

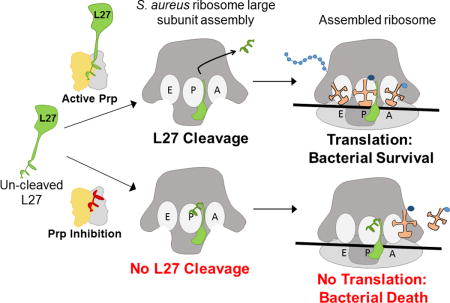
Summary
In Firmicutes and related bacteria, ribosomal large subunit protein L27 is encoded with a conserved N-terminal extension that is removed to expose residues critical for ribosome function. Bacteria encoding L27 with this N-terminal extension also encode a sequence-specific cysteine protease, Prp, which carries out this cleavage. In this work we demonstrate that L27 variants with an un-cleavable N-terminal extension, or lacking the extension (“pre-cleaved”), are unable to complement an L27 deletion in Staphylococcus aureus. This indicates that N-terminal processing of L27 is not only essential but possibly has a regulatory role. Prp represents a new clade of previously uncharacterized cysteine proteases, and the dependence of S. aureus on L27 cleavage by Prp validates the enzyme as a target for potential antibiotic development. To better understand the mechanism of Prp activity, we analyzed Prp enzyme kinetics and substrate preference using a fluorogenic peptide cleavage assay. Molecular modeling and site-directed mutagenesis implicate several residues around the active site in catalysis and substrate binding, and support a structural model in which rearrangement of a flexible loop upon binding of the correct peptide substrate is required for the active site to assume the proper conformation. These findings lay the foundation for the development of antimicrobials that target this novel, essential pathway.
Keywords: cysteine protease, ribosomal protein processing, L27, site-specific cleavage
Graphical abstract
In some bacteria, ribosomal protein L27 is produced with an N-terminal extension that is removed by the recently discovered protease Prp. We demonstrate that this cleavage is essential in Staphylococcus aureus, validating Prp as a potential new target for antibacterial drugs. This work explores structural and enzymatic features of Prp that will help to inform the design of new antibiotics against S. aureus and related pathogens.
Introduction
Recently, the Centers for Disease Control evaluated the current and impending risks posed by antibiotic-resistant bacterial pathogens. Their prioritized list includes Clostridium difficile as one of the most urgent threats and vancomycin-resistant Enterococcus (VRE), methicillin-resistant Staphylococcus aureus (MRSA), and drug-resistant Streptococcus pneumoniae as existing serious threats (CDC, 2014). It has become abundantly clear that continuous antibiotic development is of primary importance to protect patients from emerging resistant bacterial pathogens. Current antibiotic therapies are already of limited utility, yet the novel drugs that are currently being developed are often “me too” drugs that work via the same mechanism as existing antimicrobials. To create unique drugs, new therapeutic targets and new data on the function of existing targets are needed.
The eubacterial ribosome has historically been a major target for numerous antibiotics, including macrolides (e.g. erythromycin), lincosamides, oxazolidinones and tetracylines, which work by blocking aminoacyl-tRNA binding, preventing peptidyl transfer or causing premature peptidyl-tRNA release (Tejedor; Ballesta, 1986; Auerbach et al., 2002; Colca et al., 2003; Yonath, 2005). Protein synthesis and ribosome formation remain extremely important yet underdeveloped targets in the resistant pathogens that are only distantly related to the bacterial model organism Escherichia coli.
We recently discovered a novel site-specific protease in S. aureus that plays a critical role in basic ribosomal biology (Wall et al., 2015). This enzyme is also present in all of the resistant pathogens in the CDC list above, and provides an attractive new target for antibiotic development. We demonstrated that ribosomal protein L27 in Firmicutes such as S. aureus, as well as in several other bacterial phyla, is encoded as a pro-protein with an N-terminal extension that is not present in E. coli. The S. aureus L27 pro-protein undergoes a site-specific cleavage event, sometime before or during ribosomal assembly, to yield a mature protein with an N-terminus equivalent to that found in E. coli L27 (Wall et al., 2015). Failure to remove the N-terminal extension was hypothesized to occlude critical L27 residues that stabilize A and P-site tRNAs at the peptidyl transferase center (PTC), the active site of the ribosome (Maguire et al., 2005; Voorhees et al., 2009; Polikanov et al., 2014). Removal of the N-terminal extension is carried out by Prp, a site-specific cysteine protease that belongs to a family of conserved proteins of previously unknown function identified as DUF464. Structural analysis of several DUF464 family members revealed a protein fold unlike that of any other known peptidase, which has led to the assignment of Prp as the representative member of a new protein family, C108, in the MEROPS peptidase database (Rawlings et al., 2014). A better understanding of the mechanism of substrate binding and catalysis by this new family of proteases will be required for the development of specific inhibitors. In this work we confirm that the activity of the Prp protease is essential in S. aureus. We further characterize the enzyme using kinetics, structural modeling and site-directed mutagenesis. These data provide a foundation for further exploration of fundamental differences in ribosome biogenesis in this group of bacteria and constitute an initial basis for the rational design of new antimicrobials that target this essential cleavage step.
Results
L27 cleavage is essential in S. aureus
Previous work indicating that Prp and L27 are both essential in S. aureus was conducted via antisense RNA expression and transposon mutagenesis (Ji et al., 2001; Chaudhuri et al., 2009). The interpretation of these results is complicated by possible polar effects of a transposon insertion in Prp that might lead to lack of downstream L27 expression, or antisense RNA that might also have polar effects or destabilize the entire transcript, which also includes ribosomal protein L21.
In order to determine whether Prp cleavage of L27 was essential, it was necessary to examine mutants with altered L27 cleavage in S. aureus. We have previously shown that overexpression of catalytically inactive Prp that binds tightly to L27 is toxic (Wall et al., 2015). However, overexpression of un-cleavable L27 had minimal effects in a background that also expressed the wild-type protein, so it remained an open question whether the cleavage event was essential for S. aureus growth. To address this question, a plasmid-based complementation system was constructed that allowed a switch between wild-type L27 and mutants that either lacked the cleavage motif or had a modified “un-cleavable” motif. This experiment was performed as depicted in the schematic in Figure 1A.
Figure 1. Complementation by plasmid-encoded L27.
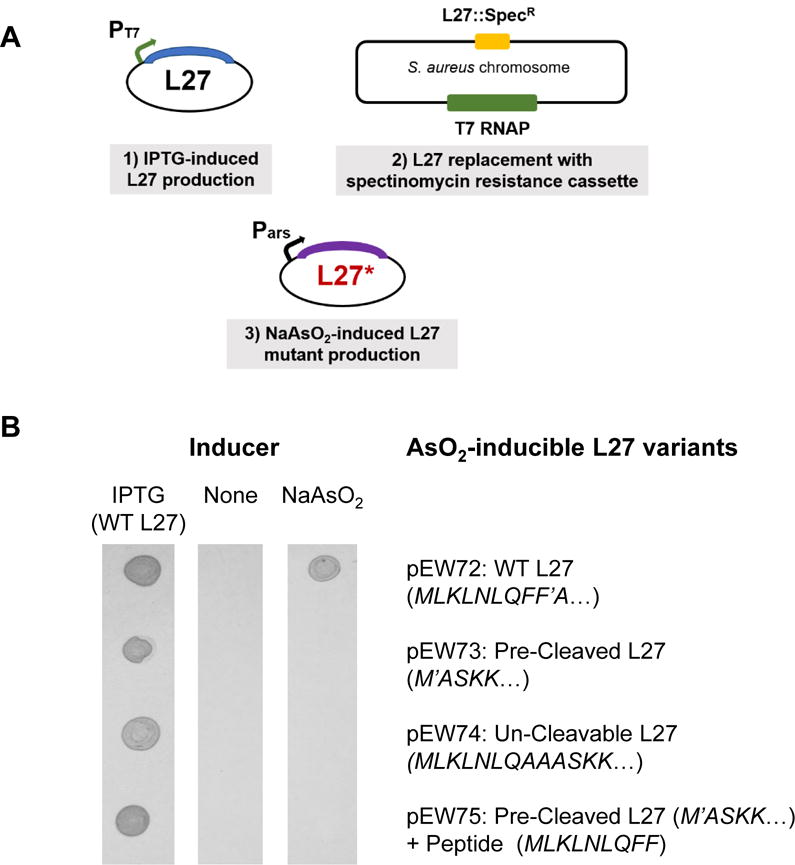
(A) Outline of the genetic elements employed in the complementation scheme. (1) Plasmid that encodes S. aureus L27 under control of the T7 promoter repressed by LacI (pEW27); (2) S. aureus host strain, derived from strain SA187RI, which contains the T7 RNA polymerase on an integrated plasmid and a spectinomycin resistance cassette in place of the gene encoding L27; (3) a compatible plasmid (pEW72–75) encoding L27 or mutants under control of an arsenite-inducible promoter. (B) Growth of cells carrying pEW27 and the different arsenite-inducible L27 mutants. Each L27 variant carried on the arsenite-inducible plasmid is listed to the left of the array. The three columns are photographs taken of the growth of bacterial cells spotted onto solid culture media containing 1 mM IPTG, no inducer, or 5 μM NaAsO2, as indicated.
In the presence of a plasmid that required IPTG to produce L27 (pEW27), the native chromosomal copy of the L27 gene (rpmA) was deleted by replacement with a spectinomycin resistance cassette. The resultant strain was dependent on IPTG for survival, demonstrating conclusively for the first time that L27 is essential in S. aureus. A compatible plasmid was subsequently introduced which encoded various alleles of L27 under control of an arsenite-inducible promoter (Liu et al., 2004). By replacing IPTG with arsenite in the medium, the mutant form of L27 was expressed in place of the wild-type copy, allowing examination of the effect of the loss of regulated L27 cleavage on S. aureus. Plasmid pEW72 expresses wild type S. aureus L27 in the presence of arsenite and it is able to complement the L27 chromosomal deletion. There is no leaky expression from either promoter; in the absence of IPTG or arsenite, the strain does not grow (Figure 1B). Plasmids pEW73-75 express variant forms of arsenite-inducible L27. Plasmid pEW73 expresses “pre-cleaved” L27 (Δ 2-9), which is missing the N-terminal cleavage motif, making it effectively equivalent to the “short” E. coli-like L27. We confirmed via Edman degradation that there is loss of the N-terminal methionine (which would occlude substrate stabilization residues ASKK), resulting a protein identical to that found in mature S. aureus ribosomes (Colca et al., 2003). Plasmid pEW74 expresses L27 F8A–F9A, an un-cleavable Prp recognition motif mutant (Wall et al., 2015). Plasmid pEW75 expresses the same pre-cleaved L27 as pEW73 and, in addition, the post-cleavage “released” peptide MLKLNLQFF, expressed from the same promoter but using a separate downstream RBS. The purpose of pEW75 was to determine whether the released peptide had some independent effect on growth.
Strains carrying pEW27 and compatible plasmids with the arsenite-inducible L27 variants were spotted onto solid media containing 1mM IPTG, no inducer, or 5 μM sodium arsenite. All grew on plates with IPTG, all failed to grow without any inducer, and only the strain containing pEW72 (WT L27) grew in the presence of arsenite (Figure 1B). This demonstrates that not only is L27 cleavage essential, it must also be tightly regulated, since the “pre-cleaved” variant failed to complement. The released peptide may yet have some role in cellular processes, but that role is insufficient to promote S. aureus growth without regulated L27 cleavage. These results confirm that L27 cleavage by Prp is an obligatory step for S. aureus viability, and validates Prp as a potential new target for antibiotics. We have accordingly undertaken further functional characterization of this important protease.
Prp purification and assay for enzymatic activity
To purify Prp for assessment of substrate binding and catalysis, and to facilitate the search for inhibitors, we developed a high throughput-capable continuous fluorescent assay that monitors Prp activity in the presence of a quenched fluorescent peptide containing the cleavage motif: 2-aminobenzoic acid-KLNLQFFASKK-dinitrophenol. When the substrate is cleaved, the N-terminal fluorophore 2-aminobenzoic acid (2-Abz) is released from its C-terminal quenching partner dinitrophenol (Dnp), resulting in fluorescence at the emission wavelength 414 nm upon excitation at 325 nm.
Prp was purified as a fusion protein with an N-terminal His6-SUMO moiety. The His6-SUMO tag was specifically removed using His6-Ulp1 protease, the mixture was dialyzed to remove imidazole, and untagged Prp was purified away from the His6-Ulp1 and the His6-SUMO tag by passage over a second nickel-NTA column (Figure S1). The dimer present in the S. aureus Prp crystal structure (PDB 2P92, revised to 4PEO; Chirgadze et al., 2015) was verified in vitro using a size-exclusion column (Figure S2).
Assay conditions were tested for efficiency of cleavage, and the optimal pH, ionic strength, and concentrations of EDTA and DTT were determined. The enzyme displayed optimal activity at pH 7.0, was relatively insensitive to EDTA concentration over a range from 0 – 5 mM or DTT over a range from 0 – 6 mM, and was slightly inhibited by the addition of NaCl (up to 600 mM). Standardized assays were performed at pH 7.0 in sodium phosphate buffer with 1.5 mM DTT and 2.35 mM EDTA. Prp readily reacts with Ellman’s reagent (5,5′-dithiobis(2-nitrobenzoic acid)) under non-denaturing conditions and is inhibited by a chloromethylketone-containing substrate peptide, Ac-KLNLQFF-CMK, consistent with its identification as a cysteine protease.
Raw assay data were corrected for background with respect to no-enzyme blank reactions and then converted to concentration of substrate cleaved using a standard curve generated by cleavage of the substrate with trypsin (Figure S3A). Cleavage of the fluorescent peptide by Prp was shown to progress to completion by comparing final fluorescence values produced by Prp and trypsin (Figure S3B). The maximum velocity for cleavage of the fluorogenic substrate was determined to be 1.960 ± 0.060 nM/s, the Km was 0.1824 ± 0.0280 μM, the turnover number (kcat) was 0.0458 ± 0.0014 s−1, and the specificity constant (kcat/Km) was 0.2511 ± 0.0393 μM−1s−1 (Figure 2). The Km of Prp for its quenched fluorescent substrate is quite low when compared to other motif directed cysteine proteases. For example, the viral nuclear inclusion A proteases like that of Tobacco Etch Virus, and the picornaviral 3C endopeptidases, have minimum Km values of 69 to 5uM, respectively, for fluorescent peptide substrates (BRENDA database; Schomburg et al., 2002).
Figure 2. Michaelis-Menten plot of Prp kinetics.
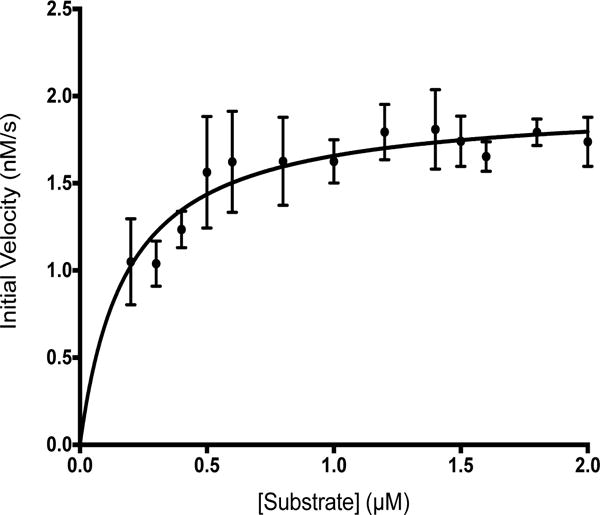
The average initial velocity at each substrate concentration is plotted. Based on fits to non-linear regression equations for Michaelis-Menten kinetics and kcat, the vmax was 1.96 ± 0.06 nM/s, the Km was 0.18 ± 0.03 μM, kcat was 0.05 ± 0.00 s−1, and the specificity constant (kcat/Km) was 0.25 ± 0.04 μM−1s−1.
To explore the feasibility of utilizing peptide derivatives as a scaffold for drug design, we examined the ability of peptides of various lengths containing the substrate cleavage motif to competitively inhibit enzymatic activity. Using 0.2 μM fluorogenic substrate and 21.38 nM enzyme (calculated as a dimer), a non-fluorescent peptide with the same 11 amino acid sequence as the fluorogenic substrate gave 50% inhibition at a concentration between 0.8 μM and 1 μM. Shorter peptides were much poorer inhibitors, and due to their low solubility we were unable to perform titrations to determine their Ki values. As an alternative, competitive peptides were assayed at 0.8 μM (the concentration that gave ~ 50% inhibition with the 11-mer) and the initial velocity for each reaction was used to calculate the percent inhibition with respect to a fluorogenic-peptide-only (no inhibitor) control. Inhibition by peptides derived from the L27 cleavage sequence dropped off rapidly with decreasing length, suggestive of a requirement for extended peptide recognition (Table 1; Figure S4). Cleavage of alternative peptide substrates derived from the phage 80α capsid protein (CP) cleavage motif (Poliakov et al., 2008) was also consistent with this overall trend. A 13-mer corresponding to the full CP N-terminal cleavage motif gave increased inhibition, while a CP 11-mer yielded results similar to the 11-mer from L27 (Table 1; Figure S4).
Table 1.
Inhibition of Prp activity by competitive peptides
| Competitive peptide | Percent Inhibition (Mean ± SD) |
|---|---|
|
L27 11mer Ac-KLNLQFFASKK-Am |
39 ± 5 |
|
L27 10mer Ac-LNLQFFASKK-Am |
25 ± 8 |
|
L27 8mer Ac-NLQFFASK-Am |
20 ± 7 |
|
L27 6mer Ac-QFFASK-Am |
19 ± 8 |
|
CP 13mer Ac-KLKLNLQHFASNN-Am |
63 ± 8 |
|
CP 11mer Ac-KLNLQHFASNN-Am |
32 ± 10 |
Molecular modeling of the active site and peptide docking
S. aureus Prp is a 23.4 kDa dimer composed of two identical chains of 106 amino acids (2P92, revised to 4PEO; Chirgadze et al., 2015). Two additional structures of Prp from bacteria encoding L27 as a pro-protein were available in the PDB, both also obtained from crystallographic consortia. These included homologs from S. pneumoniae (2IDL, unpublished) and Streptococcus mutans (2G0I, unpublished). A striking feature of these structures is their apparent lack of catalytic capacity. The catalytic residues must at some point be solvent exposed to accept substrate, and the His and Cys residues must face each other in such a way that the His can abstract a proton from the sulfhydryl group of the Cys. None of these structures had both of these features. The S. aureus Prp structure (2P92), on which our model is based, is missing part of a flexible loop that contains the active site histidine in chain A (see Fig 3A). A model of Prp docked with its substrate is needed in order to better understand the nature of the catalytic center and binding mechanism of this highly specific motif-directed protease.
Figure 3. Prp modeled and docked with a substrate peptide.
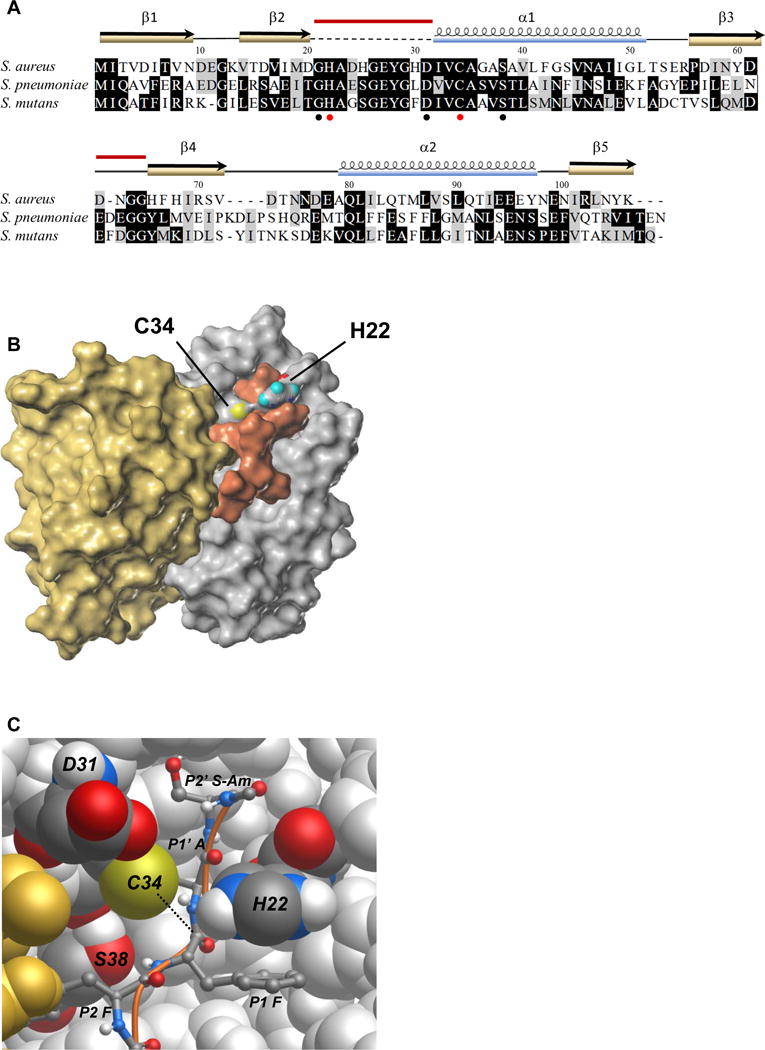
(A) Alignment of S. aureus Prp with the homologs from S. mutans and S. pneumoniae for which crystal structures have also been reported. Identical residues are highlighted in black, similar residues in gray. Conserved residues Gly 21, Asp 31, and Ser 38 surrounding the catalytic center are noted with black circles and the catalytic pair His 22 and Cys 34 are marked with red circles. The secondary structure elements from the crystal structure of S. aureus Prp are shown above the alignment. The dashed line indicates the region that was absent in the crystal structure, and the red bars indicate the two regions that were modeled in this study. PDB accession numbers: S. aureus (2P92/4PEO), S. mutans (2G0I), S. pneumoniae (2IDL). Amino acid sequences were aligned using MUSCLE and formatted in BOXSHADE through the Mobyle portal (Neron et al., 2009).
(B) Surface map of the Prp dimer and docked substrate Ac-NLQFFAS-Am. The catalytic pair residues are labeled and colored according to atomic convention (sulfur in yellow, oxygen in red, nitrogen in blue and hydrogen here in teal). Prp chain A is depicted in silver, chain B is in gold and the peptide substrate is shown in copper. In this model, the flexible loop that did not crystallize in PDB ID 2P92/4PEO is arranged in an arc above the active site. The substrate appears to associate with both chains of the enzyme.
(C) Atomic model of the Prp catalytic site. Prp chain B is in gold, chain A is silver with conserved labeled residues shown in atomic convention colors as above, with carbon shown in graphite and hydrogen in white. The peptide substrate Ac-NLQFFAS-Am is pictured as a ball and stick model with a copper backbone. In this depiction only the substrate residues FFAS-Am are visible (P2–P2′). The nucleophile (thiolate on Cys 34) is positioned to attack the carbonyl carbon of the scissile bond between P1 Phe and P1′ Ala on the substrate peptide (dotted line). The imidazolium group of His 22 is shown here π-π stacking with the phenyl group of the P1 Phe and, owing to its flexibility provided by Gly 21 (not visible), is positioned correctly to have abstracted a proton from Cys 34. The conserved hydroxyl group of Ser 38 is thought to provide a hydrogen bonding partner for the amide group between the P2 and P1 Phe residues. The role of Asp 31 is not clear in this model; its side chain protrudes near the catalytic site but appears to contact only solvent.
In cysteine protease catalysis, nucleophilic attack on the carbonyl carbon in the scissile bond of the substrate is achieved via a catalytic dyad, His and Cys, or in some cases a catalytic triad, His, Cys and Asp (or Asn) (Rawlings; Barrett, 1994). In addition to the conserved Cys and His residues, almost all members of the Prp family contain an Asp or Asn at position 31 (all numbering is with respect to S. aureus Prp), three residues prior to the catalytic Cys, raising the formal possibility of a catalytic triad in many Prp homologs. The requirements for catalysis in cysteine proteases noted above informed the structural modeling of the Prp active site.
To create the model, chain B and all the co-crystallizing waters were deleted from 2P92 so that loop refinement could be performed on monomer chain A. The modeled regions are indicated in Fig 3A. The loop from Gly 21 to Asp 31 was restored and refined into a pro-catalytic conformation. The only catalytic conformation of this loop that seemed plausible was one that resulted in a catalytic dyad comprised of Cys 34 and His 22. Asp 31 remains near the catalytic center, but it could not be oriented in such a way that it could act as a general base to polarize His 22.
The newly modeled Prp chain A was dimerized by duplication, followed by alignment of the copied chain with chain B of 2P92 to form a dimer with twofold rotational symmetry. The catalytic pair was ionized to represent their state prior to nucleophilic attack and formation of a covalent intermediate with substrate. Binding of peptide substrate very likely occurs in the cleft between the pair of α helices and the β-sheet of each monomer structure, where the active site residues lie. Access to this site required remodeling of a small loop consisting of residues 62–65 (see Fig 3A). A 7 amino acid peptide fragment with the substrate cleavage motif, acetyl (Ac)-NLQFFAS-amide (Am), was docked into this model such that the carbonyl carbon of the scissile bond was in the closest possible proximity to the Cys 34 thiolate ion. The 7-mer was the largest peptide that could be docked due to accuracy constraints on free peptide length. Residues Met 1, Leu 2, Lys 3 and Leu 4 from the full L27 cleavage motif do not appear in the model, though Leu 4 is quite conserved. After docking and molecular dynamics simulations the two phenyl side chains of the peptide substrate fit well into hydrophobic pockets near the enzyme catalytic center. The result is depicted in Figure 3B and C, with alternate views including a ribbon model that demonstrates the flexible loop clearly in Figure S5 A, and hydrophobicity maps of the Prp dimer alone and bound to substrate (Figure S5 B and C).
This model predicts the importance of some conserved Prp residues, but is ambiguous regarding others. The side chain of the Ser 38 residue on Prp, which is universally conserved as a serine or threonine among all Prp homologs, is a likely hydrogen bonding partner for the peptide nitrogen between P2 Phe and P1 Phe in the substrate backbone and may serve to orient the neighboring scissile bond for catalysis. The side chain of Asp 31 does not seem to interact with anything directly, but its conservation in more closely related Prp homologs suggests an important function, perhaps related to full-length substrate binding. The conserved location of His 22 in a flexible loop downstream of Gly 21 is consistent with changes in conformation around the active site upon substrate binding to adopt a catalytic conformation. Thus, the flexibility imposed by Gly 21 might be instrumental in the sequence specificity of Prp. The model suggests a possible pi-pi stacking interaction of catalytic His 22 on the enzyme with the P1 Phe on the substrate (Figure 3C). Given the inherent flexibility of the loop, this stacking interaction could confer stability to the active conformation of the enzyme only in the presence of its appropriate substrate, thereby linking enzyme activity directly with sequence specificity.
Prp mutants
Based on structural predictions from the model and sequence conservation data, mutations were made in the catalytic cysteine (C34), the catalytic histidine (H22), the conserved and possibly hinge-like glycine (G21), the conserved aspartic acid (D31) and the conserved serine (S38). Each of these residues was replaced by alanine with the exception of C34, which was changed to serine. Prp C34S is an isosteric mutant that had already been characterized as being able to bind but not cleave tagged L27 (Wall et al., 2015). The D31A mutation was made to formally test whether that conserved aspartic acid has a role as part of a catalytic triad, even though this model does not predict one. The H22A and C34S catalytic site mutants were expected to abrogate Prp activity while the S38A mutation was expected to affect substrate binding. The G21A hinge mutation was predicted to reduce the flexibility of the loop containing the catalytic histidine, affecting catalysis, substrate binding, or both.
Activity of selected Prp point mutants
Wild-type Prp and the mutants C34S, G21A, H22A, D31A, and S38A were assayed at 21.38 nM with 2.0 μM fluorogenic substrate. Figure 4A shows that none of the mutants had any measureable activity at this concentration. To determine whether higher concentrations of mutant enzyme would produce any measureable activity, 213.8 nM of each enzyme was tested with 2.0 μM substrate. Of these reactions, only the S38A mutant had slight activity, displaying just 2.1 ± 0.2% activity relative to the wild-type enzyme. All of the mutations made in Prp were severely detrimental to substrate binding, catalysis, or both.
Figure 4. Analysis of Prp active-site mutants.
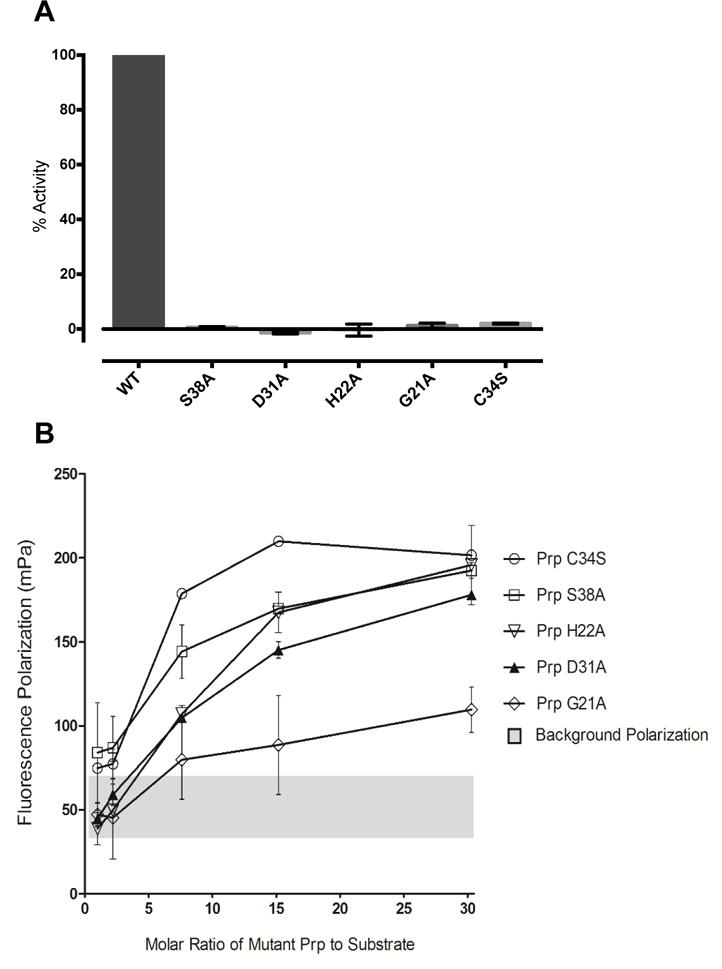
(A) Enzymatic activity. The average percent activity for each mutant is shown relative to the wild-type enzyme. Each enzyme was assayed at a concentration of 21.4 nM (dimer), as described in Experimental Procedures. Error bars indicate one standard deviation; n ≥ 3. (B) Substrate binding. Substrate binding as measured by fluorescence polarization is shown for each of the Prp point mutants, at differing molar ratios of mutant enzyme to 3 μM substrate. Error bars represent 95% CI, n=4. The grey bar summarizes the range of background fluorescence from each of the individual experiments.
To help distinguish between defects in binding and catalysis, we examined the mutants for substrate binding via fluorescence polarization. This assay measures the enzyme’s ability to bind and slow the rotation of a fluorescent substrate. Peptide 2-Abz-KLNLQFFASKK-Am (identical to the enzymatic assay substrate except for the lack of a Dnp quencher) was added at 3 μM to various molar concentration ratios of Prp. The isosteric Cys to Ser mutation was expected to eliminate catalytic activity but not substrate binding, and the C34S mutant did yield maximum polarization. All of the other mutants showed reductions in polarization, ranging from moderate in the case of S38A to drastic in the case of G21A (Figure 4B). These data suggest that the main factor causing the inactivity of these mutants is their inability to bind the peptide substrate. Prp H22A, while mutant at a catalytic residue, demonstrates a deficiency in binding as well when compared to the C34S mutation. This could be due to loss of the hypothesized pi-pi stacking interaction with the P1 Phe of the substrate, as discussed above. The S38A mutant retains a small amount of activity, which is consistent with its ability to bind slightly better than the other non-catalytic mutants, D31A and G21A. The severe loss of binding and lack of catalytic activity in the G21A mutant provides support for the hypothesis that the conserved glycine is a hinge that allows the flexible loop to adopt its bound catalytic conformation in the presence of the correct substrate.
Discussion
This work confirms the presence of a novel essential pathway in S. aureus for proteolytic processing of a precursor of ribosomal protein L27 to its functional mature form. This pathway is also likely to be essential in all Firmicutes, Synergistetes, Fusobacteria and related bacteria, including some pathogenic Tenericutes. Importantly, this L27 processing step is apparently regulated, because pre-cleaved L27 Δ 2–9 cannot complement an L27 deletion. Edman degradation confirms that the expression of the pre-cleaved L27 mutant produces a protein identical to that found in mature ribosomes (no retained N-formyl Met), indicating that its normal function of tRNA stabilization should be intact. The un-cleavable mutant was not expected to complement an L27 deletion because retention of the N-terminal extension was postulated to interfere with the activity of the PTC. Previous studies have shown that uncleaved L27 is associated with 50S ribosomal subunit precursors, as is mutant Prp that is cleavage-defective (Wall et al. 2015). We have not been able to experimentally address whether “pre-cleaved” L27 is assembled into ribosomes, due to the lethality of the L27 Δ 2–9 mutation. Thus, we cannot at this point distinguish whether the function of this cleavage process is related to ribosome biogenesis, regulation of translation, or perhaps both. However, an attractive hypothesis consistent with the current evidence is that Prp serves as a chaperone for L27 assembly. The ribosome is understood to have myriad chaperones involved during its construction, many of which are enzymes (Wilson; Nierhaus, 2007; Shajani et al., 2011; Goto et al., 2013). It could be that cleavage occurs after the L27 pro-protein has been incorporated into a large ribosomal subunit precursor in order to disengage Prp from the complex. This would also be consistent with the observed low turnover rate for this enzyme. Regardless of the biological role of L27 precursor cleavage, it is clear that the action of Prp provides an important new drug target in highly drug resistant pathogens, and Prp characterization is a necessary foundation for the possible development of new antimicrobials.
S. aureus Prp is the prototype of its novel structural clade and presents opportunities to understand the biochemistry of an entirely new group of cysteine proteases. The Prp homolog structures from S. pneumoniae and S. mutans (PDB IDs 2IDL and 2G0I, respectively) show a high degree of structural conservation despite low sequence identity - no more than 47% (see Fig 3A). Homology modeling of related Prp sequences of unknown structure are consistent with these observed structures. None of the existing crystal structures, however, is in a conformation that would allow catalytic activity – the flexible loop that contains the catalytic His is either missing or deformed in such a way that the imidazole group of the His cannot interact with the thiol group of the Cys. Therefore the model of S. aureus Prp with substrate depicted here indicates certain important structure-function relationships of conserved residues in the Prp family.
The catalytic Cys replacement with isosteric Ser was made originally to test the possibility that Ser hydroxyl could substitute as a catalytic nucleophile in this protease. This is not the case; the enzyme is catalytically inactive although it still binds substrate (Wall et al., 2015). The C34S mutant was therefore used as a model for maximum binding in the fluorescence polarization assay. The other member of the catalytic pair, His 22, is suggested by the model to play a dual role in catalysis and binding, based on an apparent pi-pi stacking interaction with the substrate P1 Phe. Enzymatic and fluorescence polarization data are consistent with this prediction, showing reduced binding in addition to a lack of activity. Both the modeling and the existing crystal structures indicate that this residue is located on a dynamic, flexible loop, possibly only interacting with Cys 34 in the presence of bound substrate. The adjacent Gly 21 appears to play a critical role in substrate binding, and our data provide support for the role of this residue as a hinge involved in correct positioning of the flexible loop.
The S38A mutant showed slight activity (2.0 ± 0.2%) relative to wild-type when both were assayed at a higher enzyme concentration. This is correlated with its ability to bind and polarize the fluorescent substrate better than the other mutants tested, aside from C34S. In the model, this residue supports substrate binding in a non-sequence specific manner, interacting only with the backbone amide nitrogen between the P2 and P1 residues. Ser 38 thus appears to make a significant contribution to enzymatic activity via substrate stabilization.
The role of conserved Asp 31 is not well understood – it resides one rung of an alpha helix (5.4 Å) above Cys 34, permanently ensuring proximity to that residue and not to His 22, making it seem unlikely that Asp 31 serves as a general base to ionize His 22. These observations may indicate that Asp 31 is involved in full-length substrate binding or organization of the flexible loop to facilitate catalysis. The substrate peptide Ac-NLQFFAS-Am contains a highly conserved P3 Gln that in this model does not appear to bind anything on the enzyme directly, and points outward into solvent. It is possible that the P3 Gln could interact with the conserved Asp 31 or other residues on the enzyme if the flexible loop draped over and became ordered around substrate.
Loop flexibility reduction upon substrate binding has precedent in other site-specific cysteine proteases. NMR studies of staphylococcal cysteine transpeptidase sortase A demonstrate that the β6/β7 loop, which is disordered in the apostructure, becomes immobile upon substrate peptide binding, forming a helix that stabilizes the enzyme-substrate interaction (Suree et al., 2009). Previous work demonstrated that mutations of this loop significantly impaired enzymatic activity and changed substrate specificity (Bentley et al., 2007; Bentley et al., 2008). Our results suggest that conserved flexibility of the loop above the catalytic center could be instrumental in substrate binding and stabilization in Prp as well.
Another notable feature in the model is a large hydrophobic patch that extends below the catalytic center and includes the region of postulated substrate binding (Figures S5 B and C). The observed failure of shorter peptides to compete with longer substrates is consistent with a requirement for extended substrate recognition. An obvious question regarding the model presented here is how a full-length substrate might interact with Prp. Based on the crystal structure, Chirgadze et al. (2015) proposed a “sandwich” interaction involving L21, Prp and L27. They noted the negative residues distributed over the exterior of Prp and suggested that they could serve as points of interaction with the positively-charged ribosomal proteins encoded in the same operon. At present our model does not indicate a purpose for this enzyme’s low isoelectric point, leaving the formal possibility of a closer association with the C-terminal region of L27. However, the pseudo two-fold rotational symmetry of Prp suggests that binding to both L21 and L27 at the same time is improbable, and L21 lacks the cleavage motif. S. aureus Prp is also known to cleave substrates that are not as small and positive as L27 – phage 80α scaffold and capsid proteins both have an isoelectric point of 4.9, and are 23 and 37 kDa respectively.
The model presented in this work does not depict the orientation of substrate residues past the P2′ serine. In fact, the amidated C-terminus of that residue seems to run directly and improbably into Prp density. The first forty residues of L27 are known to be highly flexible, only crystallizing in certain full ribosome structures (Wang et al., 2004; Voorhees et al., 2009; Polikanov et al., 2014). The long N-arm of L27 contains conserved glycines that could theoretically allow it to loop sharply. It is also possible that the large flexible loop in Prp is moved to one side during catalysis and forms a channel that accommodates the residues after the cleavage motif leading to the structured portion of L27. The amount of flexibility around the catalytic center makes it very difficult to understand how or where the enzyme interacts with the C-terminal regions of any substrate. There simply are not good predictions for the orientation of a ten residue loop around a substrate docked in silico, or possible interactions with the corresponding loop on the other monomer. A co-structure of Prp with substrate or a substrate analog of sufficient length is required to shed more light on this interaction.
Site-specific cysteine proteases have been studied as drug targets in many organisms, especially in relation to viral polyprotein processing and eukaryotic autophagy-related processes. There is precedent for smaller “drug-like” versions of the peptide cleavage motif working as competitive inhibitors for enzymes like caspase-1 (Margolin et al., 1997). However, the Turnip Mosaic Virus nuclear inclusion A (TuMV NIa) protease was unable to cleave smaller substrates, only cleaving peptides of at least six amino acids in length (Yoon et al., 2000). Similarly, shorter versions of the L27 cleavage motif failed to competitively inhibit Prp (Table 1), suggesting that small peptide mimetics are likely an ineffective route to anti-Prp drug design. The assay we have developed will allow high throughput screening of libraries to identify compounds that may inhibit by mechanisms other than competitive binding.
Prp cleavage was previously implicated in another system, also involving macromolecular assembly - capsid morphogenesis of staphylococcal phage 80α. At some point before or during procapsid formation, both the phage capsid and scaffold proteins are cleaved at a similar conserved motif (Poliakov et al., 2008). Co-expression of capsid and scaffold proteins without the N-terminal motif results in dysregulated assembly of capsomers into sheets or polyheads (Spilman et al., 2012). These data suggest that Prp targets substrates involved in very large and complex macromolecular assemblies - ribosomes and phage capsids, some of the oldest and most conserved bacterial or viral structures. It is possible that Prp has a slightly different role in these scenarios, but it also seems likely these complex assemblies have in common the requirement of some form of regulation during their formation, again suggestive of a role as a chaperone for Prp.
This work validates Prp as a novel drug target. We present a model that provides insight into substrate binding and catalysis by this new clade of sequence-specific cysteine proteases. These results suggest possible avenues for the development of new antibiotics against major drug resistant pathogens that contain Prp and the L27 N-terminal extension, including Clostridia, Staphylococci, Streptococci and Enterococci.
Experimental Procedures
Bacterial Culture
Strains and plasmids used in this study are listed in Table S1. All S. aureus strains are derivatives of the phage-cured, restriction-defective strain RN4220 (Kreiswirth et al., 1983). S. aureus strains were grown in Trypticase Soy Broth (TSB) or Brain Heart Infusion (BHI) at 37°C. Antibiotics used with S. aureus strains included tetracycline (2 μg/mL), erythromycin (5 μg/ml), chloramphenicol (15 μg/mL), spectinomycin (250 μg/mL) and kanamycin (30 μg/mL). E. coli strains were cultured in Luria-Bertani (LB) broth or on LB agar plates supplemented with ampicillin (100 μg/ml) and/or chloramphenicol (30 μg/ml), as required, and grown at 37°C overnight. Liquid cultures were grown on an orbital shaker at 200 rpm. For induction of regulated promoters, IPTG was used at 1 mM and NaAsO2 at 5 μM. For large scale expression, E. coli strains containing IPTG-inducible promoters were auto-induced using lactose-containing media.
DNA Manipulation
Polymerase chain reactions were performed using Pfu Ultra II polymerase (Agilent). Primers used in this study are listed in Table S2. PCR products were purified using the Nucleospin® Gel and PCR Clean-up Kit (Macherey-Nagel; Düren, Germany) according to manufacturer’s instructions. Cloning reactions were performed using an In-Fusion® HD Cloning Kit (Clontech Laboratories Inc.; Mountain View, CA) or by standard ligation with T4 ligase. Plasmids were introduced to S. aureus by electroporation.
Strain Creation
The S. aureus expression strain SA178RI carries a T7 RNA polymerase expression cassette under lac operator control. The plasmid used for T7 expression in SA178RI was pG164, an E. coli-S. aureus shuttle vector carrying the T7 late promoter into which a lac operator, a multiple cloning site, an optimized gram-positive ribosome binding site, and a constitutively expressed copy of the lac repressor gene were introduced (D’Elia et al., 2006). The mutant alleles were created using Gibson assembly of overlapping pieces of DNA that contained the desired nucleotide changes in the manipulated 15 base pair (bp) overlap regions. Plasmids were isolated from Stellar E. coli (Clontech-Takara) for sequencing and transformation of the appropriate S. aureus or E. coli strain. When replacement of S. aureus L27 with a spectinomycin resistance cassette was required, the mutant allele was exchanged into the S. aureus chromosome of strain ST256 using the shuttle plasmid pMAD as described previously (Arnaud et al., 2004), with the addition of 1 mM IPTG to ensure expression of plasmid-encoded L27.
Edman Degradation
Ni-NTA purified L27 Δ 2–9 His6 protein samples were loaded onto a 16.5% Tris-tricine precast gel (Biorad). The protein was electroblotted to a 0.45 μm PVDF membrane for 4 hours at 300 mA in a Criterion Tank Blotter (Biorad). The membrane was stained with Coomassie and the appropriate band was marked, coded and sent to the Iowa State University Protein Facility. There the protein samples on the membrane were washed six times with deionized water and loaded onto a 494 Procise Protein Sequencer/140C Analyzer (Applied Biosystems, Inc).
Recombinant Protein Overexpression and Purification
Hexahistidine-small ubiquitin-related modifier (His6-SUMO) tagged Prp (or Prp point mutants) and His6-tagged ubiquitin-like protein-specific protease 1 (Ulp1) were overexpressed in BL21-CodonPlus™ (DE3)-RIL (Agilent Technologies; Santa Clara, CA) and ScarabXpress T7 lac (Scarab Genomics; Madison, WI) E. coli cells, respectively. Bacteria were lysed using an Emulsiflex C3 High Pressure Homogenizer (Avestin Inc.; Ottawa, ON, CA) at approximately 20,000 psi. Lysate was clarified by centrifugation at 20,000 rpm for 30 minutes at 10°C. A nickel affinity column was prepared by adding 10–20 ml of Profinity™ IMAC Uncharged Resin (Bio-Rad; Hercules, CA), followed by 5–10 ml of 100 mM nickel sulfate (NiSO4). The column was washed with ~30 ml of wash buffer (25 mM Tris, pH 8, 300 mM NaCl, 10 mM imidazole) and the clarified supernatant was added to the column, allowing the His6-tagged protein to bind the resin. The bound protein was washed and eluted with ~100 ml elution buffer (25 mM Tris, pH 8, 300 mM NaCl, 110 mM imidazole). The fractions were then analyzed via SDS-PAGE. The fractions containing the protein of interest were pooled, sodium azide was added to 0.02% v/v, and the protein was dialyzed versus 50 mM sodium phosphate buffer, pH 7.5 with 0.02% v/v sodium azide (and 10% v/v glycerol for Ulp1). To cleave the His6-SUMO tag from purified and dialyzed His6-SUMO-Prp, His6-tagged Ulp1 was added in an approximately 1:1 ratio by weight with His6-SUMO-Prp with 150 mM NaCl at room temperature. Five microliter samples were taken at t = 0h, 1h, 2h, and 3h and analyzed for extent of cleavage via SDS-PAGE. After sufficient cleavage, the mixture was purified via nickel affinity chromatography in the same manner as the tagged protein, except fraction collection began immediately after the sample was applied to the column. After dialyzing, the purified, tagless protein in solution was filter sterilized using a Millex® 0.22 μm syringe filter (EMD Millipore; Billerica, MA) and the protein concentration was determined using Bradford reagent and A280, which gave comparable results. Glycerol was added to 40% v/v, and one milliliter aliquots were stored at −20°C. Glycerol was eliminated from purified Prp mutants via buffer exchange during concentration before fluorescence polarization experiments.
Fast Protein Liquid Chromatography (FPLC)
Fast protein liquid chromatography was performed on a mixed sample of tagless, wild-type Prp and His6-tagged Ulp1 using an ÄKTAFPLC™ system with UNICORN™ system control software and a 125 ml P-10™ size-exclusion column (20,000 MWCO; Bio-Rad; Hercules, CA) equilibrated to 50 mM sodium phosphate buffer, pH 7.5. Five milliliters of Prp:Ulp1 sample was manually injected onto the column and eluted with 50 mM sodium phosphate buffer, pH 7.5. Two-milliliter fractions were collected at a flow rate of 1 ml/min while the elution profile was monitored spectrophotometrically at 280 nm. Peak fractions were analyzed via SDS-PAGE.
Fluorogenic Peptide Cleavage Assay
Peptides used in this study were purchased from United Biosystems (Herndon, VA). Stock fluorogenic peptide (SauL2711) was made by dissolving a small amount of lyophilized peptide in DMSO. Its concentration was determined by measuring its absorbance in a 0.1 cm pathlength quartz cuvette at 325 nm with a Bio-Rad® SmartSpec™ 3000. Aliquots were frozen at −20°C until use. Competitive non-fluorescent peptide substrates used in this study were resuspended to 1 mM with DMSO. These working stocks were stored at −20°C until use. Fluorescence assays were performed using a Tecan Infinite® M1000 microplate reader with Magellan™ data analysis software (Männedorf, Switzerland) and Greiner® Bio-One black, flat-bottomed, chimney well, non-sterile, non-binding, 96-well microplates (Monroe, NC). The plate reader was set to read fluorescence intensity at an excitation wavelength of 325 nm and an emission wavelength of 414 nm with 5 nm bandwidths. Cleavage reactions were read at 8-second intervals for 10 minutes at ambient temperature. The plate reader’s gain was optimized at 255 and the Z-position height was optimized at 20,450 μm. Standardized assays had final concentrations of 1.5 mM DTT, 2.35 mM EDTA, 2.53% DMSO, and various concentrations of fluorogenic peptide and/or competitive peptide in 50 mM sodium phosphate buffer, pH 7.0.
Standard Curve Generation and Determination of the Extent of Assay Completion
To convert the raw values of relative fluorescence units (RFU) versus time from the assay data to concentration of fluorogenic substrate cleaved versus time, a standard curve of RFU versus concentration of fluorogenic substrate cleaved by trypsin was prepared (Figure S3A). Ten microliters of 1 mg/ml trypsin in 50 mM sodium phosphate buffer, pH 7.5, with 40% glycerol and 0.02% sodium azide was used in the standard cleavage assay with varying amounts of substrate (0–1.0 μM). A no-enzyme blank was used to correct for background. The blank-corrected RFU values from four completed reactions at each concentration were averaged and the results were plotted versus the concentration of substrate included in the reaction. Trypsin was also used in assays to determine the extent of assay completion by Prp. Identical assays were performed, cleaving 2 μM substrate with 10 μl of 1 mg/ml trypsin and Prp and the average absolute RFU values for four completed reactions were compared (Figure S3 B). Both reactions approached the same RFU values, indicating that both reactions proceeded to the same extent of completion.
Enzyme Activity Analysis
Assay data were analyzed using Microsoft® Excel® 2011 and GraphPad Prism® (La Jolla, CA). Data were collected and organized in Excel®, then the blank RFU value at each time point was subtracted from the corresponding time point of each trial for each assay. The blank-corrected RFU data were then converted to concentration of substrate cleaved using the equation of the standard curve (Figure S3A) and plotted using a smooth marked scatter plot. The slope of the initial, linear portion of each curve (initial velocity) was determined for each assay.
For measurement of wild type Prp enzyme kinetics, Prp was used at a final concentration of 21.38 nM (as dimer). The initial velocities for each substrate concentration were entered into Prism®, where the data were analyzed using non-linear regression fits to the Michaelis-Menten kinetics and kcat equations (ET constrained to 42.765 nM active sites; 2 active sites per dimer).
For competition data with competitive-peptide inhibitors, the percent activity and percent inhibition relative to a no-inhibitor control were calculated from the initial velocities for each inhibitor assay. These results were then entered into Prism® and analyzed using an ordinary one-way ANOVA, comparing the mean initial velocity for each inhibitor to that of the no-inhibitor control. For the Prp active-site mutants, the percent activity relative to a wild-type control was calculated from the initial velocities for each mutant. These results were then entered into Prism® and analyzed using an ordinary one-way ANOVA, comparing the mean initial velocity for each mutant to that of the wild-type enzyme.
Fluorescence Polarization
Fluorescence polarization experiments were conducted on a Flex Station 3 by Molecular Devices (Sunnyvale, CA). Polarization of the substrate 2-Abz-KLNLQFFASKK-Am was analyzed at 415nm. The reactions were formulated with 3 μM substrate in the presence of inactive Prp mutants at molar ratios of 1, 2.2, 7.6, 15.2 and 30.3 Prp mutant::substrate. Lower molar ratios of Prp mutants to fluorescent substrate produced no detectible polarization, possibly due to sensitivity limits of the polarization reader utilized in this study.
Molecular Modeling
The existing crystal structure of S. aureus Prp (PDB ID: 2P92; now 4PEO) lacks the loop that includes the active site residue His 22. The PDB file of the incomplete S. aureus Prp/YsxB homolog dimer was read into SYBYL-X version 2.1. Modeller version 9.12 (Webb; Sali, 2014) was used to model the loop that was not present in the crystal structure. One loop model was created using the loop_model.py script written by Dr. Hardik Parikh, and the loop was refined using the loop_refine.py script also written by Dr. Parikh. Over 200 models were created and read into SYBYL (Certara, Princeton, NJ), and one was chosen that contained a favorable His/Cys pro-catalytic conformation. The four residue loop (residues 62–65) had a closed conformation that partially obscured the active site of the enzyme. For this reason, the loop_refine.py script was again utilized to allow the four residue loop to adopt a conformation that did not obscure the active site. The model with greatest solvent exposure of the catalytic site was chosen. This model was read into SYBYL, along with the original crystal 2P92 which formed the template for modeling the dimer. This dimer was read into Gold 5.2, and docking was achieved using the blocked peptide Acetyl (Ac)-Asn-Leu-Gln-Phe-Phe-Ala-Ser-Amide (Am). Molecular dynamics (MD) simulations were carried out with the NAMD 2.8 package developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign (Phillips et al., 2005). CHARMM (Charmm-27) was used as the force field (MacKerell et al., 1998). Prior to simulation, the ionization states of His residues were checked using PROPKA 3.1 (Olsson et al., 2011). The analysis of the MD trajectory was done in VMD (Humphrey et al., 1996).
Supplementary Material
Acknowledgments
We are grateful to Dr. William Barton for the use of his microplate reader, Dr. Hardik Parikh for his time and effort in writing the modeling scripts, Dr. Mostafa Ahmed for assistance with molecular dynamics, Dr. Glen Kellogg and Dr. Phil Mosier for structural modeling guidance, Dr. Todd Black (Merck & Co.) for providing pG164 and SA178RI, and Dr. H. Tonie Wright for comments on the manuscript. These studies were supported by a grant from the VCU Presidential Research Quest Fund and National Institutes of Health grant R21 AI109202 (to GEC), and an AHA predoctoral fellowship (to EAW).
References
- Arnaud M, Chastanet A, Debarbouille M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol. 2004;70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach T, Bashan A, Harms J, Schluenzen F, Zarivach R, Bartels H, et al. Antibiotics targeting ribosomes: crystallographic studies. Curr Drug Targets Infect Disord. 2002;2:169–186. doi: 10.2174/1568005023342506. [DOI] [PubMed] [Google Scholar]
- Bentley ML, Gaweska H, Kielec JM, McCafferty DG. Engineering the substrate specificity of Staphylococcus aureus Sortase A. The beta6/beta7 loop from SrtB confers NPQTN recognition to SrtA. J Biol Chem. 2007;282:6571–6581. doi: 10.1074/jbc.M610519200. [DOI] [PubMed] [Google Scholar]
- Bentley ML, Lamb EC, McCafferty DG. Mutagenesis studies of substrate recognition and catalysis in the sortase A transpeptidase from Staphylococcus aureus. J Biol Chem. 2008;283:14762–14771. doi: 10.1074/jbc.M800974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. Antibiotic Resistance Threats in the United States, 2013 [WWW document] 2014 URL http://www.cdc.gov/drugresistance/threat-report-2013/
- Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, et al. Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH) BMC Genomics. 2009;10 doi: 10.1186/1471-2164-10-291. 291-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgadze YN, Clarke TE, Romanov V, Kisselman G, Wu-Brown J, Soloveychik M, et al. The structure of SAV1646 from Staphylococcus aureus belonging to a new `ribosome-associated’ subfamily of bacterial proteins. Acta Crystallogr D Biol Crystallogr. 2015;71:332–337. doi: 10.1107/S1399004714025619. [DOI] [PubMed] [Google Scholar]
- Colca JR, McDonald WG, Waldon DJ, Thomasco LM, Gadwood RC, Lund ET, et al. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J Biol Chem. 2003;278:21972–21979. doi: 10.1074/jbc.M302109200. [DOI] [PubMed] [Google Scholar]
- D’Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, et al. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol. 2006;188:4183–4189. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Muto A, Himeno H. GTPases involved in bacterial ribosome maturation. J Biochem. 2013;153:403–414. doi: 10.1093/jb/mvt022. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Ji Y, Zhang B, Van SF, Horn, Warren P, Woodnutt G, et al. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science. 2001;293:2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, Novick RP. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- Liu J, Dehbi M, Moeck G, Arhin F, Bauda P, Bergeron D, et al. Antimicrobial drug discovery through bacteriophage genomics. Nat Biotechnol. 2004;22:185–191. doi: 10.1038/nbt932. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Maguire BA, Beniaminov AD, Ramu H, Mankin AS, Zimmermann RA. A protein component at the heart of an RNA machine: the importance of protein L27 for the function of the bacterial ribosome. Mol Cell. 2005;20:427–435. doi: 10.1016/j.molcel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ. Substrate and inhibitor specificity of interleukin-1 beta-converting enzyme and related caspases. J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- Minamino T. Protein export through the bacterial flagellar type III export pathway. Biochim Biophys Acta. 2014;1843:1642–1648. doi: 10.1016/j.bbamcr.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Neron B, Menager H, Maufrais C, Joly N, Maupetit J, Letort S, et al. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of chemical theory and computation. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliakov A, Chang JR, Spilman MS, Damle PK, Christie GE, Mobley JA, Dokland T. Capsid size determination by Staphylococcus aureus pathogenicity island SaPI1 involves specific incorporation of SaPI1 proteins into procapsids. J Mol Biol. 2008;380:465–475. doi: 10.1016/j.jmb.2008.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Steitz TA, Innis CA. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat Struct Mol Biol. 2014;21:787–793. doi: 10.1038/nsmb.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ. Families of cysteine peptidases. Methods Enzymol. 1994;244:461–486. doi: 10.1016/0076-6879(94)44034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42:D503–9. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg I, Chang A, Schomburg D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res. 2002;30:47–49. doi: 10.1093/nar/30.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem. 2011;80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- Shin DH, Lou Y, Jancarik J, Yokota H, Kim R, Kim SH. Crystal structure of TM1457 from Thermotoga maritima. J Struct Biol. 2005;152:113–117. doi: 10.1016/j.jsb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Spilman MS, Damle PK, Dearborn AD, Rodenburg CM, Chang JR, Wall EA, et al. Assembly of bacteriophage 80alpha capsids in a Staphylococcus aureus expression system. Virology. 2012;434:242–250. doi: 10.1016/j.virol.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suree N, Liew CK, Villareal VA, Thieu W, Fadeev EA, Clemens JJ, et al. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J Biol Chem. 2009;284:24465–24477. doi: 10.1074/jbc.M109.022624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor F, Ballesta JP. Reaction of some macrolide antibiotics with the ribosome. Labeling of the binding site components. Biochemistry. 1986;25:7725–7731. doi: 10.1021/bi00371a066. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall EA, Caufield JH, Lyons CE, Manning KA, Dokland T, Christie GE. Specific N-terminal cleavage of ribosomal protein L27 in Staphylococcus aureus and related bacteria. Mol Microbiol. 2015;95:258–269. doi: 10.1111/mmi.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Takemoto CH, Murayama K, Sakai H, Tatsuguchi A, Terada T, et al. Crystal structure of ribosomal protein L27 from Thermus thermophilus HB8. Protein Sci. 2004;13:2806–2810. doi: 10.1110/ps.04864904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- Wilson DN, Nierhaus KH. The weird and wonderful world of bacterial ribosome regulation. Crit Rev Biochem Mol Biol. 2007;42:187–219. doi: 10.1080/10409230701360843. [DOI] [PubMed] [Google Scholar]
- Yonath A. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu Rev Biochem. 2005;74:649–679. doi: 10.1146/annurev.biochem.74.082803.133130. [DOI] [PubMed] [Google Scholar]
- Yoon HY, Choi KY, Song BD. Fluorometric assay of turnip mosaic virus NIa protease. Anal Biochem. 2000;277:228–231. doi: 10.1006/abio.1999.4398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.