

Abstract
Detailed herein is the photochemical organocatalytic enantioselective α‐alkylation of aldehydes with (phenylsulfonyl)alkyl iodides. The chemistry relies on the direct photoexcitation of enamines to trigger the formation of reactive carbon‐centered radicals from iodosulfones, while the ground‐state chiral enamines provide effective stereochemical control over the radical trapping process. The phenylsulfonyl moiety, acting as a redox auxiliary group, facilitates the generation of radicals. In addition, it can eventually be removed under mild reducing conditions to reveal methyl and benzyl groups.
Keywords: enamines, organocatalysis, photochemistry, radicals, reaction mechanisms
The stereocontrolled α‐alkylation of carbonyl compounds using alkyl halides as electrophiles is a fundamental synthetic process to forge a new carbon–carbon bond while setting a stereogenic center.1 While different chiral‐auxiliary‐based approaches are available,2 developing effective asymmetric catalytic variants has proven difficult.3, 4 In addition to phase‐transfer catalytic strategies,5 the field of enantioselective enamine‐mediated catalysis6 has recently provided some solutions for the direct α‐alkylation of unmodified aldehydes with organic halides. A few examples of SN2‐type alkylation have been reported,7 while most methods have combined enamine catalysis with cationic (SN1‐type)8 or radical reaction pathways.9 Despite this progress, catalytic protocols for stereoselectively introducing either a methyl or a benzyl group at the α‐position of aldehydes remain rare (Figure 1 a). While a single example of formal α‐methylation by an SN1 pathway has been reported,10 direct α‐benzylation methods generally require highly electron‐poor benzyl bromides to facilitate the generation of radical intermediates upon single‐electron transfer (SET) reduction.9b,9c A stereocontrolled SN2 polar manifold for the alkylation of enamines with benzyl bromide was recently shown to be feasible,11 but this chemistry could only be applied to α‐branched aldehydes to afford exclusively quaternarized products. The paucity of methodologies for the catalytic enantioselective α‐methylation and α‐benzylation of aldehydes stands in sharp contrast to the prominence of these stereogenic units. Indeed, they are common motifs in medicinal agents, agrochemicals, and natural product isolates.12 In particular, methylation is a fundamental process in medicinal chemistry, since introducing this small alkyl fragment can strongly modulate a molecule's biological and physical properties.12a
Figure 1.
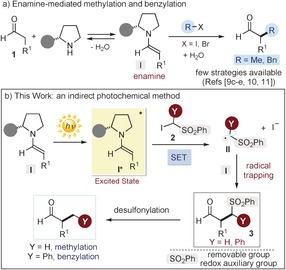
a) The catalytic enantioselective α‐methylation and α‐benzylation of aldehydes: a challenge for enamine‐mediated catalysis. b) Our photochemical approach uses enamines (I) to 1) drive the photochemical generation of the radicals (II) upon direct‐light excitation, and 2) intercept the radical to forge the stereogenic center. The phenylsulfonyl moiety within the substrates 2 plays a dual essential role since it 1) facilitates SET reduction and the generation II, acting as a redox auxiliary group, while 2) unveiling the methyl and benzyl groups upon easy desulfonylation of 3. The grey circle represents the chiral aminocatalyst scaffold.
Herein, we report a simple photochemical organocatalytic method to install either a methyl or a benzyl moiety onto simple aldehydes with high stereoselectivity. The chemistry exploits the ability of chiral enamines (I) to directly reach an electronically excited state upon light absorption13 and then generate carbon‐centered radicals (II) by SET reduction of the (phenylsulfonyl)alkyl iodides 2 (Figure 1 b). At the same time, the ground‐state chiral enamines (I) provide effective stereochemical control over the enantioselective radical‐trapping process. The resulting (phenylsulfonyl)alkylated intermediates 3 are then easily desulfonylated to reach the target products.
In implementing the α‐alkylation of aldehydes with α‐iodosulfones (2), we were inspired by our recent studies on the direct photoexcitation of enamines and their potential for generating radicals under mild reaction conditions.13 We demonstrated that I, upon light absorption, can reach an excited state (I*) to become a strong reductant, as implied by its reduction potential, which was estimated as about −2.0 V (vs. Ag/Ag+ in CH3CN).13a Thus, I* could trigger the formation of radicals through SET reduction of easily reducible bromomalonates (E p red diethyl bromomalonate=−1.69 V vs. Ag/Ag+ in CH3CN). We surmised that a similar photochemical mechanism could generate (phenylsulfonyl)alkyl radicals (II) from 2. A critical design element was the phenylsulfonyl moiety, which would act as a redox auxiliary group14 to facilitate the reductive cleavage of the C−I bond by an SET mechanism (Figure 1 b). In consonance with this scenario, we measured, by cyclic voltammetry, a reduction potential for the iodomethyl phenyl sulfone (2 a; Y=H in Figure 1 b) as low as −1.49 V (E p red vs. Ag/Ag+ in CH3CN). This experiment suggested 2 a as a viable precursor of phenylsulfonyl methyl radicals.15
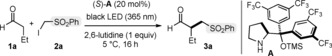
To test the feasibility of our design plan, we focused on the reaction between butanal (1 a) and 2 a (Table 1). The experiments were conducted at 5 °C in toluene and under irradiation by a single black‐light‐emitting diode (black LED, λ max=365 nm). When adding the chiral secondary amine catalyst A 16 (20 mol %), the desired (phenylsulfonyl)methylated aldehyde 3 a was formed in good enantioselectivity but with moderate chemical yield (entry 1). Despite extensive experimentation, we could not increase the yield of the photochemical alkylation, which reached a standstill at about 40 % of conversion and could not evolve further.
Table 1.
Optimization studies.[a]
| Entry | Solvent | Additives and Conditions | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|
| 1 | toluene | – | 43 | 80 |
| 2 | toluene | I2 (10 mol %) | 13 | 80 |
| 3 | toluene | Na2S2O3 (10 mol %) | 50 | 80 |
| 4 | toluene/hexanes/H2O (1:1:2 ratio) | Na2S2O3 (50 mol %) | 99 (76)[d] | 82 (80) |
| 5 | toluene/hexanes/H2O (1:1:2 ratio) | Na2S2O3 (50 mol %), in the dark | <5 | – |
| 6 | toluene/hexanes/H2O (1:1:2 ratio) | Na2S2O3 (50 mol %), O2 or TEMPO (1 equiv) | <5 | – |
| 7 | toluene/hexanes/H2O (1:1:2 ratio) | Na2S2O3 (50 mol %), band pass @ 400 nm | 92 | 82 |
[a] Reactions performed over 16 h on a 0.1 mmol scale using 3 equiv of 1 a and a single black LED (λ max=365 nm) to illuminate the reaction vessel. [b] Yield determined by NMR spectroscopy using 1,1,2‐trichloroethene as the internal standard. [c] Enantiomeric excess determined by HPLC analysis on the corresponding alcohol after in situ NaBH4 reduction of 3 a. [d] Yield and ee of the isolated aldehyde 3 a are shown within parentheses. TMS=trimethylsilyl.
A useful insight into tackling this reactivity issue came from the optical absorption spectra of the reaction components, and confirmed our expectation that I, generated upon condensation of the catalyst A with 1 a, could absorb up to the visible region (blue line in Figure 2 a; absorption band up to λ=415 nm). In addition, we noticed that 2 a could also absorb light up to λ=390 nm (red line). This observation prompted us to investigate the photochemical stability of 2 a under the reaction conditions by irradiating a toluene solution of 2 a at λ=365 nm over 16 hours. The colorless solution developed a yellow color over time (Figures 2 b i and ii). Although we recovered 95 % of 2 a, the color change suggested the possibility of a photoinduced homolytic cleavage of the C−I bond within 2 a, a minor pathway which could generate small amounts of intensely colored molecular iodine (2 I.→I2). Diiodine, even in traces, is a well‐known inhibitor of radical‐chain reactions,17 as it can trap carbon‐centered radicals at very high rates (rate constants of about 109 m −1 s−1).17b In addition, the intense light absorption of molecular iodine may also interfere with other photochemical steps by an inner filter effect. A positive standard iodine detection test confirmed the generation of I2 from 2 a under the reaction conditions:18 adding a starch solution, sodium iodide, and water to the photolyzed yellow solution of 2 a, depicted in Figure 2 b ii, produced the characteristic dark‐blue color (Figure 2 b iii). Finally, the solution switched back to achromatic (Figure 2 b iv) when adding solid sodium thiosulfate (Na2S2O3, E p red +0.28 vs. Ag/Ag+ in H2O),18a which can easily reduce I2 to iodide (Figure 2 d). To evaluate if the presence of iodine could be detrimental for the photo‐organocatalytic process, we performed the model reaction by adding I2 (10 mol %), and it resulted in a profoundly reduced reactivity (Table 1, entry 2).
Figure 2.
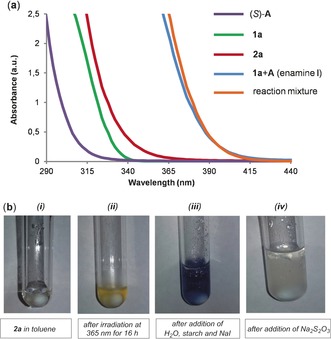
a) Optical absorption spectra acquired in toluene in 1 cm path quartz cuvettes: [1 a]=1.5 m; [2 a]=0.5 m; (S)‐A=0.1 M. b) Evaluating the photostability of 2 a: (i) aspect of a 0.25 m solution of 2 a in degassed toluene; (ii) the same solution after black LED irradiation at λ=365 nm for 16 hours; (iii) the same solution after the addition of water, starch, and NaI; (iv) final view of the solution after the addition of Na2S2O3.
On the basis of these results, we sought to implement reaction conditions which could remove any trace of diiodine. Adding solid Na2S2O3 (10 mol %) to the reaction mixture increased the yield of 3 a (50 %; Table 1, entry 3). Further optimizations revealed that using a toluene/hexanes/water solvent mixture (in a 1:1:2 ratio) and adding 50 mol % of Na2S2O3 led to the formation of 3 a in high yield with good enantioselectivity (76 % yield of the isolated 3 a, 80 % ee; entry 4).
Control experiments were conducted to obtain more mechanistic clues. Carefully excluding light completely suppressed the process (Table 1, entry 5). Reactivity was also inhibited under an aerobic atmosphere and in the presence of TEMPO (1 equiv; entry 6). Performing the reaction using a 300 W Xenon lamp equipped with a band‐pass filter at λ=400 nm (entry 7) did not alter the overall reactivity of the model reaction. Since I is the only photoabsorbing compound at λ=400 nm, the experiment indicates that the direct photoexcitation of the enamine triggers the radical generation from 2 a. On the basis of these observations, we propose the mechanism depicted in Figure 3. The photoexcited enamine Ia* induces the reductive cleavage of the C−I bond in 2 a by SET, which affords the electrophilic radical IIa. The ground‐state chiral enamine Ia then traps IIa in a stereocontrolled fashion. Considering the redox properties of the resulting α‐amino radical III (E p red V/III estimated to be ca. −0.95 V vs. Ag/Ag+ in CH3CN),13b which make it incapable of reducing 2 a by SET (E p red of 2 a=−1.49 V vs. Ag/Ag+ in CH3CN), and according to the tendency of aminoalkyl radicals toward halogen abstraction,19 we propose that III abstracts an iodine atom from 2 a, thereby regenerating IIa. The resulting adduct IV is not stable and evolves to an iodine/iminium ion pair (V), which eventually hydrolyzes to release 3 a and A. Overall, the chemistry can be considered an example of an enantioselective catalytic atom‐transfer radical addition (ATRA).20 In consonance with the classical ATRA mechanism, the present reaction proceeds through a self‐propagating radical chain initiated by the photochemical activity of the enamines, which feeds in radicals from outside the chain. This scenario is congruent with the quantum yield (Φ) of 3.9 measured for the model reaction (λ=400 nm in CH3CN, using potassium ferrioxalate as the actinometer). In addition, it well explains the striking effect of diiodine, which is a powerful inhibitor of iodine atom transfer chains.17
Figure 3.
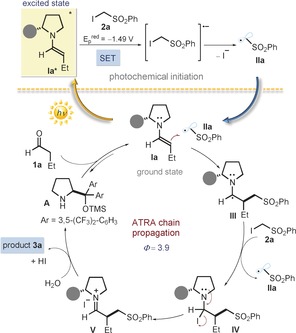
The proposed chain propagation mechanism.
Using the optimized reaction conditions described in entry 4 of Table 1, we then demonstrated that our (phenylsulfonyl)methylation reaction (Figure 4 a) is quite general in scope with respect to the aldehyde component. It could satisfactorily tolerate aldehydes bearing either long alkyl fragments or of significant steric bulk, a terminal olefin, and heteroatom moieties (Figure 4 b). These experiments afforded chiral aldehyde products which were isolated as the corresponding alcohols 4 a–j after in situ NaBH4 reduction in moderate to high yield and enantiomeric excess. We could also install substituents other than phenyl at the sulfone moiety, since α‐iodosulfones containing either a tosyl or a mesyl group actively participated in the enantioselective alkylation, leading to the products 4 k and 4 l, respectively (Figure 4 c).
Figure 4.
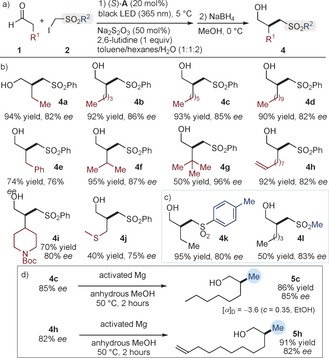
a) The photo‐organocatalytic enantioselective (phenylsulfonyl)methylation of aldehydes. Survey of the aldehydes (b) and α‐iodosulfones (c) which can participate in the reaction. Yields and enantiomeric excesses of the isolated alcohols are indicated below each entry. d) Desulfonylation reveals the methyl group.
The main goal of our studies was to provide an efficient method for stereoselectively installing a methyl group, and we used the synthetic versatility of the phenylsulfonyl moiety for this purpose. Desulfonylation of 4 c and 4 h was easily achieved under reducing conditions (Mg in MeOH) to unveil the methyl group and afford the desired adducts 5 c and 5 h, respectively, without eroding the enantiomeric purity (Figure 4 d). To infer the (S) absolute configuration of 2‐methyloctan‐1‐ol (5 c), we compared it with the optical rotation value reported in literature.10
We then evaluated the possibility of applying our strategy to the α‐aryl‐substituted iodosulfones 2 (Figure 5 a), to develop a formal α‐benzylation of aldehydes. A new cycle of optimization revealed that using a para‐fluoro phenyl substituent at the sulfone moiety along with a different solvent mixture (CH2Cl2/H2O 1:1) was essential for reactivity, thus leading to the corresponding derivatives 6 a–e in moderate yield but with high enantiocontrol (Figure 5 b). The low diastereoselectivity generally observed did not affect the overall process, since this stereochemical information is erased upon desulfonylation. Indeed, 6 a (mixture of two diastereoisomers) was transformed into the corresponding enantioenriched benzylated derivative 7 a in good yield and high enantioselectivity using samarium iodide as a reducing agent (Figure 5 c). As detailed in Sections D1 and D2 of the Supporting Information, the majority of products 4 and 6 have been successfully desulfonylated under reducing conditions using either magnesium‐ or samarium‐based methods.
Figure 5.
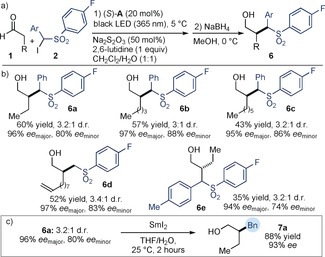
a) The photo‐organocatalytic enantioselective (phenylsulfonyl)benzylation of aldehydes. b) Survey of the aldehydes and the α‐iodosulfones which can participate in the reaction. Yields and enantiomeric excesses of the isolated products are indicated below each entry. c) Desulfonylation reveals the benzyl group. THF=tetrahydrofuran.
In summary, we have reported a photochemical strategy for the enantioselective catalytic formal α‐methylation and α‐benzylation of aldehydes. This two‐step method uses mild reactions conditions and easily available substrates and catalysts to provide compounds which are difficult to synthesize with other catalytic approaches. The chemistry exploits the phenylsulfonyl moiety within the radical precursor 2, which facilitates the generation of open‐shell species acting as a redox auxiliary group, while unveiling the methyl and benzyl groups upon easy desulfonylation of the alkylation products. Key for reaction development was also the ability of chiral enamines to generate radicals upon light excitation and then trap them in a stereocontrolled fashion.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the CERCA Programme (Generalitat de Catalunya) and the European Research Council (ERC 681840—CATA‐LUX). G.F. thanks the Severo Ochoa Excellence Accreditation 2014–2018 (MINECO, SEV‐2013‐0319) for a predoctoral fellowship. We thank Xin Huang for preliminary investigations and Professor Oliver Kappe for useful discussions.
G. Filippini, M. Silvi, P. Melchiorre, Angew. Chem. Int. Ed. 2017, 56, 4447.
Contributor Information
Giacomo Filippini, http://www.iciq.org/research/research_group/prof‐paolo‐melchiorre/.
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Modern Carbonyl Chemistry (Ed.; J. Otera), Wiley-WCH, Weinheim, 2000; [Google Scholar]
- 1b. Principles of Asymmetric Synthesis, 2nd ed. (Eds. R. E. Gawley, J. Aubé), Elsevier, Amsterdam, 2012, chap. 3. [Google Scholar]
- 2.
- 2a. Enders D., Eichenauer H., Tetrahedron Lett. 1977, 18, 191–194; [Google Scholar]
- 2b. Enders D., Eichenauer H., Chem. Ber. 1979, 112, 2933–2960; [Google Scholar]
- 2c. Schmierer R., Grotemeier G., Helmchen G., Selim A., Angew. Chem. Int. Ed. Engl. 1981, 20, 207–208; [Google Scholar]; Angew. Chem. 1981, 93, 209–211; [Google Scholar]
- 2d. Evans D. A., Ennis M. D., Mathre D. J., J. Am. Chem. Soc. 1982, 104, 1737–1739; [Google Scholar]
- 2e. Oppolzer W., Moretti R., Thomi S., Tetrahedron Lett. 1989, 30, 5603–5606. [Google Scholar]
- 3.
- 3a. Imai M., Hagihara A., Kawasaki H., Manabe K., Koga K., J. Am. Chem. Soc. 1994, 116, 8829–8830; [Google Scholar]
- 3b. Doyle A. G., Jacobsen E. N., J. Am. Chem. Soc. 2005, 127, 62–63; [DOI] [PubMed] [Google Scholar]
- 3c. Fischer C., Fu G. C., J. Am. Chem. Soc. 2005, 127, 4594–4595. [DOI] [PubMed] [Google Scholar]
- 4. Melchiorre P., Angew. Chem. Int. Ed. 2009, 48, 1360–1363; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1386–1389. [Google Scholar]
- 5. Hashimoto T., Maruoka K., Chem. Rev. 2007, 107, 5656–5682. [DOI] [PubMed] [Google Scholar]
- 6. Mukherjee S., Yang J. W., Hoffmann S., List B., Chem. Rev. 2007, 107, 5471–5569. [DOI] [PubMed] [Google Scholar]
- 7.For examples of intramolecular SN2-type alkylation, see:
- 7a. Vignola N., List B., J. Am. Chem. Soc. 2004, 126, 450–451; [DOI] [PubMed] [Google Scholar]
- 7b. Enders D., Wang C., Bats J. W., Angew. Chem. Int. Ed. 2008, 47, 7539–7542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7649–7653. [Google Scholar]
- 8.For selected examples, see:
- 8a. Shaikh R. R., Mazzanti A., Petrini M., Bartoli G., Melchiorre P., Angew. Chem. Int. Ed. 2008, 47, 8707–8710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8835–8838; [DOI] [PubMed] [Google Scholar]
- 8b. Cozzi P. G., Benfatti F., Zoli L., Angew. Chem. Int. Ed. 2009, 48, 1313–1316; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1339–1342; [Google Scholar]
- 8c. Brown A. R., Kuo W.-H., Jacobsen E. N., J. Am. Chem. Soc. 2010, 132, 9286–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Nicewicz D., MacMillan D. W. C., Science 2008, 322, 77–80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Shih H.-W., VanderWal M. N., Grange R. L., MacMillan D. W. C., J. Am. Chem. Soc. 2010, 132, 13600–13603. For organocatalytic methods, in the absence of any external photoredox catalyst, see:20831195 [Google Scholar]
- 9c. Arceo E., Jurberg I. D., Álvarez-Fernández A., Melchiorre P., Nat. Chem. 2013, 5, 750–756. [DOI] [PubMed] [Google Scholar]
- 10.The previous formal α-methylation of aldehydes did not use an organic halide but 1,3-benzodithiolylium tetrafluoroborate, which served as a carbocation precursor. See: Gualandi A., Emer E., Guiteras Capdevila M., Cozzi P. G., Angew. Chem. Int. Ed. 2011, 50, 7842–7846; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7988–7992. [Google Scholar]
- 11. List B., Čorić I., Grygorenko O. O., Kaib P. S. J., Komarov I., Lee A., Leutzsch M., Chandra Pan S., Tymtsunik A. V., van Gemmeren M., Angew. Chem. Int. Ed. 2014, 53, 282–285; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 286–289. [Google Scholar]
- 12.
- 12a. Schönherr H., Cernak T., Angew. Chem. Int. Ed. 2013, 52, 12256–12267; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12480–12492; [Google Scholar]
- 12b. Leung C. S., Leung S. S. F., Tirado-Rives J., Jorgensen W. L., J. Med. Chem. 2012, 55, 4489–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Silvi M., Arceo E., Jurberg I. D., Cassani C., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 6120–6123; [DOI] [PubMed] [Google Scholar]
- 13b. Bahamonde A., Melchiorre P., J. Am. Chem. Soc. 2016, 138, 8019–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Yoshida J., Kataoka K., Horcajada R., Nagaki A., Chem. Rev. 2008, 108, 2265–2299; [DOI] [PubMed] [Google Scholar]
- 14b. Tyson E. L., Farney E. P., Yoon T. P., Org. Lett. 2012, 14, 1110–1113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Beatty J. W., Douglas J. J., Cole K. P., Stephenson C. R., Nat. Commun. 2015, 6, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For reports on the generation of phenylsulfonyl methyl radicals, see:
- 15a. Masnyk M., Tetrahedron Lett. 1991, 32, 3259–3262; [Google Scholar]
- 15b. Gui J., Zhou Q., Pan C., Yabe Y., Burns A. C., Collins M. R., Ornelas M. A., Ishihara Y., Baran P. S., J. Am. Chem. Soc. 2014, 136, 4853–4856; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Liu F., Li P., J. Org. Chem. 2016, 81, 6972–6979. [DOI] [PubMed] [Google Scholar]
- 16. Donslund B. S., Johansen T. K., Poulsen P. H., Halskov K. S., Jørgensen K. A., Angew. Chem. Int. Ed. 2015, 54, 13860–13874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14066–14081. [Google Scholar]
- 17.
- 17a. Curran D. P., Chang C.-T., Tetrahedron Lett. 1990, 31, 933–936; [Google Scholar]
- 17b. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]
- 18.
- 18a. Jeffery G. H., Bassett J., Mendham J., Denney R. C., Vogel's Quantitative Chemical Analysis, 5th ed., Longman Scientific & Technical, Harlow Essex, 1989; [Google Scholar]
- 18b. Ono S., Tsuchihashi S., Kuge T., J. Am. Chem. Soc. 1953, 75, 3601–3602. [Google Scholar]
- 19. Lalevée J., Allonas X., Fouassier J. P., Chem. Phys. Lett. 2008, 454, 415–418. [Google Scholar]
- 20. Pintauer T., Matyjaszewski K., Encyclopedia of Radicals, Vol. 4, Wiley, Hoboken, 2012, p. 1851. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary