

Abstract
Mitochondrial dysfunction is responsible for the toxicity of a number of drugs. Current isolated mitochondria or cellular monoculture mitochondrial respiration measurement systems lack physiological relevance. Using a tissue engineering rather than cell- or mitochondria-based approach enables a more physiologically relevant detection of drug-induced mitochondrial impairment. To probe oxygen consumption and mitochondrial health, we assayed the bioenergetic profile of engineered three-dimensional human skeletal muscle myobundles derived from primary myoblasts. Through experimental and computational techniques, we did not find external or internal oxygen transport limiting the engineered myobundles in the commercial O2k system to measure oxygen consumption. In response to the complex I inhibitor rotenone, myobundle basal respiration decreased dose dependently with an IC50 of 9.24 ± 0.03 nM. At a 20 nM concentration of rotenone, myobundle maximal respiration decreased by 44.4% ± 9.8%. Respiratory depression by rotenone suggests that cultured myobundles rely heavily on the complex I pathway for ATP synthesis during times of both basal and increased energy demand. To address whether these decrements in mitochondrial function corresponded to alterations in physiological muscle function, we determined fatigue susceptibility that revealed a 46.0% ± 7.4% depression at 20 nM rotenone. The bioenergetic health index, which is a measure of normal oxidative mitochondrial function, was inversely correlated with the extent of fatigue. The human myobundles reproduce normal muscle metabolism under both basal and maximal energy demand conditions enabling the detection of drug-induced mitochondrial toxicity.
Keywords: : tissue engineering, human skeletal muscle, oxygen consumption rate, bioenergetics, mitochondrial toxicity, drug testing
Introduction
Despite encouraging preclinical studies, 90% of potential therapeutics fail in clinical trials.1 Furthermore, serious adverse effects are discovered in half of FDA-approved pharmaceuticals. Many of these effects are detected only after approval.2 These negative outcomes are due to the inherent limitations of current preclinical models to indicate reliably drug efficacy and safety.3,4 Mitochondrial toxicity is a key “off-target” effect of many drugs.5 Numerous commonly prescribed therapeutics, such as statins, compromise mitochondrial function by uncoupling or inhibiting oxidative phosphorylation leading to organ dysfunction.6 Mitochondrial toxicity led to the market withdrawal of troglitazone and cerivastatin and severe restrictions placed on tolcapone.7,8 Due to the small subset of patients who experience adverse effects, detection of mitochondrial dysfunction is difficult in clinical trials.7 These rare occurrences are hypothesized to be a function of genetic and environmental factors.9 These candidate drugs may have been ceased earlier in development if toxicity had been discovered in preclinical models. Unfortunately, current in vitro preclinical methods lack physiologically relevant, functional cellular oxidative stress indicators.4,9 Previous efforts to create a preclinical, mitochondrial toxicity platform have focused on measuring alterations in the oxygen consumption rate of cells or mitochondria either in solution10,11 or in monoculture.12 The challenge to determine human mitochondrial toxicity earlier has inspired a drive for the pharmaceutical industry to develop better methods and models for detection of drug-induced mitochondrial dysfunction.13
Tissue-engineered human skeletal muscle is an ideal candidate to assess mitochondrial function and drug-induced impairment in vitro. Skeletal muscle has a high ATP flux due to high energy demands, constitutes 40% of total body mass,14 and receives a significant fraction of blood flow.15,16 Due to this, muscle comes into frequent contact with systemically administered drugs and commonly experiences mitochondrial off-target effects.17
In this study, we used engineered skeletal myobundles that replicate the normal function of the human skeletal muscle.18 To supplement our current assessment of engineered muscle function by contractile force,18 we assessed oxygen consumption to detect drug-induced mitochondrial toxicity and impairment. Rates of cellular respiration are one of the most informative techniques for assessing mitochondrial function.19,20 Pharmaceutical-induced respiratory alterations in either magnitude or kinetics are interpreted as quantitative changes in oxidative phosphorylation. Oxygen consumption rate measurements expand the myobundle functional assessments to include an indirect measure of oxidative ATP production and thus a functional cellular mitochondrial stress indicator.
To demonstrate that our engineered skeletal muscle model could identify drug-induced mitochondrial toxicity, we tested our model for toxicity induced by the mitochondrial complex I inhibitor, rotenone. We first characterized the myobundle bioenergetic profile by probing the system with mitochondrial inhibitors and an uncoupler. We then measured the changes in oxygen flux, force production, and muscle fatigue due to the complex I inhibitor rotenone. Our data show that rotenone-treated myobundles displayed decreased basal and maximal oxygen consumption rates, decreased force production, and decreased fatigue resistance.
Materials and Methods
Human myoblast culture
Human skeletal muscle cells were isolated according to previously described, Duke IRB-approved methods.18,21 Cells were cultured in human growth media (hGM) containing low-glucose (LG; 1 g/L glucose) DMEM (GIBCO/Invitrogen) supplemented with 8% fetal bovine serum (Hyclone), 1× antibiotic–antimycotic, and SkGM SingleQuots (Lonza) without insulin or gentamicin. Myoblasts were either differentiated on well plates into myotubes or used for the generation of three-dimensional myobundles.
Two-dimensional culture of human myotubes
After the myoblasts reached approximately 80% confluence, the media were switched to differentiation media (hDM) consisting of LG-DMEM supplemented with 2% adult horse serum (HS), 100 μM fatty acids (1:1 oleate:palmitate) conjugated to 0.14% BSA, 100 μM carnitine, and 1× antibiotic–antimycotic. Myotubes were differentiated for 14 days. On day 14, myotubes were enzymatically removed from the well plate using 0.025% trypsin-EDTA, centrifuged, and resuspended in hDM.
Human tissue-engineered skeletal muscle bundle (myobundle) culture
The myobundle assembly procedure was used as previously described.18,22 Briefly, myoblasts encapsulated in a fibrin/Matrigel matrix were seeded on a Cerex frame. The matrix was fabricated by mixing a cell solution (per bundle, 7.5 × 105 cells in 17.2 μL media + 2 μL of 50 U/mL thrombin in 0.1% BSA in PBS) and an ice-cold gelling solution (11 μL media + 10 μL Matrigel + 10 μL of 20 mg/mL fibrinogen in DMEM); then pipetting the mixture in a custom-made Teflon mold placed between the beams of the Cerex frame. The myobundles polymerized in the mold and to the frame for 30 min and then were cultured in hGM containing 6-aminocaproic acid (ACA) on a rocker (0.33 Hz) at 37°C. On day 4, the media were switched to hDM supplemented with 2 mg/mL of ACA.
Human skeletal muscle myobundle and isolated myotube respirometry
After 2 weeks of differentiation, mitochondrial respiratory function was determined in myobundles and myotubes in solution prepared by dissociating myotubes with 0.025% trypsin-EDTA. Oxygen consumption was determined polarographically with a Clark electrode within an Oxygraph-O2k high-resolution respirometer (Oroboros Instruments). The respirometer consists of two closed chambers each equipped with magnetic stirring and an electrode. The oxygen consumption rates were calculated as the negative time derivative of oxygen concentration using DatLab software. For the myobundles, a Viton O-ring was added to the chamber to inhibit interaction with the stir bar. The engineered tissue or enzyme-dissociated myotubes in solution (1 × 106 cells/chamber) were then transferred into the respiration chamber in 2 mL of hDM. Oxygen flux was monitored in real time following standardized instrumental and chemical background calibrations using DatLab software.23,24
O2k measurement validation
Respirometry data were collected at atmospheric conditions (200.5 ± 0.56 nmol/mL) and hyperbaric conditions (400 nmol/mL). Measurements were also made at two different stir speeds: 750 rpm and 900 rpm (maximum). After initial validation experiments, all respirometry data were collected at atmospheric oxygen conditions at 37°C with a stir speed of 750 rpm.
Mitochondrial function was evaluated utilizing established protocols.23,25 Mitochondrial inhibitor and uncoupler, oligomycin and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), were titrated to achieve optimized leak and maximal respiration (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tec). Final concentrations of inhibitors and uncoupler added to the chamber were 11 μM oligomycin, 8 μM FCCP, and 10 μM antimycin A. Mitochondrial respiration was corrected for oxygen flux due to instrumental background and for residual oxygen consumption after complex III inhibition and normalized by the number of nuclei. ATP production is defined as the portion of oligomycin-sensitive basal respiration. These rates were measured for the entire bundle and reported as r, mole s−1. For modeling, the reaction rate r was divided by the bundle volume V to obtain R, mole m3 s−1.
Cellular respiratory flux control ratios were determined by normalizing oxygen consumption rates (basal [R], leak [L], ATP production [net R]) to the maximal respiration (E). Flux control ratios (routine [R/E], leak [L/E], and net routine [net R/E]) were calculated as described by Gnaiger.26
Rotenone respiration experiments
Rotenone respiration dose–response curves were collected by first measuring oxygen consumption under uninhibited basal conditions. Increasing concentrations of rotenone were added (0–1000 nM) until no further decreases in respiration were observed. Uninhibited basal respiration was set at 100%, to which all other data were related. A nonlinear regression model was fit with the equation, Y = Φ + (β − Φ)/{1 + 10^[(Log(X) − LogIC50)]}, to determine the IC50 of rotenone. Where Φ is the bottom plateau, β is the top plateau of the curve, and IC50 is the rotenone concentration that inhibits half of basal respiration.
One concentration below the IC50 (8 nM rotenone), one above the IC50 (20 nM rotenone), and the vehicle control (0.1% ethanol) were chosen to characterize the influence of rotenone on all respiration states. Either ethanol or rotenone was added before adding inhibitors and uncoupler.
The bioenergetic health index (BHI) was calculated using the following equation: BHI = {[(reserve capacity) × (ATP-linked OCR)]/[(nonmitochondrial OCR) × (proton leak)]} as previously described.27 This quantity represents the ratio of positive benefits of oxidative metabolism to factors that shunt oxygen from these beneficial aspects.
Computational model
A finite element method (FEM)-based approach was implemented in COMSOL Multiphysics 5.0. Oxygen transport is described by the general form of the conservation of mass for dilute solutions in an incompressible fluid28:
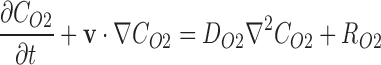 |
Where CO2 denotes the oxygen concentration [mol·m3], DO2 is the diffusion coefficient [m2·s−1] of oxygen, RO2 is the reaction rate [mol·m3·s−1], and v is the velocity field [m·s−1]. Based on O2k stir speed data presented in the Results section, we assumed that the solution was well mixed and the myobundle external surface was initially set at atmospheric oxygen concentrations. The velocity field within the myobundle was assumed to be zero and oxygen consumption was assumed to follow the Michaelis–Menten-type kinetics29–31: RO2 = (Rbasal × CO2)/(Km + CO2). Where Rbasal is the maximum reaction rate under basal conditions obtained from the measurements in the O2k system, and Km is the Michaelis constant (1.33 × 10−3 mol/m3).28 The Michaelis constant, diffusion coefficient, and porosity were obtained from the literature (Table 1). The diffusion coefficient was assumed to be that of oxygen in tissue at 37°C,32 and the porosity was assumed to be that of a fibrin gel.33,34 The area was determined from a representative myobundle by taking an aerial image and sketching the outline using SolidWorks (Dassault Systems). The myobundle thickness was measured using ImageJ (FIJI) from three different donors (0.36–0.73 mm). The volume was calculated as the product of the area times the thickness. The effectiveness factor is defined as the ratio of average reaction rate with diffusion to the reaction rate in the absence of mass transfer limitations.35 This was calculated using R as (average reaction rate in myobundle)/(reaction rate in myobundle if the O2 concentration within the bundle equaled the surface concentration).
Table 1.
Computational Model Parameters
| Parameter | Value or equation | Description | Reference |
|---|---|---|---|
| rBasal | −2.67 × 10−11 [mol O2/s] | Basal respiration maximum reaction rate | Measured |
| rFCCP | −5.40 × 10−11 [mol O2/s] | FCCP-induced respiration maximum reaction rate | Measured |
| CSA | 1.102 × 10−5 [m2] | Cross-sectional area of representative myobundle | Measured |
| t | 2.5 × 10−4–7.5 × 10−4 [m] | Thickness | Measured |
| Volume | CSA*t | Volume of myobundle | |
| RBasal | rBasal/Volume | Volumetric basal respiration maximum reaction rate | Measured |
| RFCCP | rFCCP/Volume | Volumetric FCCP-induced maximum reaction rate | Measured |
| Km | 1.33 × 10−3 [mol/m3] | Michaelis constant, substrate concentration at which the reaction rate is half of Rmax | 28 |
| DO_muscle | 2.0 × 10−9 [m2/s] | Diffusion coefficient | 32 |
| T | 293.15 [K] | Temperature | Measured |
| Ep | 0.99 | Porosity | 33 |
FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity in the media was determined after 30-min incubations with the vehicle control, 8 nM rotenone, or 20 nM rotenone in hDM as an index of cell viability. The LDH concentration was quantified using the Pierce LDH cytotoxicity assay kit (Thermo Scientific) by following the manufacturer's recommended protocol. The results are presented as the percent of the positive control (lysed myobundle) using the following equation: LDH release = (sample LDH − media control)/(positive control − media control) × 100%.
Contractile function assessment
After two weeks of differentiation, myobundles were loaded into a custom-made force measurement apparatus as previously described.18 Samples were stimulated (40 V/cm) to recreate twitch (1 Hz for 10 ms) and tetanic (20 Hz for 1 s) contraction. Contractile force traces were analyzed for peak twitch and tetanus force using a custom MATLAB program.18 To evaluate muscle fatigue, either vehicle or rotenone (8, 20 nM) was added 30 min before force testing. The force decline was recorded over the first 30 s of constant 20 Hz simulation. From this, the percent of peak force was calculated: (Peak force [mN]/Force at 30 s [mN]) × 100%.
DNA isolation and quantification
To efficiently determine the number of nuclei per myobundle, human myoblasts were used to create a standard curve based on absorbance readings. Standard curves were created in triplicate from ten samples with increasing, known numbers of myoblasts from each of the seven donors. Samples were lysed in a 9:1 buffer, including the ATL/proteinase K solution (Qiagen). Each sample or standard was combined with an equal volume of Hoechst dye in a 96-well plate. After a 5-min incubation period at room temperature, sample absorbances were measured: 350 nm (excitation) and 486 nm (emission). Nuclei quantification was determined by comparing absorbances of the donor-specific linear standard curve with readings of the bundle samples with subtraction of the matrix background readings.
Viability imaging and quantification
Cell viability was quantified by incubating each bundle with ethidium homodimer (dead nuclei; 561 nm), Calcein AM (live cytoplasm; 488 nm), and Hoechst dye (all double-stranded DNA; 405 nm). Six representative images per bundle were taken using a confocal microscope. A spot counting algorithm was developed to report the total number of nuclei and the number of dead nuclei per image. Cell viability was determined using the following equation: Cell viability = (1–[dead nuclei/total nuclei]) × 100%.
Statistical analyses
The data are presented as mean ± SEM. N represents the number of donors tested per experiment with three to six biological replicates per condition. Statistical analyses were carried out using one-way ANOVA, two-way ANOVA, or unpaired t-test. A Bonferroni post hoc test was used for intergroup comparisons (GraphPad Prism 5). The BHI and fatigue relationship was analyzed via linear regression. Statistical significance is shown as *p < 0.05, **p < 0.01, and #p < 0.001.
Results
Characterization of myobundle cellularity
The myobundle cellularity was characterized by measuring the number of nuclei per bundle. This measure was used for normalize the respirometry data and account for differences in myobundle cell density between bundles. This normalization method resulted in a reduction of variance in the respiration measurements. Nuclei quantification also served as a measure to characterize cell growth over time in the myobundles. At the onset of differentiation (day 4), some cells likely were already committed to proliferate, explaining the continued increase in nuclei after day 4. The proliferation slowed and eventually plateaued as fibers continue to mature (Fig. 1A). The percent viability as assessed by live/dead staining did not significantly decrease over time in culture when compared to day 1 (Fig. 1B).
FIG. 1.

Characterization of engineered myobundle cellularity. (A) Change in the number of nuclei over days of growth (day 1–4) and differentiation (day 5–18) in 3D human myobundles as assessed by Hoechst staining. Results represent mean ± SEM analyzed in duplicate of four donors with four biological replicates. (B) Myobundles were assessed for viability over days of growth and differentiation using a live/dead cytotoxicity assay. Results represent mean ± SEM of four donors with four biological replicates and six representative frames of view per sample.
High-resolution respirometry validation experiments for 3D constructs
We performed bioenergetics characterization of the myobundles in the O2k (Fig. 2A). To determine whether oxygen at air saturation imposes a limit on maximal respiration, we tested bundles under air saturation and hyperoxic conditions. Maximal respiration values did not significantly differ when comparing air saturation to hyperoxia (Fig. 2B). These data suggest that oxygen concentrations above air saturation are not required to obtain maximal respiration under these experimental conditions.
FIG. 2.

OROBOROS O2k validation. (A) Modifications to OROBOROS O2k system for engineered bundle respirometry measurements. A Viton O-ring was added to the O2k closed chambers consisting of a glass reservoir, stopper with drug port, electrode, and magnetic stir bar. The O-ring provided a myobundle resting platform during magnetic stirring. (B) FCCP-induced maximal respiration at both atmospheric and hyperbaric oxygen tensions is not statistically different. Results represent mean ± SEM of five independent experiments and two separate donors. (C) Basal and maximal oxygen consumption rates remain unchanged when stir rate is at maximum speed (900 rpm). Results represent mean ± SEM of five independent experiments. FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone.
In addition, we examined whether oxygen uptake was limited by external mass transport. The stir bar frequency of rotation was set to both the optimal value, 750 rpm, and the maximum value, 900 rpm; stirring frequency above 750 rpm did not have an effect on basal or maximal oxygen uptake rate. Thus, at an atmospheric oxygen concentration and stir rate of 750 rpm, external oxygen transport to myobundles was not limiting (Fig. 2C) providing support for the choice of these conditions for all subsequent assays.
Computational model predictions of oxygen concentration profile and average reaction rate
To assess whether myobundle oxygen consumption is diffusion limited, a theoretical model was built using COMSOL. To estimate the average reaction rate per volume, the volume was calculated by multiplying the cross-sectional area (Fig. 3A, B) by the thickness (Fig. 3C). The oxygen concentration profiles then were modeled for constructs with thicknesses ranging from 0.25 mm to 0.75 mm (Fig. 3D), which encompassed the range of measured myobundle thickness. The model was used to predict the oxygen concentration throughout the center of the myobundle; the dissolved oxygen concentration is a function of spatial distance from the center of the engineered tissue (Fig. 3E).
FIG. 3.

Computational model of engineered muscle oxygen consumption. (A) Image of engineered myobundle after 2 weeks of differentiation in culture. (B) SolidWorks sketch of a representative myobundle area. (C) SolidWorks generated model of the myobundle volume with t representing the myobundle thickness. (D) Concentration profiles in myobundles with differing reaction values and thicknesses. (E) Oxygen concentration profile within myobundles of varying thicknesses during basal and maximal respiration. (F) The high effectiveness factors across all changes in myobundle thickness and respiration rates suggest that there are no oxygen diffusion limitations for the conditions used in this study.
To assess if lower core concentrations resulted in significant decreases in the overall myobundle oxygen consumption rate, internal myobundle mass transport was characterized in terms of the effectiveness factor, which is the ratio of the actual rate of reaction to the rate of reaction in the absence of internal mass transport limitations.28 When comparing the variation in oxygen concentration of the largest bundle to the smallest bundle during both basal and maximal respiration, the change in effectiveness factor was only 0.2% and 0.8%, respectively (Fig. 3F). The high effectiveness factor in all conditions suggests that the myobundle thicknesses used in this study did not lead to internal mass transport limitations.
Characterization of human myobundle bioenergetic profile
A bioenergetic profile was constructed by probing changes in oxygen consumption rate under established respiratory states.19,23 Specifically, after measuring basal oxygen consumption in complete media, the ATP synthase inhibitor oligomycin was added to inhibit oxidative phosphorylation. Under these conditions, the remaining oxygen consumption would be due primarily to proton leak across the inner mitochondrial membrane. A subsequent addition of FCCP uncoupled electron transport from oxidative phosphorylation and provided an index of maximal respiratory chain activity. Finally, inhibition of complex III by antimycin A stopped electron flow through the electron transport chain (ETC) allowing assessment of nonmitochondrial oxygen consumption (Fig. 4A).
FIG. 4.

Characterization of human-engineered bundles' bioenergetic profiles. (A) Schematic of inhibitors utilized to probe bioenergetic respiration states. (B) Portion of basal mitochondrial respiration due to ATP production and proton leak as measured by inhibition of ATP synthase (ATP turnover) and complex III (proton leak). (C) Bioenergetic profile of engineered bundles assessed by addition of mitochondrial inhibitors. (D) Cell respiratory ratios of engineered myobundles reported as ratios of respiration states. For all graphs, data are expressed as mean ± SEM of seven donors with two biological replicates.
When comparing normalized myotube respiration in monolayer culture relative to culture in the 3D myobundle architecture, basal and maximal oxygen consumption rates were significantly higher in cells cultured in 2D. Interestingly, although basal oxygen consumption was higher in 2D, the ratios of ATP turnover relative to basal oxygen consumption were not significantly different (Table 2).
Table 2.
Mitochondrial Respiration in Intact Skeletal Muscle Cells (Control, Nondiseased)
| Basal | ATP | Leak | Maximal | Nonmitochondrial | Cell type | Ref. |
|---|---|---|---|---|---|---|
| OCR (pmol O2/s/106 nuclei) | ||||||
| 49.4 ± 12.6 | 24.9 ± 11.6 | 24.5 ± 1.0 | 138.7 ± 34.7 | 1.9 ± 1.4 | Primary human skeletal muscle myotubes in solution differentiated for 14 days | This study |
| 19.5 ± 1.4 | 9.8 ± 0.8 | 9.7 ± 1.0 | 45.1 ± 1.8 | 3.4 ± 1.0 | 3D primary human skeletal muscle myobundles | This study |
| OCR (pmol O2/s/106 cells) | ||||||
| 75 | 53 | 22 | 500 | n/d | Primary human myotubes in solution differentiated for 8–10 days* | 37γ |
| 7.5 | 4.2 | 3.3 | 20.83 | n/d | 2D human rhabdomyosarcoma cells (HG DMEM +10% FBS)* | 44γ |
| 5.5 | 3.5 | 2 | 20 | n/d | L6 myotubes in solution (DMEM serum free) | 45Δ,γ |
| 42.6 ± 0.8 | n/d | n/d | 157 ± 8.2 | n/d | L6 myoblasts in solution (DMEM serum free) | 46 |
| 40 | 20 | 20 | 75 | 8 | Primary mouse myoblasts differentiated for 6 h and transfected with an empty vectorλ | 47γ |
γ, Value estimated from figure; Δ, value corrected for nonmitochondrial respiration; *, values calculated assuming cells double over culture period; λ, values also normalized to citrate synthase activity.
n/d, no data.
The trends in myobundle respiration rates are consistent with current literature (Table 2). Myobundle leak respiration was 50% ± 5% of basal respiration (Fig. 4B), similar to the 52% of basal respiration in rat skeletal muscle at rest.36 The chemically induced maximal respiration was 231% ± 9% of average basal respiration, comparable to the 222% increase of human skeletal muscle myotubes in solution.37 All other respiration states (Fig. 4C) were also in agreement with current literature.
The values illustrated in Figure 4C were used to calculate the respiratory control ratios depicted in Figure 4D. The routine flux control ratio (R/E) was 0.37. This indicates that about 40% of maximal respiration was used for basal metabolism; this relationship leaves a high reserve capacity to increase oxidative ATP production during periods of high energy demand, for example, exercise. The leak flux control ratio (L/E) at 0.14 indicates a tight coupling of oxidative phosphorylation. The net routine respiratory flux ratio (netR/E) was measured to be 0.23. Therefore, 23% of the oxygen consumed at ETC maximal capacity was allocated for ATP production.38 These ratios were in agreement to those reported of human myotubes in solution: 0.45, 0.13, and 0.32 for R/E, L/E, and netR/E, respectively.37
Cellular respiration of rotenone-treated myobundles
Rotenone acutely decreases cellular oxygen consumption by inhibition of complex I of the ETC.39 Rotenone decreased myobundle basal oxygen uptake rate in a dose-dependent manner (Fig. 5A). To find rotenone's half-maximal inhibitory concentration (IC50), the experimental dose–response data were fit by a nonlinear regression model (R2 = 0.98): Oxygen consumption rate as a percent of basal uptake = 33.21 + 68.99/{1 + 10^[(Log[Rotenone(nM)]) − 0.9655]}. The IC50 of rotenone derived from the fit was 9.24 ± 0.03 nM.
FIG. 5.

Complex I inhibition by rotenone dose dependently inhibits oxygen consumption rates of engineered muscle. (A) Basal oxygen consumption rates of human myobundles in the presence of increasing concentrations (0–1000 nM) of rotenone. Results represent mean ± SEM of four donors normalized to the vehicle control. (B) Relative fold changes of basal, leak, and maximal respiration states due to rotenone normalized to the no treatment control. (C) Bioenergetic health index calculated from the non-normalized respiration states in (B) is demonstrated. Results represent mean ± SEM of four to five donors. *p < 0.05, **p < 0.01, #p < 0.001.
To measure the effect of rotenone on myobundle respiration states, basal, leak, and maximal respiration were measured in the presence of the vehicle control, 8 nM or 20 nM rotenone. Rotenone at 8 nM and 20 nM significantly decreased basal respiration, ATP turnover, and maximal respiration rates (Fig. 5B). This respiratory depression suggests that cultured myobundles rely heavily on complex I-linked substrates for ATP synthesis during times of both basal and increased energy demand. Our results agree with other studies where rotenone decreased skeletal muscle ATP production to 35% of the control.12 As an additional test of the impact of rotenone, the BHI, a measure of mitochondrial function, was assessed. As shown in Figure 5C, the BHI is significantly different (p = 0.035) between the vehicle myobundles and those exposed to 20 nM rotenone. The decreased BHI is indicative of bioenergetic dysfunction.
Rotenone impairment of force production in engineered myobundles
Twitch and tetanus forces of myobundles treated with rotenone displayed a downward trend with increasing rotenone concentration for three different donors (Fig. 6A, B). This drop in force production was not due to decreased cell viability (Supplementary Fig. S2). To probe whether rotenone-induced mitochondrial dysfunction would impair muscle performance at sustained maximal contraction, the myobundles were fatigued by continuous electrical stimulation at 20 Hz for 30 s (Fig. 6C). Rotenone decreased the force production after 30 s in a dose-dependent manner (Fig. 6D). In addition, the vehicle, 8 nM and 20 nM rotenone values for BHI and fatigue were fitted with a linear regression. The BHI was inversely correlated with the extent of fatigue (Fig. 6E) (p < 0.01).
FIG. 6.

Rotenone decreases force production and increases engineered muscle fatigue. Myobundles were treated with either vehicle or rotenone (8, 20 nM) for 30 min after 2 weeks of differentiation. (A, B) Twitch and tetanus force production of human myobundles in the presence of 8 and 20 nM rotenone measured after electrical stimulation at 1 Hz and 20 Hz. Results represent mean ± SEM of four separate donors. (C, D) Representative traces and averaged data of relative changes in tetanus force production during a 30-s 20 Hz stimulation of myobundles with and without rotenone. *p < 0.05, **p < 0.01. (E) Correlation between bioenergetic health index (BHI) and percent relative force after 30 s of electrical stimulation at 20 Hz. Corresponding rotenone concentrations are adjacent to each point and the 95% confidence intervals for the linear regression line are shaded in gray. Results represent mean ± SEM of four separate donors.
Discussion
The goal of this work was to develop a platform to measure drug-induced mitochondrial toxicity in human skeletal muscle with distinct advantages over existing models. We characterized the bioenergetic profile of 3D human myobundles and challenged the platform with the well-characterized complex I inhibitor, rotenone. Previous efforts to create a preclinical, mitochondrial toxicity platform have focused mainly on measuring alterations in the oxygen consumption rate of cells or mitochondria either in solution10,11 or in monoculture.12 Some in vitro work successfully recapitulated clinical side effects and determined pharmaceuticals that are toxic to isolated mitochondria.11 To date, none of these methods measured mitochondrial drug toxicity in a three-dimensional platform capable of detecting mitochondrial impairment using two functional assays: respiration rate and muscle fatigue.
Our model greatly enhances the physiological relevance of the measurements by assessing respiration using engineered muscle tissue rather than isolated mitochondria or individual cells. The study of mitochondrial health with the in vivo complexity of cell–cell and cell–matrix interactions could previously only be studied in animal or human subjects; these studies are now possible in the three-dimensional engineered myobundles. An added advantage to the tissue-engineered myobundle model is the presence of a physiologic cellular architecture, including aligned myotubes that are grown and tested under passive tension. Furthermore, the myobundles do not require nonphysiologic processing, such as permeabilization, mitochondrial isolation, or enzymatic separation, which may alter function. These advantageous features may have significant impact on metabolic and bioenergetic behavior. Our results suggest that the contribution of these factors may be important since maximal and basal respiration was significantly different when comparing myotubes in solution and three-dimensional myobundles. In addition, the myobundle cellular environment was relatively undisturbed versus that of permeabilized bundles and isolated mitochondria. Mitochondria were exposed to a relevant mix of substrates and ions. However, the ability to test isolated substrates is limited. Although beyond the scope of these studies, direct testing of mitochondria could be achieved by cell membrane permeabilization of the myobundles.
These respiration measurements and analysis provide insight into the oxygen concentrations to which the skeletal muscle myotubes within three-dimensional engineered skeletal muscle are exposed. Within a scaffold, oxygen transport is often a concern since myotubes rely on passive diffusion. Overcoming oxygen transport limitations is highly important to ensure myotube survival, mimicry of in vivo structure, and myobundle functionality. A better understanding of the relationship between construct thickness, respiration rate, and myobundle oxygen concentration helps to guide construct design and implantation. In addition, this work provides important input parameters for both developing theoretical models of oxygen transport through tissue-engineered constructs and optimization of the bioreactor conditions for skeletal muscle tissue engineering. This study demonstrates the utility of using human, tissue-engineered myobundles for mitochondrial respiration measurements. Overall, these engineered myobundles maintained high viability and matured by 18 days after preparation. Due to the lack of internal vasculature, a major obstacle in tissue engineering is overcoming external and internal oxygen transport limitations. Through experimental and computational techniques, we did not find limitations in external or internal oxygen transport. These data suggest that oxygen concentrations above air saturation are not required to obtain maximal respiration under the experimental conditions we used.
To gain more insight into the metabolic phenotype of the human myobundles, we assessed myobundle bioenergetic profiles. Basal oxygen consumption rates of the myobundles indicate the aerobic energetic demands of the myotubes under baseline conditions. Basal respiration was 2.5 times higher in cells in solution than cells cultured in the myobundles. Others have observed that monolayer cultures have higher oxygen consumption rates than nontransport limited 3D cultures.40 This could be attributed to either the change in configuration from an in vivo 3D architecture to a monolayer, or the result of enzymatically removing cells from a surface. These attributes of cells in monoculture or solution could induce a mechanically stressed state resulting in an increased oxygen consumption rate.40 In addition, differences in respiration could also be due to variations in myogenic maturation between the myobundles and the myotubes grown in monolayer culture.
The bioenergetic profile and cell respiratory ratios of the human myobundles followed similar trends to differentiated myotubes in solution41 and in monolayer culture.37 Our data show that ∼50% of myobundle basal respiration is allocated to ATP production. This is in agreement with previous literature measuring ATP production at 48% of basal respiration in rat skeletal muscle at rest.36 The variation in respiration between donors may be due to differences in genetics and environment factors, or donor-specific differences in myotube development. Although beyond the scope of these studies, myotube maturation could be assayed by accessing myogenic regulatory proteins such as myogenin and myogenic factor 6.
The myobundles' maximal respiration rate is more than twice the basal respiration rate, indicating that our myobundles have the capacity to substantially increase ATP production in the event of cellular stress, a feature consistent with skeletal muscle and other metabolic tissues.15 The maximal respiration induced by FCCP uncoupling might not perfectly correlate to a physiological energy demand; the uncoupling may not induce a replenishment of the substrate supply necessary to match the demand, yielding a lower maximal respiration rate.19 However, both physiological and FCCP uncoupling lead to rapid oxidation of substrates to restore mitochondrial membrane potential; also, the average myobundle basal respiration per volume (2.92 ± 0.23 nmol/cm3/s) and maximal respiration per volume (6.26 ± 0.28 nmol/cm3/s) were within the range of normal human muscle at rest and during exercise.16 These data suggest human myobundle basal and uncoupled maximal respiration model human adult respiration during these conditions.
The myobundles recapitulated the in vitro and in vivo responses to rotenone. Rotenone dose dependently decreased human myobundle basal and maximal respiration. Decreased maximal respiration is due to a reduction in flux of electron entry into the ETC. Since fewer electrons are available to reduce oxygen to H2O, oxygen consumption rate is reduced. We also quantified the BHI to assess the impact of rotenone on the composite mitochondrial profile for each condition (Fig. 5C). The lowest BHI values corresponded to the highest concentrations of rotenone; the BHI was suppressed over 65% in the 20 nM rotenone condition, suggesting significant bioenergetic dysfunction. Taken together, these results validate that our model is mimicking previously reported responses to rotenone.
Importantly, in addition to changes in respiratory kinetics, rotenone subtly affected contractile force production modulated by reduction of ATP production. Rotenone reduces L6 myotube ATP levels by 65%.12 In this study, the human myobundles exhibited a downward trend in both twitch and tetanus force when exposed to rotenone in three out of four donors. On the donor level, there was a direct relationship between sensitivity to rotenone as measured by a decrease in twitch force and decrease in tetanus force. This functional measure in combination with the myobundle bioenergetic profiles would enable a personalized measurement of pharmaceutical effects on an individual's mitochondria; this is especially important where the majority of individuals benefit from a medication, while others experience severe contraindications.42
Unlike twitch and tetanus force, rotenone caused a dose-dependent increase in muscle fatigue, defined here as the reduction in capacity to generate force after 30 s of electrical stimulation. The decline in performance might not be immediately apparent when measuring twitch or tetanus force because the myobundles can engage anaerobic metabolism to meet ATP demands. Increasing the duration of the myobundle contraction increases total ATP demand, requiring a higher contribution of ATP from cellular oxidative phosphorylation.43 This is apparent after inhibiting mitochondrial respiration with 20 nM rotenone resulting in a 46.0% ± 7.4% drop in force production. We found a correlation between the decreases in bioenergetic and functional data due to rotenone. The results suggest that the mitochondrial dysfunction, quantified by a reduction in BHI, produced the fatigue. BHI and fatigue can serve as complementary, sensitive indicators of mitochondrial toxicity in the human myobundles.
Conclusions
This study demonstrated the utility of using engineered human myobundles to assess mitochondrial toxicity. The myobundles experience negligible mass transfer inhibition as confirmed by the computational model, and there was little donor-to-donor variation in the bioenergetic profiles. Furthermore, the toxicity of rotenone was detected at low doses using oxygen consumption rate and muscle fatigue. To our knowledge, this is the first human myobundle model enabling the coupling of energetic measurements to a physiologic outcome and may serve as an important conduit between in vitro and in vivo testing in the drug development pipeline.
Supplementary Material
Acknowledgments
The authors thank Tracy Cheung, Megan Kondash, and Samantha Lasater for excellent technical discussions and the laboratory of Deborah Muoio for support, both in equipment and teaching. This work was supported by an NSF Graduate Research Fellowship to BNJD and NIH grants UH2TR000505, 4UH3TR000505, and the NIH Common Fund for the Microphysiological Systems Initiative to GAT.
Disclosure Statement
The authors declare no conflict of interest.
References
- 1.FDA. FDA Issues Advice to Make Earliest Stages of Clinical Drug Development More Efficient. Silver Spring, MD: FDA; 2006 [Google Scholar]
- 2.(GAO), U.S.G.A.O. FDA Drug Review Postapproval Risks 1976–1985. Report to the Chairman, Subcommittee on Human Resources and Intergovernmental Relations, Committee on Government Operations, House of Representatives Washington, DC,1990
- 3.Olson H., Betton G., Robinson D., Thomas K., Monro A., Kolaja G., Lilly P., Sanders J., Sipes G., Bracken W., Dorato M., Van Deun K., Smith P., Berger B., and Heller A. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol 32, 56, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Astashkina A., Mann B., andGrainger D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol Ther 134, 82, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Astashkina A., and Grainger D.W. Critical analysis of 3-D organoid in vitro cell culture models for high-throughput drug candidate toxicity assessments. Adv Drug Deliv Rev 69–70, 1, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Nadanaciva S., Bernal A., Aggeler R., Capaldi R., and Will Y. Target identification of drug induced mitochondrial toxicity using immunocapture based OXPHOS activity assays. Toxicol In Vitro 21, 902, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Dykens J.A., and Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today 12, 777, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Valiyil R., and Christopher-Stine L. Drug-related myopathies of which the clinician should be aware. Curr Rheumatol Rep 12, 213, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dykens J.A., and Will Y. Biomarkers. In: Vaidya V.S., and Bonventre J.V., eds. Medicine, Drug Discovery, and Environmental Health. Hoboken, NJ: John Wiley & Sons, 2010 [Google Scholar]
- 10.Kaufmann P., Torok M., Zahno A., Waldhauser K.M., Brecht K., and Krahenbuhl S. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci 63, 2415, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hynes J., Marroquin L.D., Ogurtsov V.I., Christiansen K.N., Stevens G.J., Papkovsky D.B., and Will Y. Investigation of drug-induced mitochondrial toxicity using fluorescence-based oxygen-sensitive probes. Toxicol Sci 92, 186, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Dott W., Mistry P., Wright J., Cain K., and Herbert K.E. Modulation of mitochondrial bioenergetics in a skeletal muscle cell line model of mitochondrial toxicity. Redox Biol 2, 224, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadanaciva S., and Will Y. New insights in drug-induced mitochondrial toxicity. Curr Pharm Des 17, 2100, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Janssen I., Heymsfield S.B., Wang Z., and Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol 89, 81, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Burton D.A., Stokes K., and Hall G.M. Physiological effects of exercise. Contin Educ Anaesth Critic Care Pain 4, 185, 2004 [Google Scholar]
- 16.Cheng C.S., Davis B.N., Madden L., Bursac N., and Truskey G.A. Physiology and metabolism of tissue-engineered skeletal muscle. Exp Biol Med (Maywood, NJ) 239, 1203, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owczarek J., Jasinska M., and Orszulak-Michalak D. Drug-induced myopathies. An overview of the possible mechanisms. Pharmacol Rep 57, 23, 2005 [PubMed] [Google Scholar]
- 18.Madden L., Juhas M., Kraus W.E., Truskey G.A., and Bursac N. Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. eLife 4, e04885, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand M.D., and Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem J 435, 297, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warburg O. Über sauerstoffatmende Körnchen aus Leberzellen und über Sauerstoffatmung in Berkefeld-Filtraten wässriger Leberextrakte. Pflüger's Arch 154, 599, 1913 [Google Scholar]
- 21.Cheng C.S., El-Abd Y., Bui K., Hyun Y.E., Hughes R.H., Kraus W.E., and Truskey G.A. Conditions that promote primary human skeletal myoblast culture and muscle differentiation in vitro. Am J Physiol Cell Physiol 306, C385, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng C.S., Ran L., Bursac N., Kraus W.E., and Truskey G.A. Cell Density and Joint microRNA-133a and microRNA-696 Inhibition Enhance Differentiation and Contractile Function of Engineered Human Skeletal Muscle Tissues. Tissue Eng Part A 22, 573, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pesta D., and Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol (Clifton, NJ) 810, 25, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 41, 1837, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Hütter E., Unterluggauer H., Garedew A., Jansen-Dürr P., and Gnaiger E. High-resolution respirometry–a modern tool in aging research. Exp Gerontol 41, 103, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Gnaiger E. Mitochondrial Pathways and Respiratory Control. Innsbruck, Austria: OROBOROS MiPNet Publications; 2nd ed. Steiger Druck GmbH: OROBOROS INSTRUMENTS Corp.; 2009 [Google Scholar]
- 27.Chacko B.K., Kramer P.A., Ravi S., Benavides G.A., Mitchell T., Dranka B.P., Ferrick D., Singal A.K., Ballinger S.W., Bailey S.M., Hardy R.W., Zhang J., Zhi D., and Darley-Usmar V.M. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (London, England: 1979) 127, 367, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Truskey G.A., Yuan F., and Katz D.F. Transport Phenomena in Biological Systems. Upper Sadie River, NJ: Pearson Prentice Hall, 2004 [Google Scholar]
- 29.Wilson D.F., Rumsey W.L., Green T.J., and Vanderkooi J.M. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem 263, 2712, 1988 [PubMed] [Google Scholar]
- 30.Avgoustiniatos E.S., and Colton C.K. Effect of external oxygen mass transfer resistances on viability of immunoisolated tissue. Ann N Y Acad Sci 831, 145, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Buchwald P. FEM-based oxygen consumption and cell viability models for avascular pancreatic islets. Theoretical Biol Med Model 6, 5, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fournier R.L. Basic Transport Phenomena in Biomedical Engineering. Boca Raton, FL: CRC Press, 1998 [Google Scholar]
- 33.Hsu Y.-H., Moya M.L., Abiri P., Hughes C.C.W., George S.C., and Lee A.P. Full range physiological mass transport control in 3D tissue cultures. Lab Chip 13, 81, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helm C.L., Zisch A., and Swartz M.A. Engineered blood and lymphatic capillaries in 3-D VEGF-fibrin-collagen matrices with interstitial flow. Biotechnol Bioeng 96, 167, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Weisz P.B. Diffusion and Chemical reaction: an interdisciplinary excursion. Science (New York, NY) 179, 433, 1973 [DOI] [PubMed] [Google Scholar]
- 36.Rolfe D.F., Newman J.M., Buckingham J.A., Clark M.G., and Brand M.D. Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. Am J Physiol 276, C692, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Nisr R.B., and Affourtit C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim Biophys Acta 1837, 270, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gnaiger E. Mitochondrial Pathways and Respiratory Control. Innsbruck: OROBOROS MiPNet Publications; 2007 [Google Scholar]
- 39.Gondal J.A., and Anderson W.M. The molecular morphology of bovine heart mitochondrial NADH—ubiquinone reductase. Native disulfide-linked subunits and rotenone-induced conformational changes. J Biol Chem 260, 12690, 1985 [PubMed] [Google Scholar]
- 40.Patzer J.F., 2nd. Oxygen consumption in a hollow fiber bioartificial liver—revisited. Artif Organs 28, 83, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Henique C., Mansouri A., Fumey G., Lenoir V., Girard J., Bouillaud F., Prip-Buus C., and Cohen I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J Biol Chem 285, 36818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parr R.L., and Martin L.H. Mitochondrial and nuclear genomics and the emergence of personalized medicine. Hum Genomics 6, 3, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker J.S., McCormick M.C., and Robergs R.A. Interaction among Skeletal Muscle Metabolic Energy Systems during Intense Exercise. J Nutr Metab 2010, 13, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaughan R.A., Garcia-Smith R., Bisoffi M., Conn C.A., and Trujillo K.A. Conjugated linoleic acid or omega 3 fatty acids increase mitochondrial biosynthesis and metabolism in skeletal muscle cells. Lipids Health Dis 11, 142, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casanova E., Baselga-Escudero L., Ribas-Latre A., Arola-Arnal A., Blade C., Arola L., and Salvado M.J. Epigallocatechin gallate counteracts oxidative stress in docosahexaenoxic acid-treated myocytes. Biochim Biophys Acta 1837, 783, 2014 [DOI] [PubMed] [Google Scholar]
- 46.Park J., Pak Y.K., and Pak J.J. A microfabricated reservoir-type oxygen sensor for measuring the real-time cellular oxygen consumption rate at various conditions. Sensors Actuators B Chem 147, 263, 2010 [Google Scholar]
- 47.De Palma, C., Falcone S., Pisoni S., Cipolat S., Panzeri C., Pambianco S., Pisconti A., Allevi R., Bassi M.T., Cossu G., Pozzan T., Moncada S., Scorrano L., Brunelli S., and Clementi E. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ 17, 1684, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.