

Abstract
Lower Lewis acidity boranes demonstrate greater tolerance to combinations of water/strong Brønsted bases than B(C6F5)3, this enables Si−H bond activation by a frustrated Lewis pair (FLP) mechanism to proceed in the presence of H2O/alkylamines. Specifically, BPh3 has improved water tolerance in the presence of alkylamines as the Brønsted acidic adduct H2O–BPh3 does not undergo irreversible deprotonation with aliphatic amines in contrast to H2O–B(C6F5)3. Therefore BPh3 is a catalyst for the reductive amination of aldehydes and ketones with alkylamines using silanes as reductants. A range of amines inaccessible using B(C6F5)3 as catalyst, were accessible by reductive amination catalysed by BPh3 via an operationally simple methodology requiring no purification of BPh3 or reagents/solvent. BPh3 has a complementary reductive amination scope to B(C6F5)3 with the former not an effective catalyst for the reductive amination of arylamines, while the latter is not an effective catalyst for the reductive amination of alkylamines. This disparity is due to the different pK a values of the water–borane adducts and the greater susceptibility of BPh3 species towards protodeboronation. An understanding of the deactivation processes occurring using B(C6F5)3 and BPh3 as reductive amination catalysts led to the identification of a third triarylborane, B(3,5‐Cl2C6H3)3, that has a broader substrate scope being able to catalyse the reductive amination of both aryl and alkyl amines with carbonyls.
Keywords: frustrated Lewis pairs, boron, protodeboronation, reductive amination, water tolerance
Introduction
Considerable progress in frustrated Lewis pair (FLP) chemistry has been achieved in the last decade principally using tris(pentafluorophenyl)borane, B(C6F5)3.1 Compared to BPh3, the presence of fluorine atoms dramatically increases the Lewis acidity.2 While high Lewis acidity is essential in enabling certain FLP reactivity, it also poses challenges including the compatibility of FLPs with water (e.g. from unpurified reactants/solvents or as a reaction by‐product)/ base combinations, a topic which has attracted recent attention.3, 4, 5, 6 A fluorinated triarylborane with a high Lewis acidity towards hydride (which is desirable for H−H and Si−H bond activations) also has considerable oxophilicity, with the corresponding triarylborane–water adduct exhibiting much greater Brønsted acidity than water itself.7 Indeed, the Brønsted acidity of H2O–B(C6F5)3 was determined by Parkin and co‐workers (pK a=8.4 in MeCN) to be comparable to that of HCl (8.5 in MeCN).7a This poses a limit to the water tolerance of these fluorinated arylboranes in the presence of certain Brønsted bases because irreversible deprotonation of the borane–water adduct yields an inactive (for FLP chemistry) hydroxytriarylborate anion.
Ashley, Stephan, and co‐workers pioneered ROH‐tolerant FLP reactions and demonstrated that B(C6F5)3 could be used for the hydrogenation of carbonyls (Scheme 1 A). Importantly, the alcohol–borane adducts are not irreversibly deprotonated under these weakly basic conditions (which use ethereal solvents such as 1,4‐dioxane as Lewis bases to activate H2 via an FLP mechanism).3, 8 Demonstration of the water tolerance of B(C6F5)3 was subsequently reported proving that the hydrogenation of ketones could be performed using non‐purified, “wet” reactants and solvents (H2O–B(C6F5)3 also is not irreversibly deprotonated by ethereal solvents).4 Recently, we reported the water tolerance of a B(C6F5)3‐catalysed system involving more basic arylamines (conjugate acid pK a ca. 11 in MeCN, Scheme 1 B).5 In particular we found that B(C6F5)3 is able to catalyse the reductive amination of aldehydes and ketones with anilines using 1.2 equivalents of silane as reductant.9 This proceeds in the presence of a super‐stoichiometric amount of water derived from imine formation and the use of non‐purified solvents. An elegant extension of this approach was recently reported using B(C6F5)3 to catalyse the tandem Meinwald rearrangement and reductive amination of epoxides with anilines and silanes.10 However, in the latter, as in our work, reductive amination could not be extended to alkylamines (conjugate acid pK a ≥ 16 in MeCN) due to the irreversible deprotonation of H2O–B(C6F5)3. Thus, the compatibility of H2O–B(C6F5)3 with bases appears to be limited to those bases with conjugate acids that have pK a values ≤12 (in MeCN). A broader amine scope catalytic reductive amination methodology using a simple triarylborane is desirable as a one‐pot method (thus preferable from an efficiency perspective) to rapidly access amines that are ubiquitous functionalities in natural products, pharmaceuticals and agrochemicals.
Scheme 1.
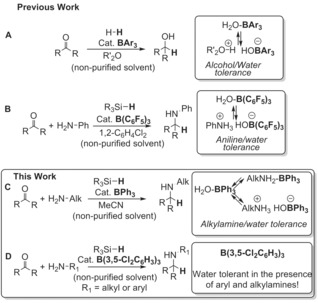
Previous work (top and middle): alcohols and anilines tolerated by fluorinated‐triarylborane–water adducts; this work (inset): alkylamines tolerated by the BPh3–OH2 adduct and both alkyl and arylamine/H2O combinations tolerated by B(3,5‐Cl2C6H3)3.
To circumvent the limitation of B(C6F5)3 towards water/strong Brønsted base combinations, Lewis acids that are less oxophilic are required. These could be “hydride selective” Lewis acids, such as Group 14 based Lewis acids (which maintain high hydridophilicity but have lower oxophilicity)11 or Lewis acids that are globally less Lewis acidic (e.g., less oxophilic and less hydridophilic).12 The latter approach was utilised by Papai, Soós and co‐workers who employed less Lewis acidic partially halogenated triarylboranes for example, (2,3,5,6‐C6F4H)2B(2,6‐C6H4Cl2), for the catalytic hydrogenation of carbonyls in ethereal solvents, with some water tolerance demonstrated.6 Taking this approach further, the non‐halogenated triarylborane BPh3 should have enhanced tolerance to water and strong base combinations due to its lower Lewis acidity. BPh3 does however still possess sufficient hydridophilicity to be useful as a catalyst in FLP‐type reactions as recently demonstrated.13, 14 While H2O–B(C6F5)3 is well documented,7 the corresponding H2O–BPh3 adduct is less studied, particularly its ability to act as a Brønsted acid.16, 17, 18, 19 Herein we report an extension to the water and base tolerance of boranes to strong amine bases, focusing, in particular on the triarylborane‐catalysed reductive amination of aldehydes/ketones with alkylamines using silanes as reducing agents. This demonstrates that BPh3 is an effective catalyst for the reductive amination of alkylamines and carbonyls (Scheme 1 C), including examples challenging to reduce with borohydride salts (e.g., [(OAc)3BH]−). Furthermore, B(3,5‐Cl2C6H3)3 is effective for the reductive amination of carbonyls and both aryl and alkylamines without requiring any inert atmosphere techniques or solvent/reagent purification (Scheme 1 D).
Results and Discussion
To determine if H2O–BPh3 protonates alkylamines, BnNH2 (conjugate acid pK a=16.6 in MeCN)8 was added to a solution of H2O–BPh3 in [D3]‐MeCN. 1H NMR spectroscopy showed coordination of BnNH2 to BPh3, as indicated by a 2H integral resonance at δ=5.3 ppm (for BnNH 2) shifted downfield from free BnNH 2 in [D3]‐MeCN (1.5 ppm). Identical 1H NMR resonances are observed for Ph3B–N(H)2Bn formed under anhydrous conditions in [D3]‐MeCN (for both δ 11B=−1.7 ppm). Coordination of BnNH2 to BPh3 is reversible at room temperature as addition of benzaldehyde led to rapid imine formation, thus the absence of any observable [HO–BPh3]− is attributed to the lower Brønsted acidity of H2O–BPh3. In contrast, the addition of BnNH2 to H2O–B(C6F5)3 led to formation of [HO–B(C6F5)3]− as the major product (by 11B and 19F NMR spectroscopy) as expected based on relative pK a values. With no observable deprotonation of H2O–BPh3 with BnNH2, the utility of BPh3 as a catalyst was explored in the reductive amination of benzaldehyde (1.0 equiv) with benzylamine (1.2 equiv), under air using non‐purified BPh3, non‐purified solvents, and silane as reductant (Table 1). In this reaction, upon imine formation, water is produced as a by‐product, so both excess (relative to BPh3) water and a good Brønsted base (BnNH2, used in slight excess to favour imine formation) are present in the reaction mixture.
Table 1.
Initial optimization of BPh3‐catalysed reductive amination.
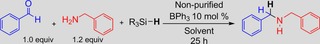
| Entry | Solvent | Silane | Equiv. Silane | Temp. [°C] | Yield [%][a] |
|---|---|---|---|---|---|
| 1 | o‐DCB | PhMe2SiH | 1.2 | 100 | <5 |
| 2 | MeCN | PhMe2SiH | 1.2 | 100 | 33 |
| 3 | o‐DCB | PhMe2SiH | 3.5 | 100 | <5 |
| 4 | MeCN | PhMe2SiH | 3.5 | 100 | 87 (80)[b] |
| 5c | MeCN | PhMe2SiH | 3.5 | 100 | 35 |
| 6 | MeCN | PhMe2SiH | 3.5 | 60 | 6 |
| 7 | MeCN | Ph2SiH2 | 3.5 | 100 | 86 |
| 8 | MeCN | Ph2MeSiH | 3.5 | 100 | 8 |
| 9 | MeCN | PhMeSiH2 | 3.5 | 100 | 55 |
| 10 | MeCN | PhSiH3 | 3.5 | 100 | 56 |
Reactions performed in sealed tubes. [a] Yield by 1H NMR spectroscopy versus mesitylene as internal standard. [b] Isolated yield. [c] Reaction at 5 mol % catalyst loading.
For a direct comparison with our previous work using B(C6F5)3,5 we initially performed the reaction in ortho‐dichlorobenzene (o‐DCB) using 1.2 equivalents of silane. Under these conditions imine formation proceeds but no reduction was observed using 10 % mol BPh3 (Table 1, entry 1). Okuda and co‐workers reported that BPh3 is a more effective catalyst for (de)hydrosilylation reactions in polar solvents such as MeCN or nitromethane.13 Changing the solvent from o‐DCB to MeCN now resulted in the desired product being obtained in moderate yield. On increasing the amount of silane from 1.2 to 3.5 equivalents, dibenzylamine was obtained in good yield (87 % NMR yield and 80 % isolated yield). The requirement for excess silane is due to imine reduction and H2O/silanol dehydrosilylation occurring concurrently. The activity of this system is not due to initial consumption of all H2O by excess silane and then imine reduction proceeding under anhydrous conditions as indicated by the absence of any induction period in this reductive amination. This was further confirmed by analysis of the reaction mixture after 3 hours at 100 °C, at which point considerable imine reduction had occurred (ca. 30 %) but significant water and PhMe2SiOH were still present.20 Decreasing the catalyst loading to 5 mol % resulted in a lower yield (entry 5), while 100 °C was found to be critical (entry 6). The applicability of other silanes was then investigated: while Ph2SiH2 was viable in the reductive amination (entry 7), the increase in the steric hindrance of the silane going from PhMe2SiH to Ph2MeSiH, resulted in a significant drop in imine reduction (entry 4 vs. 8). When smaller silanes were employed (entries 9 and 10), dibenzylamine was the major component among multiple products, including EtNH2 presumably deriving from MeCN reduction.
With the compatibility of BnNH2 and H2O–BPh3 mixtures confirmed by the successful reductive amination of benzaldehyde and BnNH2, a direct comparison between B(C6F5)3 and BPh3 was performed. In our previous work we found that B(C6F5)3 catalysed reductive aminations of anilines and aldehydes in o‐DCB at 100 °C, but not the more basic alkylamines due to irreversible deprotonation of H2O–B(C6F5)3.5 To avoid any disparities arising from the solvent employed, comparative reductive aminations using benzaldehyde and aniline or benzylamine with B(C6F5)3 or BPh3 as catalyst were performed in MeCN (Table 2). Although the coordination of MeCN to B(C6F5)3 is well documented,21 the reductive amination of benzaldehyde and aniline still proceeded to high yield (96 %) in 1 h at 100 °C on replacing o‐DCB with MeCN. As previously reported, 1.2 equivalents of silane is sufficient using anilines with imine reduction occurring preferentially to water dehydrosilylation. Interestingly, on replacing B(C6F5)3 with BPh3 under identical conditions, minimal (8 %) imine reduction and minimal water dehydrosilylation were observed after 1 h on heating at 100 °C. A similar outcome was observed using 0.1 equivalent BPh3 loading and 3.5 equivalents of silane (entry 2) with a low reductive amination conversion even after 25 h. In contrast, in the reductive amination of benzaldehyde/benzylamine under identical conditions the use of BPh3 results in an excellent conversion, whilst B(C6F5)3 is effectively inactive (entry 3).
Table 2.
Reductive amination catalysed by BPh3 or B(C6F5)3.

| Entry | R | Mol % Catal. | Equiv. silane | Time [h] | Yield [%][a] B(C6F5)3 BPh3 | |
|---|---|---|---|---|---|---|
| 1 | Ph | 5 | 1.2 | 1 | 96 | 8 |
| 2 | Ph | 10 | 3.5 | 25 | >96 | 35 |
| 3 | Bn | 10 | 3.5 | 25 | <5 | 87 |
Reactions performed in sealed tubes. [a] Yield by 1H NMR spectroscopy versus mesitylene.
Notably, during reductive aminations using BPh3 as catalyst four‐coordinate boron species (such as imine→BPh3 and amine→BPh3) and 11B resonances consistent with Ph2BOH and PhB(OH)2 are all observed. Importantly, attempts to catalyse the reductive amination of benzaldehyde/benzylamine with PhB(OH)2, Ph2B(OH) or Ph3BOH− (whilst not observed the latter is feasibly present in low concentration through a small degree of H2O–BPh3 deprotonation) in place of BPh3 led to very low conversions (e.g., ca. 10 % using Ph2BOH) after 25 h at 100 °C in MeCN. The use of Brønsted acids such as HCl and HNO3 also resulted in minimal reductive amination. Combined these control reactions indicate the importance of the triarylborane as the catalyst in this process, presumably for activation of the silane via established (for B(C6F5)3) mechanistic pathways.22
To better understand the disparities between PhNH2 and BnNH2 in reductive aminations catalysed by BPh3, a number of control reactions were performed. A solution of BPh3 in anhydrous MeCN was heated at 100 °C sealed under air, with no significant reaction (e.g., protodeboronation) observed. However, adding 10 equivalents of water to this solution led to significant protodeboronation after 2 hours at 100 °C (PhB(OH)2, Ph2B(OH) and PhH observed by 1H and 11B NMR spectroscopy) presumably via an intramolecular protodeboronation process from H2O–BPh3 as recently calculated for H2O–B(C6F5)3.23 Having identified that H2O–BPh3 can undergo protodeboronation to produce catalytically inactive products the effect of amine basicity on protodeboronation was investigated. The addition of 10 equivalents of PhNH2 to a solution of H2O–BPh3 (made by mixing 1 equivalents of BPh3 with 10 equivalents of water in MeCN to approximate the catalysis conditions) did not prevent protodeboronation on heating. Notably, when 10 equivalents of the more basic amine BnNH2 was added to an identical solution containing H2O–BPh3, protodeboronation proceeded to a significantly lower extent (by monitoring the appearance of benzene in the 1H NMR spectrum and by 11B NMR spectroscopy). Even upon heating at 100 °C for 20 hours (Figure 1) four‐coordinate L→BPh3 compounds were still the dominant species with BnNH2 in contrast to that with PhNH2.
Figure 1.
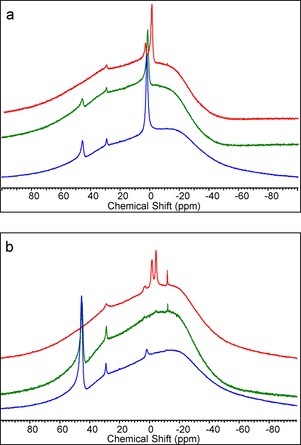
11B NMR spectra of H2O–BPh3 or H2O–BPh3/amine 1:10:10 immediately on mixing (top) and after heating at 100 °C for 20 h (bottom) in [D3]‐MeCN. Blue (no amine), green (+PhNH2), red (+BnNH2).
The disparity between PhNH2 and BnNH2 in reductive amination catalyzed by BPh3 will be due to different amine (or imine) basicity, however this will affect a number of processes, therefore to identify the origin of this disparity a number of control reactions were performed. The disparity is not due to the less nucleophilic imine derived from aniline/benzaldehyde leading to a significantly greater barrier to an SN2 type reaction with the R3Si–H–BPh3 species. This was confirmed by the fact that under anhydrous conditions using catalytic BPh3 and stoichiometric PhMe2SiH, N‐benzylidene aniline and N‐benzylidene benzylamine were both reduced (Scheme 2, left). However, under catalytic reductive amination conditions the key electrophile could be the silylated iminium cation (if the BPh3 activated silane is directly attacked by the imine) or the protonated iminium cation (via imine protonation by [R3Si–OH2][HBPh3] formed from initial attack by H2O on R3Si–H–BPh3). Although no silylated amine was observed during reductive amination, the exact nature of the iminium cation could not be unambiguously defined in this process due to the fast hydrolysis of silylated amine under these conditions. Nevertheless, further control reactions showed that both protonated N‐benzylidene aniline and N‐benzylidene benzylamine were reduced by [HBPh3]− (consistent with Okuda and co‐workers report on imine hydroboration catalyzed by [HBPh3]− salts). 24 There was no evidence for differing degrees of side reactions (such as evolution of PhH (by protodeboronation)) or significant differences in the rate of reduction during the control reactions with the iminium cations (Scheme 2, right). Whilst the iminium cations derived from N‐benzylidene aniline do undergo slower reductions (than those derived from N‐benzylidene benzylamine) this should only result in longer reaction times being required for complete reductive amination using PhNH2/benzaldehyde under BPh3 catalysis. However, this is not observed, as no further increase in conversion is observed on longer reaction times in reductive aminations. Combined these observations indicate that the difference in reactivity is due to more rapid catalyst decomposition in the presence of PhNH2 relative to BnNH2 and not any intrinsic barrier to N‐benzylidene aniline reduction.
Scheme 2.
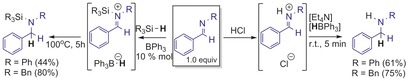
N‐benzylidene amines reduction.
As BPh3 decomposition most probably proceeds via H2O–BPh3 (based on its fast protodeboronation), reducing the concentration of this species in solution should be key to provide enhanced catalytic activity. At least two scenarios are feasible for achieving this: i) the more basic species (BnNH2 or its derived imine) retards protodeboronation by deprotonating H2O–BPh3 resulting in a different catalyst resting state, [HO–BPh3]− , that is more stable to protodeboronation; ii) the more nucleophilic amine/imine (e.g., BnNH2 or its derived imine) forms a Lewis adduct L→BPh3, which is more stable to protodeboronation than Ph3B–OH2. Based on the in situ NMR data for H2O–BPh3/BnNH2 the latter is more probable as only Bn(H)2 n–BPh3 is observed with no [Ph3B–OH]− detectable. In contrast, with the less basic/nucleophilic aniline, the adduct Ph(H)2 n–BPh3 (which when formed under anhydrous conditions has a characteristic integral 2H singlet in the 1H NMR spectrum at δ=5.7 ppm for the NH2 group) reacts with equimolar water as indicated by a drastic shift in the 1H NMR spectrum to a broad resonance at δ=2.1 ppm (integral four for the combined NH2/OH2 resonance). This suggests an equilibrium between Ph(H)2 n–BPh3 and H2O–BPh3 consistent with the more rapid protodeboronation observed. The 11B NMR spectra are inconclusive for this system as H2O–BPh3 and Ph3B–N(H)2Ph have extremely similar chemical shifts, whilst the slow exchange regime is not reached even at −38 °C in [D3]‐MeCN.
With the disparity between BnNH2 and PhNH2 in reductive aminations catalyzed with BPh3 clarified, we next investigated the highly Brønsted basic but less nucleophilic amine tBuNH2. Significantly, tBuNH2 and PhNH2 have similar Mayr nucleophilicity values in MeCN (N=12.35 and 12.64, respectively),25 but the conjugate acid of tBuNH2 has a pK a of 18.4. Under standard conditions (3.5 equiv. silane, 10 mol % BPh3, MeCN), the reductive amination of tBuNH2 and benzaldehyde proceeded to a 93 % conversion after 25 h at 100 °C. Again the 11B NMR spectrum after 25 h was dominated by four‐coordinate boron species with minimal PhB(OH)2 and Ph2B(OH) observed. To investigate the origin of the enhanced stability of BPh3 in the presence of tBuNH2, the 1H and 11B NMR spectra of BPh3/tBuNH2/H2O mixtures was examined, which revealed broad resonances at 25 °C, (e.g., a 1H resonance at δ=3.7 ppm) shifted downfield with respect to tBuNH 2 and H 2O–BPh3 (δ=1.3 and 2.6 ppm, respectively). Cooling this solution to below −10 °C resulted in the appearance of tBuN(H)2–BPh3, however, this was a minor component (ca. 10 %). The major resonance in the 1H NMR spectrum was still broad with a chemical shift not consistent with H2O–BPh3 or free tBuNH2, instead it is assigned as H2O–BPh3 and [HOBPh3][H3NtBu] in fast exchange, a process which was not frozen out at −38 °C in [D3]‐MeCN. Based on these observations feasible key processes occurring in situ in the reductive amination reactions are summarised in Scheme 3.
Scheme 3.
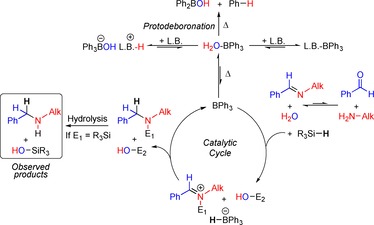
Feasible key reactions in reductive amination reaction mixtures. L.B.=Lewis bases. E1=H, E2=R3Si or E1=R3Si, E2=H.
Upon heating, enough BPh3 is generated from a Lewis adduct or the hydroxyborate to activate the silane to nucleophilic attack. Nucleophilic attack leads to the formation of [HBPh3]− that in turn would reduce the iminium cation (either silylated or protonated) by hydride transfer thus regenerating the catalyst. The protodeboronation pathway deactivates the catalyst, and is a process which most probably proceeds from H2O–BPh3. The concentration of this species can be minimized in solution by using stronger bases/nucleophiles which lead to formation of LB→BPh3 or [LB–H][HOBPh3] (LB=amine or imine). Notably, in the presence of both BnNH2 and N‐benzylidene benzylamine, BPh3 binds the former preferentially. As the optimal catalysis conditions uses a slight excess of amine, the continued presence of free amine presumably helps reduce the quantity of H2O–BPh3 present and thus limit protodeboronation.
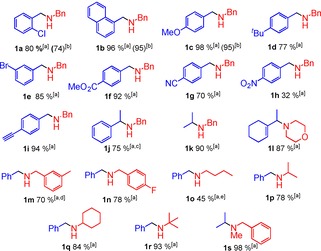
With an understanding of the limitations of using BPh3 for catalytic reductive amination, the substrate scope was then explored with the reactions performed under air, using non‐purified solvent and reactants with everything combined at the start in an operationally simple process (Table 3).
Table 3.
Substrates screening of the reductive amination.

|
|---|
|
Reactions performed in sealed tubes. [a] 1H NMR yields versus mesitylene. [b] Isolated yield. [c] 48 h. [d] 40 h. [e] 30 h.
A range of functionalised benzaldehydes were amenable in the reductive amination with benzylamine, with good in situ conversions and isolated yields (1 a–e). It is noteworthy that ester and cyano substituents were compatible, with no evidence for their reduction under these conditions (1 f, g). However, the reaction was less tolerant to nitro substituents (due to trans‐imination and formation of dibenzylamine observed as the major by‐product). It is noteworthy that when electron‐withdrawing groups are present in the para position of benzaldehyde (e.g. ‐CO2Me or ‐CN), minimal siloxane (and silanol) were observed after 25 h (by 1H and 29Si NMR spectroscopy), with significant reduction of the imine still occurring. Furthermore >50 % imine reduction to 1 f was observed with only 1.2 equivalents of silane after 25 h. This indicates that more electrophilic imines effectively out compete H2O for reaction with the borohydride, whereas with less electrophilic imines the rates of water/silanol dehydrosilylation and iminium cation reduction are comparable hence excess silane is required. Reductive amination also proceeded in the presence of a terminal C−C triple bond without significant reduction of the latter (1 i), or any observable side reactivity, for example, dehydroboration.1d When aliphatic aldehydes (n‐butyraldehyde and propionaldehyde) were used, full consumption of the in situ formed imine was observed, but the desired product was only a minor component due to over‐alkylation to the tertiary amine or enamine isomerization reactions, as reported for B(C6F5)3.5 However, when ketones were utilised, the reaction was successful, allowing a secondary carbon centre to be attached to the nitrogen (1 j,k). Notably, the reductive amination of acetophenone and benzylamine is challenging with widely used reducing agents such as Na[triacetoxyborohydride] (Na[(OAc)3BH], 55 % yield after 10 days),26 in contrast using BPh3/silane 1 j is produced in higher yield in shorter reaction times. The reductive amination of 1‐acetyl‐1‐cyclohexene and morpholine to yield 1 l is also challenging using [(OAc)3BH]− (only 10 % yield after 4 days),26 but it proceeds to 87 % yield using BPh3/silane. This demonstrates that the BPh3‐catalysed process is applicable to systems where established borohydride reductive amination approaches struggle. Furthermore, the formation of 1 l shows the compatibility of this methodology with C−C double bonds. The inclusion of substituents on benzylamine, as well as the use of nBuNH2 as another C‐primary amine, was also realized (e.g. 1 m–o), although using the latter amine over‐alkylation also occurred to some extent (e.g. forming nBu2NBn). C‐secondary amines, such as cyclo‐hexylamine and isopropylamine, or a C‐tertiary amine tBuNH2, gave good conversions to the desired products (1 p–r). It is noteworthy that a common product could be formed from a different combination of aldehyde/amine (e.g. 1 k and 1 p), offering two retrosynthetic strategies. Finally, when a secondary amine such as BnN(H)Me was used in combination with an enolizable ketone the reaction still proceeds successfully to form 1 s in excellent yield. It should be emphasized that these amines are not accessible by reductive amination using B(C6F5)3 as catalyst due to it being limited to aniline derivatives. To demonstrate scalability the reductive amination of benzaldehyde and 1‐adamantylamine was performed on gram‐scale under air, using 10 mol % of unpurified BPh3 in non‐purified acetonitrile and using PhMe2SiH as reductant (Scheme 4). Combining all the reactants at the start and heating the reaction mixture at 100 °C for 25 hours enabled the desired product to be isolated in a 90 % isolated yield (1.1 g).
Scheme 4.

Gram‐scale synthesis of N‐benzyl‐1‐adamantylamine.
The results discussed above indicate that B(C6F5)3 and BPh3 have complementary tolerance to water/amine combinations in reductive aminations (Figure 2). B(C6F5)3 is a viable catalyst for aryl amines (conjugate acids pK a<12 in MeCN) but not alkylamines (conjugate acids pK a>16 in MeCN) due to irreversible deprotonation of H2O–B(C6F5)3 with the latter. In contrast, BPh3 is a viable reductive amination catalyst for alkylamines but not arylamines due to more rapid protodeboronation in the presence of the latter. We were thus interested in exploring an amine with an intermediate pK a, specifically the reductive amination of benzaldehyde and benzhydrylamine (conjugate acid pK a 15 in MeCN)27 was performed with both these boranes using 10 mol % catalyst loading. In all cases the in situ conversions were only moderate at best (less than 30 %) under a range of conditions with both boranes (e.g., in MeCN or o‐DCB at 100 °C), indicating that an amine whose conjugate acid has a pK a between 12–16 is particularly challenging for both boranes. Again in situ analysis revealed that with BPh3 significant protodeboronation proceeded upon heating (by 11B NMR spectroscopy), whilst with B(C6F5)3 the deactivation was due to the effectively irreversible deprotonation of H2O–B(C6F5)3 (by 11B/19F NMR spectroscopy).
Figure 2.
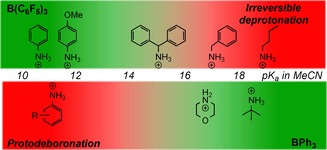
Water/amine tolerance of B(C6F5)3 and BPh3 under the reductive amination reaction conditions.
Given the respective limitations of B(C6F5)3 and BPh3, a single triarylborane that is a viable catalyst for the reductive amination of both aryl and alkyl amines (including benzhydrylamine) was targeted. To have a broad amine scope, the triarylborane must form a H2O–BAryl3 adduct that is both more resistant to protodeboronation than H2O–BPh3 and less Brønsted acidic than H2O–B(C6F5)3. Furthermore, a triarylborane that does not contain ortho‐halogen aryl substituents is desirable, as ortho substituents increase the steric bulk around boron and thus can significantly hinder amine/imine coordination to boron.12 The latter is actually desired in this process as it reduces the concentration of H2O–BAryl3 in solution, thus also helping to limit protodeboronation. Given these requisites B(3,5‐Cl2C6H3)3 was selected and its synthesis via the protolytic decomposition of its tetraarylborate salt was utilised as the borate salt is air and moisture stable as a solid in contrast to the free triarylboranes (see subsequent discussion). Tetraarylborate anion decomposition has significant precedence for [BPh4]− salts which react with Brønsted acids to release BPh3 compounds.28 Furthermore, we recently observed decomposition of Na[B(3,5‐Cl2C6H3)4] (termed Na[BArCl] herein) in wet solvents on heating. To confirm that Na[BArCl] decomposition by protonolysis generates B(3,5‐Cl2C6H3)3 species, the strong Brønsted acid HNTf2 was added to NaBArCl. This resulted in the appearance of a major new resonance at δ=67 ppm in the 11B NMR spectrum assigned as B(3,5‐Cl2C6H3)3, with this chemical shift consistent with other reported tri(chloroaryl)boranes.29 Applying this in situ B(3,5‐Cl2C6H3)3 generation procedure (using an excess of Na[BArCl] relative to HNTf2 to preclude any trace Brønsted acid remaining as strong Brønsted acids can also activate Si−H bonds),30 B(3,5‐Cl2C6H3)3 catalyzed the reductive amination of benzaldehyde and benzhydrylamine to give the desired product in good yield (Scheme 5). The use of both B(C6F5)3 and BPh3 as catalysts under these conditions gave low conversions.
Scheme 5.
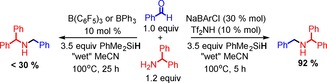
Reductive amination with benzaldehyde and benzylhydrylamine using B(C6F5)3, BPh3 or B(3,5‐C6H3Cl2)3 (generated in situ) as catalyst.
Seeking an operationally simpler process, the decomposition of Na[BArCl] by action of H2O was investigated as a route to generate B(3,5‐Cl2C6H3)3 in situ.31, 32 This approach was successful for the catalytic reductive amination of benzhydrylamine and benzaldehyde using 10 mol % Na[BArCl] in o‐DCB (Scheme 6), with all manipulations performed in air using non‐purified solvent/reagents. Weakly coordinating solvents are essential as attempts using MeCN as solvent led to no reductive amination. The solvent dependency is attributed to the formation of [(H2O)xNa]+ species in o‐DCB that have enhanced Brønsted acidity (relative to H2O) and are thus key to effecting anion protodeboronation and generation of the triarylborane, as previously discussed for NaBPh4.28 In contrast in MeCN, the solvent is presumably solvating the Na cations, resulting in a less Brønsted acidic solution and no anion protodeboronation.
Scheme 6.
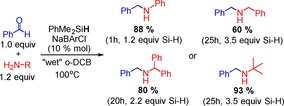
Reductive aminations under air employing Na[BArCl] as precursor catalyst.
With an in situ catalyst generation protocol in hand, a brief amine substrate scope exploration was undertaken. Most notably, the triarylborane derived in situ from Na[BArCl] was able to catalyse the reductive amination of benzaldehyde with PhNH2, BnNH2, and tBuNH2 amines whose conjugate acids span the pK a range from 10.6 to 18.4 in MeCN. This indicates a reduced acidity of the corresponding H2O–B(3,5‐Cl2C6H3)3 adduct (relative to that of H2O–B(C6F5)3) and an improved stability of B(3,5‐Cl2C6H3)3 species to protodeboronation (relative to BPh3). The amount of silane required for good conversion to the reductive amination product was explored and again found to depend on the imine electrophilicity, with the more electrophilic imine (derived from aniline) reduced using only 1.2 equivalents of silane, whilst the less electrophilic imines again required an excess of silane due to competitive dehydrosilylation reactions.
The ability to use Na[BArCl] as a precursor to the active triarylborane catalyst has practical advantages since it is readily synthesized and is bench stable for at least 6 months. In contrast, whilst BPh3 is commercially available its storage as a solid under ambient atmosphere leads to gradual decomposition (even after only 14 days significant PhB(OH)2 and Ph2B(OH) are observed by 11B NMR spectroscopy). This negatively impacts conversion; for example using pristine BPh3 gives 87 % conversion of benzaldehyde and benzylamine to the reductive amination product whereas the same batch of BPh3 stored as a solid in air for 2 weeks results in only 52 % conversion when used as the catalyst under otherwise identical conditions. In contrast, Na[BArCl] stored as a solid for 6 months in air shows no deterioration in reductive amination catalytic activity. Thus Na[BArCl] is a useful bench‐stable catalyst precursor for reductive aminations, with its utility further demonstrated in the rapid synthesis of the more complex drug molecule Piribedil (used in the treatment of Parkinson's disease)33 in good yield (Scheme 7) under air using non‐purified reagents/solvents.
Scheme 7.

Synthesis of Piridebil by reductive amination.
Conclusions
In summary, BPh3 has a higher tolerance to H2O and alkylamine combinations than B(C6F5)3, due to the lower Brønsted acidity of H2O–BPh3. This extends the water/base tolerance of FLP systems to strong bases (conjugate acid pK a=18.5). This enables the utilisation of BPh3 as a catalyst for the reductive amination of aldehydes and ketones with many different aliphatic amines, ranging from C‐primary to C‐tertiary. This system is even effective for the reductive amination of substrates that are challenging with conventional borohydrides (e.g., [(OAc)3BH]−). BPh3 and B(C6F5)3 exhibit complementary amine scope in reductive aminations, with the former limited by the protodeboronation of H2O–BPh3 in the presence of weaker amine Brønsted bases/nucleophiles, while the latter is limited by H2O–B(C6F5)3 undergoing irreversible deprotonation by stronger Brønsted basic amines such as alkylamines. Finally, a third triarylborane, B(3,5‐Cl2C6H3)3, of intermediate Lewis acidity, was shown to be effective for the reductive amination of a range of amines whose conjugate acids span pK a values of 10.6 to 18.5 in MeCN. Furthermore, in situ tetraarylborate anion decomposition by H2O in non‐coordinating solvents represents a simple route to generate the active triarylborane catalyst from a readily accessible bench‐stable precursor. The reductive amination methodologies presented herein are operationally simple (e.g. no purification of any materials/solvent is required and the reactions are performed under air) and are applicable to gram‐scale and complex molecule synthesis.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was made possible by financial support from the University of Manchester, the EPSRC (EP/M023346/1 and EP/K03099X/1), and the Royal Society (for the award of a University Research Fellowship). Dr. Alex Pulis is thanked for useful discussions.
V. Fasano, M. J. Ingleson, Chem. Eur. J. 2017, 23, 2217.
References
- 1.For recent reviews on FLP chemistry see:
- 1a. Oestreich M., Hermeke J., Mohr J., Chem. Soc. Rev. 2015, 44, 2202–2220; [DOI] [PubMed] [Google Scholar]
- 1b. Stephan D. W., J. Am. Chem. Soc. 2015, 137, 10018–10032; [DOI] [PubMed] [Google Scholar]
- 1c. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2015, 54, 6400–6441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6498–6541; [Google Scholar]
- 1d. Topics in Current Chemistry, Vol. 332, Frustrated Lewis Pairs I II (Eds.: G. Erker, D. W. Stephan), Springer, Heidelberg, 2013. [Google Scholar]
- 2.For the Lewis acidity of perfluoroarylboranes see:
- 2a. Timoshkin A. Y., Frenking G., Organometallics 2008, 27, 371–380; [Google Scholar]
- 2b. Morgan M. M., Marwitz A. J. V., Piers W. E., Parvez M., Organometallics 2013, 32, 317–322; [Google Scholar]
- 2c. Sivaev I. B., Bregadze V. I., Chem. Soc. Rev. 2014, 270, 75–88. [Google Scholar]
- 3.For alcohol tolerant FLP reductions see:
- 3a. Scott D. J., Fuchter M. J., Ashley A. E., J. Am. Chem. Soc. 2014, 136, 15813–15816; [DOI] [PubMed] [Google Scholar]
- 3b. Mahdi T., Stephan D. W., J. Am. Chem. Soc. 2014, 136, 15809–15812; [DOI] [PubMed] [Google Scholar]
- 3c. Mahdi T., Stephan D. W., Angew. Chem. Int. Ed. 2015, 54, 8511–8514; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8631–8634. [Google Scholar]
- 4. Scott D. J., Simmons T. R., Lawrence E. J., Wildgoose G. G., Fuchter M. J., Ashley A. E., ACS Catal. 2015, 5, 5540–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fasano V., Radcliffe J. E., Ingleson M. J., ACS Catal. 2016, 6, 1793–1798. [Google Scholar]
- 6. Gyömöre A., Bakos M., Földes T., Papai I., Domjá N. A., Soós T., ACS Catal. 2015, 5, 5366–5372. [Google Scholar]
- 7.For Brønsted acidity of water-borane adduct see:
- 7a. Bergquist C., Bridgewater B. M., Harlan C. J., Norton J. R., Friesner R. A., Parkin G., J. Am. Chem. Soc. 2000, 122, 10581–10590; [Google Scholar]
- 7b. Di Saverio A., Focante F., Camurati I., Resconi L., Beringhelli T., D′Alfonso G., Donghi D., Maggioni D., Mercandelli P., Sironi A., Inorg. Chem. 2005, 44, 5030–5041. [DOI] [PubMed] [Google Scholar]
- 8.For example the pK a of protonated Et2O is 0.2 in MeCN. For the pK a values cited in the article see:
- 8a. Morris R. H., Chem. Rev. 2016, 116, 8588–8654; [DOI] [PubMed] [Google Scholar]
- 8b. Kolthoff I. M., Chantooni M. K., Bhowmik S., J. Am. Chem. Soc. 1968, 90, 23–28; [Google Scholar]
- 8c. Muckerman J. T., Skone J. H., Ning M., Wasada-Tsutsui Y., Biochim. Biophys. Acta Bioenerg. 2013, 1827, 882–891. [DOI] [PubMed] [Google Scholar]
- 9.Fu and co-workers reported the reductive amination of hexanal/aniline with large excess of Si–H (5 equiv) to afford the amine in moderate (42 %) conversion. Fu M. C., Shang R., Cheng W.-M., Fu Y., Angew. Chem. Int. Ed. 2015, 54, 9042–9046; [Google Scholar]; Angew. Chem. 2015, 127, 9170–9174. [Google Scholar]
- 10. Tiddens M. R., Gebbink R. J. M. K., Otte M., Org. Lett. 2016, 18, 3714–3717. [DOI] [PubMed] [Google Scholar]
- 11.For select examples of less oxophilic Lewis acids in FLP Si–H and H–H activation chemistry see:
- 11a. Scott D. J., Phillips N. A., Sapsford J. S., Deacy A. C., Fuchter M. J., Ashley A. E., Angew. Chem. Int. Ed. 2016, 55, 14738–14742; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14958–14962; [Google Scholar]
- 11b. Clark E. R., Ingleson M. J., Angew. Chem. Int. Ed. 2014, 53, 11306–11309; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11488–11491. For a review on the use of Lewis acids that are not based on boron see: [Google Scholar]
- 11c. Weicker S. A., Stephan D. W., Bull. Chem. Soc. Jpn. 2015, 88, 1003–1016. [Google Scholar]
- 12.
- 12a. Erös G., Mehdi H., Pápai I., Rokob T. A., Király P., Tárkanyi G., Soós T., Angew. Chem. Int. Ed. 2010, 49, 6559–6563; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6709–6713; [Google Scholar]
- 12b. Courtemanche M.-A., Pulis A. P., Rochette E., Legaré M.-A., Stephan D. W., Fontaine F.-G., Chem. Commun. 2015, 51, 9797–9800; [DOI] [PubMed] [Google Scholar]
- 12c. Légaré M.-A., Rochette E., Lavergne J. L., Bouchard N., Fontaine F.-G., Chem. Commun. 2016, 52, 5387–5390. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Mukherjee D., Shirase S., Mashima K., Okuda J., Angew. Chem. Int. Ed. 2016, 55, 13326–13329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13520–13523; [Google Scholar]
- 13b. Mukherjee D., Sauer D. F., Zanardi A., Okuda J., Chem. Eur. J. 2016, 22, 7730–7733. [DOI] [PubMed] [Google Scholar]
- 14. Li H., Aquino A. J. A., Cordes D. B., Hung-Low F., Hase W. L., Krempner C., J. Am. Chem. Soc. 2013, 135, 16066–16069. [DOI] [PubMed] [Google Scholar]
- 15. Mummadi S., Unruh D. K., Zhao J., Li S., Krempner C., J. Am. Chem. Soc. 2016, 138, 3286–3289. [DOI] [PubMed] [Google Scholar]
- 16. Blake A. J., Greig J. A., Schröder M., J. Chem. Soc. Dalton Trans. 1988, 2645–2647. [Google Scholar]
- 17. Fowler D. L., Kraus C. A., J. Am. Chem. Soc. 1940, 62, 1143–1144. [Google Scholar]
- 18. Kelsen V., Vallee C., Jeanneau E., Bibal C., Santini C. C., Chauvin Y., Olivier-Bourbigou H., Organometallics 2011, 30, 4284–4291. [Google Scholar]
- 19. Menye-Biyogo R., Delpech F., Castel A., Pimienta V., Gornitzka H., Rivière P., Organometallics 2007, 26, 5091–5101. [Google Scholar]
- 20.As for B(C6F5)3, (Ref. [5]) the presence of amine retards water dehydrosilylation, so its actual rate during the reductive amination is slower.
- 21. Jacobsen H., Berke H., Döring S., Kehr G., Erker G., Fröhlich R., Meyer O., Organometallics 1999, 18, 1724–1735. [Google Scholar]
- 22.For the mechanism of silane activation with B(C6F5)3 see:
- 22a. Parks D. J., Blackwell J. M., Piers W. E., J. Org. Chem. 2000, 65, 3090–3098; [DOI] [PubMed] [Google Scholar]
- 22b. Rendler S., Oestreich M., Angew. Chem. Int. Ed. 2008, 47, 5997–6000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6086–6089; [Google Scholar]
- 22c. Hermeke J., Mewald M., Oestreich M., J. Am. Chem. Soc. 2013, 135, 17537–17546; [DOI] [PubMed] [Google Scholar]
- 22d. Houghton A. F., Hurmalainen J., Mansikkamaki A., Piers W. E., Tuononen H. M., Nat. Chem. 2014, 6, 983–988; [DOI] [PubMed] [Google Scholar]
- 22e. Piers W. E., Marwitz A. J. V., Mercier L. G., Inorg. Chem. 2011, 50, 12252–12262. [DOI] [PubMed] [Google Scholar]
- 23.S. K. Pati, S. Das, Chem. Eur. J 2016, DOI: 10.1002/chem.201602774.
- 24. Mukherjee D., Shirase S., Spaniol T. P., Mashimab K., Okuda J., Chem. Commun. 2016, 52, 13155–13158. [DOI] [PubMed] [Google Scholar]
- 25. Kanzian T., Nigst T. A., Maier A., Pichl S., Mayr H., Eur. J. Org. Chem. 2009, 6379–6385. [Google Scholar]
- 26. Abdel-Magid A. F., Mehrman S. J., Org. Process Res. Dev. 2006, 10, 971–1031. [Google Scholar]
- 27.The pK a of benzhydrylamine in MeCN has been estimated starting from its pK a value in water (7.5), as reported in: Acids and bases: solvent effects on acid–base strength (Ed.: B. G. Cox), Oxford University Press, Oxford, 2013. [Google Scholar]
- 28.For the decomposition of [BPh4]− by Brønsted acids see:
- 28a. Chisholm M. H., Gallucci J. C., Yin H., Dalton Trans. 2007, 4811–4821; [DOI] [PubMed] [Google Scholar]
- 28b. Stenzel O., Raubenheimer H. G., Esterhuysen C., Dalton Trans. 2002, 1132–1138. [Google Scholar]
- 29. Ashley A. E., Herrington T. J., Wildgoose G. G., Zaher H., Thompson A. L., Rees N. H., Krämer T., O'Hare D., J. Am. Chem. Soc. 2011, 133, 14727–14740. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Doyle M. P., West C. T. J., J. Org. Chem. 1975, 40, 3835–3838; [Google Scholar]
- 30b. Chen Q.-A., Klare H. F. T., Oestreich M., J. Am. Chem. Soc. 2016, 138, 7868–7871. [DOI] [PubMed] [Google Scholar]
- 31. Fasano V., Radcliffe J. E., Curless L., Ingleson M. J., Chem. Eur. J. 2017, 23, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Numerous attempts (under a range of conditions) to synthesise and isolate B(3,5-Cl2-C6H3)3 starting from the aryl Grignard have all been unsuccessful, producing the desired compound in low conversion in mixtures that proved intractable in our hands.
- 33.Piribedil was recently produced by the B(C6F5)3-catalysed N-alkylation of amines using a carboxylic acid, but this process does not proceed via the imine but instead via the amide and required >4 equivalents of the carboxylic acid. See
- 33a.Ref. [9]; and
- 33b. Zhang Q., Fu M.-C., Yu H.-Z., Fu Y., J. Org. Chem. 2016, 81, 6235–6243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary