guest for the interaction with the host, thus destabilizing the complex,25 whereas these salt effects are absent in the solvent (D2O) used for the 1H‐NMR experiments. In agreement with ITC, CB[8] binds more strongly with the shorter peptide PheGly2 (3.0×1011 m −2) than the longer PheGly6 (7.1×109 m −2, Table 1). The optimal fits gave K 1/K 2=0.5 and 1.2, for PheGly2 and PheGly6, respectively, indicative of non‐cooperative or slightly positive cooperative binding.
Figure 3.
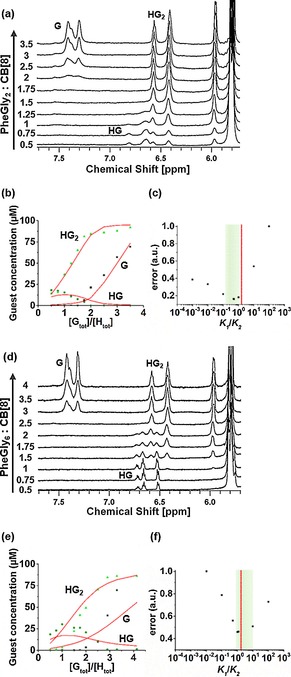
1H‐NMR titrations of CB[8] (50 μm) with PheGly2 (a) and PheGly6 (d) in D2O at 25 °C. Experimental [G] in G, HG, and HG2 (data points) are simultaneously fitted (see also Figure S7) to a model varying K 1 and K 2 (solid lines) for (b) PheGly2 and (e) PheGly6. Plots of the normalized fit error calculated at fixed values of the ratio K 1/K 2 for (c) PheGly2 and (f) PheGly6. Red vertical lines in c and f indicate the non‐cooperative value of K 1/K 2=2. Green areas indicate the acceptable ranges of K 1/K 2 within 20 % of the minimum error.
To assess the sensitivity of the degree of cooperativity, the graphs in Figure 3 c, f were obtained by optimizing K 2 (and the correlated K 1) at chosen values of the ratio K 1/K 2. The values of the least‐squares error for each K 1/K 2 ratio are reported for each peptide (Figure 3 c, f). A cut‐off value of 20 % from the minimum fit error was arbitrarily chosen to find the acceptable range of degree of cooperativity. The values of K 1/K 2 are in a range between 0.2 and 1 for the shorter peptide PheGly2, and between 0.6 and 10 for the longer PheGly6. Notably, the minima by 1H NMR are within the range of K 1/K 2 obtained by calorimetry, indicating a non‐cooperative system. Taken together, these results confirm a most probable scenario in which the ternary complexation between the peptides and CB[8] is non‐cooperative.
It should be noted that these ternary CB[8]‐peptide complexes cannot be compared directly to, for example, the cooperativity observed in hemoglobin, because in the former case, the first guest does not occupy one of two identical, well‐spaced binding sites, but resides somewhere in the same cavity to which also the second one binds in the next step. As a result, the second guest experiences interactions with the first guest directly, as witnessed by the correlation between enthalpy and entropy.
In conclusion, combining the pieces of evidence from calorimetric and 1H‐NMR titrations shown in this work, the most probable scenario to describe the homoternary complexation of phenylalanine‐based peptides by CB[8] is a non‐cooperative mode of interaction. This is independent of the tail length of the peptides studied in this work. Remarkably, whereas the second guest experiences a stronger interaction with the host after the first complexation step, there appears to be a counterbalancing entropic contribution that leads to an overall non‐cooperative behavior in affinity. This contrasts the normal non‐cooperative behavior of well‐separated binding sites, in which case the binding enthalpies of all steps are equal, and entropy differences arise solely from differences in statistical pre‐factors. The binding behavior of the homoternary peptide complexes resembles that observed for heteroternary complexes. The PheGly binding motif offers the synthetic flexibility and biocompatibility of peptides, and can have an active role in natural functional structures as well, such as in nuclear membrane pores.27 The insights in the complexation between peptides and CB[8] allow for a rational design of more complex self‐assembled systems built on this powerful interaction motif.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
B. H. M. Ruel and A. Juan Ruiz del Valle are acknowledged for technical support. Starting grant from the ERC (259183) to P.J. and E.C. is acknowledged for financial support.
E. Cavatorta, P. Jonkheijm, J. Huskens, Chem. Eur. J. 2017, 23, 4046.
Contributor Information
Prof. Pascal Jonkheijm, Email: p.jonkheijm@utwente.nl.
Prof. Jurriaan Huskens, Email: j.huskens@utwente.nl.
References
- 1. Reinhoudt D. N., Crego-Calama M., Science 2002, 295, 2403–2407. [DOI] [PubMed] [Google Scholar]
- 2. Oshovsky G. V., Reinhoudt D. N., Verboom W., Angew. Chem. Int. Ed. 2007, 46, 2366–2393; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2418–2445. [Google Scholar]
- 3. Uhlenheuer D. A., Petkau K., Brunsveld L., Chem. Soc. Rev. 2010, 39, 2817–2826. [DOI] [PubMed] [Google Scholar]
- 4. Aida T., Meijer E. W., Stupp S. I., Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Lagona J., Mukhopadhyay P., Chakrabarti S., Isaacs L., Angew. Chem. Int. Ed. 2005, 44, 4844–4870; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4922–4949; [Google Scholar]
- 5b. Lee J. W., Samal S., Selvapalam N., Kim H.-J., Kim K., Acc. Chem. Res. 2003, 36, 621–630; [DOI] [PubMed] [Google Scholar]
- 5c. Masson E., Ling X., Joseph R., Kyeremeh-Mensaha L., Lua X., RSC Adv. 2012, 2, 1213–1247; [Google Scholar]
- 5d. Barrow S. J., Kasera S., Rowland M. J., Del Barrio J., Scherman O. A., Chem. Rev. 2015, 115, 12320–12406. [DOI] [PubMed] [Google Scholar]
- 6. Kim J., Jung I.-S., Kim S.-Y., Lee E., Kang J.-K., Sakamoto S., Yamaguchi K., Kim K., J. Am. Chem. Soc. 2000, 122, 540–541. [Google Scholar]
- 7. Kim H.-J., Heo J., Jeon W. S., Lee E., Kim J., Sakamoto S., Yamaguchi K., Kim K., Angew. Chem. Int. Ed. 2001, 40, 1526–1529; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1574–1577. [Google Scholar]
- 8. Rauwald U., Scherman O. A., Angew. Chem. Int. Ed. 2008, 47, 3950–3953; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4014–4017. [Google Scholar]
- 9. McKee J. R., Appel E. A., Seitsonen J., Kontturi E., Scherman O. A., Ikkala O., Adv. Funct. Mater. 2014, 24, 2706–2713. [Google Scholar]
- 10. Zhang J., Coulston R. J., Jones S. T., Geng J., Scherman O. A., Abell C., Science 2012, 335, 690–694. [DOI] [PubMed] [Google Scholar]
- 11. Stoffelen C., Voskuhl J., Jonkheijm P., Huskens J., Angew. Chem. Int. Ed. 2014, 53, 3400–3404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3468–3472. [Google Scholar]
- 12. An Q., Brinkmann J., Huskens J., Krabbenborg S., De Boer J., Jonkheijm P., Angew. Chem. Int. Ed. 2012, 51, 12233–12237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12399–12403. [Google Scholar]
- 13. Heitmann L. M., Taylor A. B., Hart P. J., Urbach A. R., J. Am. Chem. Soc. 2006, 128, 12574–12581. [DOI] [PubMed] [Google Scholar]
- 14. Sonzini S., Ryan S. T. J., Scherman O. A., Chem. Commun. 2013, 49, 8779–8781. [DOI] [PubMed] [Google Scholar]
- 15. Hou C., Li J., Zhao L., Zhang W., Luo Q., Dong Z., Xu J., Liu J., Angew. Chem. Int. Ed. 2013, 52, 5590–5593; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5700–5703. [Google Scholar]
- 16. Sankaran S., De Ruiter M., Cornelissen J. J. L. M., Jonkheijm P., Bioconjugate Chem. 2015, 26, 1972–1980. [DOI] [PubMed] [Google Scholar]
- 17. Cavatorta E., Verheijden M. L., van Roosmalen W., Voskuhl J., Huskens J., Jonkheijm P., Chem. Commun. 2016, 52, 7146–7149. [DOI] [PubMed] [Google Scholar]
- 18. Bosmans R. P. G., Briels J. M., Milroy L. G., de Greef T. F. A., Merkx M., Brunsveld L., Angew. Chem. Int. Ed. 2016, 55, 8899–8903; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9045–9049. [Google Scholar]
- 19. Ercolani G., J. Am. Chem. Soc. 2003, 125, 16097–16103. [DOI] [PubMed] [Google Scholar]
- 20. Ackers G. K., Doyle M. L., Myers D., Daugherty M. A., Science 1992, 255, 54–63. [DOI] [PubMed] [Google Scholar]
- 21. Biedermann F., Uzunova V. D., Scherman O. A., Nau W. M., De Simone A., J. Am. Chem. Soc. 2012, 134, 15318–15323. [DOI] [PubMed] [Google Scholar]
- 22. Biedermann F., Vendruscolo M., Scherman O. A., De Simone A., Nau W. M., J. Am. Chem. Soc. 2013, 135, 14879–14888. [DOI] [PubMed] [Google Scholar]
- 23. Miskolczy Z., Biczók L., Phys. Chem. Chem. Phys. 2014, 16, 20147–20156. [DOI] [PubMed] [Google Scholar]
- 24. Yi S., Kaifer A. E., J. Org. Chem. 2011, 76, 10275–10278. [DOI] [PubMed] [Google Scholar]
- 25. Ling X., Saretz S., Xiao L., Francescon J., Masson E., Chem. Sci. 2016, 7, 3569–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang T., Sun S., Liu F., Fan J., Pang Y., Sun L., Peng X., Phys. Chem. Chem. Phys. 2009, 11, 11134–11139. [DOI] [PubMed] [Google Scholar]
- 27. Frey S., Richter R. P., Görlich D., Science 2006, 314, 815–817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary