

Abstract
The valosin-containing protein (VCP) participates in signaling pathways essential for cell homeostasis in multiple tissues, however, its function in the heart in vivo remains unknown. Here we offer the first description of the expression, function and mechanism of action of VCP in the mammalian heart in vivo in both normal and stress conditions. By using a transgenic (TG) mouse with cardiac-specific overexpression (3.5-fold) of VCP, we demonstrate that VCP is a new and powerful mediator of cardiac protection against cell death in vivo, as evidenced by a 50% reduction of infarct size after ischemia/reperfusion versus wild type. We also identify a novel role of VCP in preserving mitochondrial respiration and in preventing the opening of mitochondrial permeability transition pore in cardiac myocytes under stress. In particular, by genetic deletion of inducible isoform of nitric oxide synthase (iNOS) from VCP TG mouse and by pharmacological inhibition of iNOS in isolated cardiac myocytes, we reveal that an increase of expression and activity of iNOS in cardiomyocytes by VCP is an essential mechanistic link of VCP-mediated preservation of mitochondrial function. These data together demonstrate that VCP may represent a novel therapeutic avenue for the prevention of myocardial ischemia.
Acute myocardial ischemia (MI) and coronary artery disease remain among the top ranking causes of death and disability worldwide1. Mitochondrial dysfunction contributes to cell damage during ischemia/reperfusion (IR) and is central to cardiomyocyte death2,3,4. It has been shown that ischemic preconditioning (IPC), the gold standard for cardioprotection, attenuated the decline of mitochondrial function induced by ischemic injury, including oxidative phosphorylation, respiratory chain coupling and mitochondrial efficiency5,6,7,8,9. There is also increasing evidence that the mitochondrial permeability transition pore (mPTP) opening plays a central role in mediating both the necrotic and apoptotic components of IR injury, particularly at the onset of reperfusion, and inhibition of mPTP opening is considered to be a critical target of cardioprotection by IPC3,10,11,12,13,14.
We identified previously that the valosin-containing protein (VCP), a member of a family that includes ATP-binding proteins, promotes a significant reduction in apoptosis in isolated cardiomyocytes under cell stress15. These data support the hypothesis that over-expression of VCP in vivo in the heart may provide protection against ischemic injury.
To test such hypothesis, we generated a transgenic (TG) mouse with cardiac-specific overexpression of VCP. Our results demonstrate that overexpression of VCP significantly reduced the infarct size (IS) by 50% after IR compared to wild type (WT) mice. We also showed that VCP exhibited a new role on activation of mitochondrial respiration efficiency and inhibition of the mPTP opening. Using both genetic and pharmacological approaches, we also show that the effect of VCP on the cardiac mitochondrial function was dependent upon the increase of inducible isoform of nitric oxide synthase (iNOS) conferred by VCP.
Results
Cardiac specific overexpression of VCP in a TG mouse did not alter cardiac structure and function at baseline
The construct used to generate the TG mouse is shown in Fig. 1a. Different levels of VCP overexpression (from 3.5-fold to 10-fold) were found among the mice lines. In accordance with our previous data in vitro15, cardiac-specific overexpression of VCP dose-dependently increased iNOS expression in the hearts (from 2.5-fold to 10-fold, data not shown). Further characterization was performed on the VCP TG line with a 3.5-fold overexpression (Fig. 1b), since it increased iNOS expression by 2.5-fold in protein level when compared to WT (p < 0.01 vs. WT, Fig. 1d), which best reproduces the range of iNOS expression found in the second window of preconditioning (SWOP)16,17. Overexpression of VCP in this mouse line also increased the mRNA level of iNOS by 3.5 fold and the activity of iNOS by 1.8-fold compared to WT (p < 0.05 vs. WT, Fig. 1c and e). There was no change in eNOS expression in TG mouse heart compared to WT (Fig. 1f).
Figure 1. Cardiac-specific overexpression of VCP increases iNOS expression and activity in TG mouse hearts compared to WT.

(a) VCP TG mouse construct. (b) Representative immunoblots and protein level of VCP overexpression in mouse hearts, n = 8 per group. (c) mRNA level of iNOS in mouse hearts by qPCR, n = 5 per group. (d) Representative immunoblots and protein level of iNOS in mouse hearts, n = 8 per group. (e) iNOS activity in mouse heart tissues, n = 5 per group. (f) Protein level of eNOS in mouse hearts, n = 4 per group. GAPDH was used as loading control. *p < 0.05.**p < 0.01 vs. WT. Data are the mean ± SEM.
Compared to WT mice, the VCP TG mouse exhibited similar left ventricle (LV) weight-to-tibia length ratio (LV/TL), LV-to-body weight ratio (LV/BW), and the lung weight-to-tibia length ratio (LW/TL) in basal conditions (Table 1). VCP overexpression had no significant effect on myocyte cross-sectional area, collagen accumulation, and apoptosis (Table 1). No difference on structural and functional echocardiographic parameters was observed in VCP TG mice when compared to WT in terms of heart rate, LV end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD), wall thickness, LV ejection fraction (LVEF) and fractional shortening (FS) (Table 1).
Table 1. Physiological and morphological characteristics of VCP TG mice compared to WT littermates.
| WT (n = 6) | TG (n = 6) | |
|---|---|---|
| Body weight (g) | 24.4 ± 0.4 | 24.6 ± 0.9 |
| LV/body weight (mg/g) | 3.2 ± 0.1 | 3.3 ± 0.2 |
| LV/tibial length (mg/mm) | 3.6 ± 0.1 | 3.8 ± 0.1 |
| Ling weight/tibial length (mg/mm) | 8.0 ± 0.4 | 8.0 ± 0.5 |
| LV Ejection fraction (%) | 71.9 ± 0.9 | 71.2 ± 1.2 |
| LV Fractional shortening (%) | 34.5 ± 0.7 | 34.0 ± 0.9 |
| Heart rate (bpm) | 481 ± 4.7 | 486 ± 5.7 |
| LV End-diastolic septal wall thickness (mm) | 0.72 ± 0.02 | 0.72 ± 0.01 |
| LV End-diastolic posterior wall thickness (mm) | 0.65 ± 0.01 | 0.66 ± 0.01 |
| LV End-diastolic diameter (mm) | 3.1 ± 0.02 | 3.1 ± 0.01 |
| LV End-systolic diameter (mm) | 2.0 ± 0.02 | 2.1 ± 0.02 |
| Myocyte cross-sectional area (µm2) | 328 ± 15 | 329 ± 16 |
| Apoptosis positive cells (‰ Nuclei) | 0.2 ± 0.07 | 0.2 ± 0.03 |
| Collagen (% surface) | 3.5 ± 0.9 | 3.4 ± 0.7 |
Overexpression of VCP in a TG mouse model reduces IS during IR
To test whether VCP is cardioprotective in vivo, 3 to 4-month old WT and TG mice were submitted to a 45 min ischemia followed by a 24 hours reperfusion as described in the Methods and illustrated in Fig. 2a. Although the area-at-risk (AAR) was similar, there was a 50% reduction of IS in TG mice compared with WT (p = 0.006 vs. WT, Fig. 2b and c), providing a cardioprotection against IR that is comparable to that observed in SWOP16,17.
Figure 2. Overexpression of VCP reduces myocardial infarct size in TG mice.
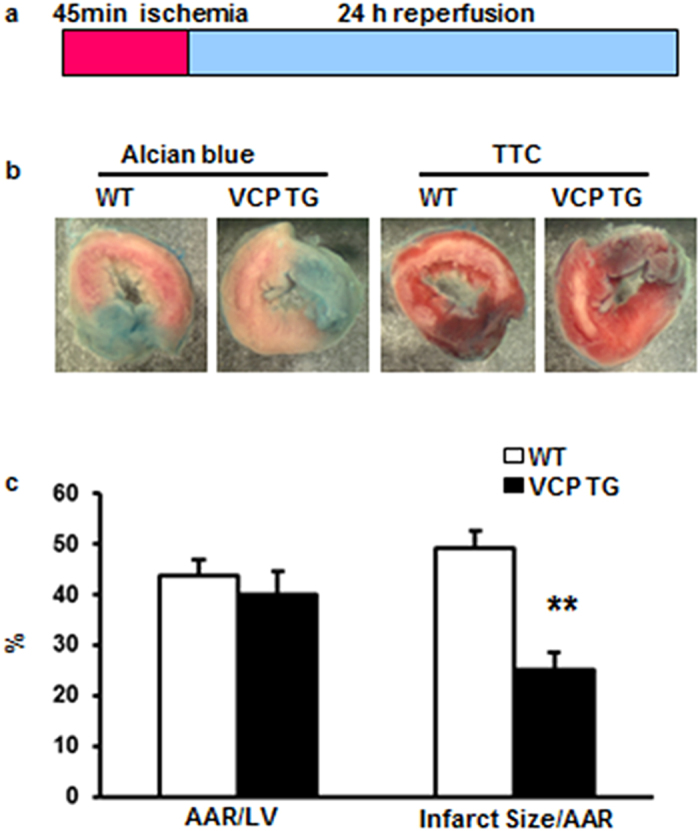
(a) Schema of the ischemia/reperfusion protocol. (b) Representative staining of left ventricle by Alcian blue and Triphenyl Tetrazolium Chloride (TTC). (c) Quantitation of AAR/LV and IS/AAR in WT and TG mouse hearts (n = 5 for TG and 7 for WT group). Data are the mean ± SEM. **p = 0.0006 vs. WT.
Mitochondrial distribution of VCP and iNOS was increased in VCP TG hearts
Our previous study showed that not only the increase of total endogenous iNOS expression but also its subsequent translocation to the mitochondria in cardiac myocytes is crucial for its effects on both mitochondrial respiration and cardioprotection18. To further test this concept in VCP TG mouse, subcellular fractions were extracted from both VCP TG and WT mouse hearts as described in the Methods. To determine the cellular distribution of iNOS as accurately as possible, the purity of each cellar fraction was confirmed by specific protein markers (Fig. 3a). As shown in Fig. 3b, VCP and iNOS exhibited a similar subcellular distribution in which both VCP and iNOS were localized primarily in the mitochondria in both WT and TG mouse hearts. Compared with WT, TG mice showed a 3.1-fold increase in VCP and 4.5-fold increase in iNOS in the mitochondrial fraction (P < 0.01 vs. WT, Fig. 3c and d). iNOS activity in isolated mitochondria from VCP TG mouse hearts was also increased by 2.1-fold compared to WT mice, indicating its functional activity in heart mitochondria in VCP TG mice (Fig. 3e).
Figure 3. Sub-cellular distribution of VCP and iNOS in mouse heart.

(a) Specific markers show the purity of the subcellular fractions: voltage-dependent anion channel (VDAC) for mitochondria (Mit); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for cytosol (Cyto), histone 1 for nucleus (Nu). (b) Representative subcellular distribution of VCP and iNOS in WT and VCP TG mouse hearts. GAPDH was used as a marker to exclude the contamination of cytosal in mitochondrial fraction. (c) Immunoblot of the expression of VCP and iNOS in mitochondrial fraction in TG and WT mouse hearts. VDAC was used as loading control of mitochondria. Negative staining of GAPDH and histone1 confirm the purity of mitochondrial fractions. (d) Ratio of VCP/VDAC and iNOS/VDAC in mitochondria in VCP TG mice compared to WT. n = 7/group. *p < 0.01 vs WT. (e) iNOS activity in isolated mitochondria from mouse hearts. n = 5/group. *p = 0.02 vs WT. Data are the mean ± SEM.
ADP-dependent oxygen consumption was enhanced in VCP in TG mouse heart
Since the majority of VCP is localized in mitochondria, we tested the effect of VCP on mitochondrial respiration in the heart. We first tested the complex I-dependent mitochondrial respiration by measuring oxygen consumption in the presence of pyruvate and malate, two Complex I substrates, with and without ADP, as illustrated in Fig. 4a. Respiration rates of states 2 to 4 were determined by the oxygen consumption per minute normalized by mitochondrial proteins. As shown in Fig. 4b, although there was a modest increase in state 2 respiration rate in the mitochondria from VCPTG mice vs WT, the difference did not reach statistical significance. Mitochondria from VCP TG mouse heart tissues showed a significant increase in oxygen consumption at the ADP-dependent state 3 under complex I stimulation compared to WT, while no significant difference was seen in oxygen consumption states 4 (upon the addition of oligomycin, a known inhibitor of the ATP synthase) (Fig. 4b). The efficiency of mitochondrial respiration, as measured by the respiratory control ratio (RCR: state 3/state 4) was significantly increased in VCP TG mice versus WT (Fig. 4c). In addition, VCP TG mice also exhibited a significant increase in maximum respiration capacity versus WT as measured after the addition of the uncoupling agent carbonyl cyanide 4-trifluoro methoxyphenylhydrazone (FCCP) (Fig. 4d).
Figure 4. Effects of VCP overexpression on mitochondrial respiration.

(a) An representative plot of relative oxygen concentration from each group (% of initial oxygen concentration nmol/ml). (b) Measurement of mitochondrial respiration rate (states 2 to 4) of mouse heart tissue measured by Clark electrode. State 2 and state 4 represent the substrate-dependent respiration rate measured under the addition of pyruvate and malate in the absence of exogenous ADP; State 3 represents ADP-stimulated respiration rate measured under the addition of ADP; (c) Efficiency of mitochondrial respiration as measured by the respiratory control ratio (RCR: State3/State4). (d) Maximum respiration capacity as measured after the addition of the uncoupler FCCP. n = 6 per group for b to d. *p < 0.05, **p < 0.01 vs. WT; #p < 0.01 vs. VCP TG. (e) Representative tracing of oxygen consumption rate of isolated mitochondria from mouse hearts during a stepwise protocol (plots 1 to 7 indicating the order of treatment as 1. glutamate/malate, 2. ADP, 3. cytochrome c, 4. succinate, 5. rotenone, 6. inhibitor of cytochrome oxidase, KCN and 7. oligomycin). (f) Average oxygen consumption rates of panel e. n = 4–5 per group *p < 0.01 vs. WT; #p < 0.01 vs. VCP TG. (g) iNOS activity and (h) Mitochondrial oxygen consumption rate (OCR) in RNCMs with or without iNOS inhibitor, 1400 W. n = 6/group for g to h. *p < 0.05 vs. Ad-ß-Gal; #p < 0.05 vs. Ad-VCP. Data are the mean ± SEM.
In addition, we tested the effect of VCP on complex II dependent respiration in the isolated mitochondria by adding succinate, a known Complex II substrate, in the presence of ADP, with and without the Complex I inhibitor rotenone. As shown in Fig. 4e and f, there was a remarkable increase in respiration rate from both WT and VCP TG mice after the addition of succinate compared to Complex I alone, suggesting that respiration under Complex II was additive. However, the difference of respiration rates between VCP TG and WT observed under stimulation of Complex I (Plot 2 in Fig. 4e and f) lost significance upon the simulation of Complex I and II after the addition of succinate (Plot 4 in Fig. 4e and f). We tested Complex II-specific respiration by adding the Complex I inhibitor, rotenone, and found that there was no longer a difference in respiration rate between VCP TG and WT (Plot 5 in Fig. 4e and f). These data together suggest that VCP may be predominantly affecting Complex I dependent respiration in VCP TG mice.
To determine whether the increase of oxygen consumption in VCP TG mice was the result of enhanced mitochondrial respiration, we used Cytochrome C to test the integrity of the mitochondrial membrane. As shown in Plot 3 of Fig. 4e and f, addition of cytochrome C did not increase the respiration rates of either group’s samples, indicating a high membrane integrity. Next, we added potassium cyanide (KCN), a known inhibitor of cytochrome oxidase. Our data showed that addition of KCN inhibited the respiration rates for both WT and VCP TG in a similar manner, indicating a direct effect on mitochondrial respiratory chain (Plot 6 in Fig. 4e and f). Moreover, there were no significant difference observed between VCP TG and WT upon the addition of oligomycin after the inhibition of cytochrome oxidase by KCN (Plot 7 in Fig. 4e and f).
Genetic deletion of iNOS abolished VCP-mediated increase in mitochondrial respiration in vivo
We have shown that VCP dose-dependently increases the expression of iNOS in myocytes15. However, it is unknown if iNOS mediates the effect of VCP on mitochondrial respiration. To further determine whether the stimulation of mitochondrial respiration observed in VCP TG mouse heart is mediated by iNOS in vivo, a bigenic VCP TG/iNOS KO−/− mouse was generated, in which iNOS was deleted from VCP TG mice (see Methods for mating strategy). Oxygen consumption was measured in these VCP TG/iNOS KO−/− mice and compared with their litter-matched VCP TG/iNOS+/+ mice and WT/iNOS+/+ counterparts. As shown in Fig. 4a to c, the increase in mitochondrial respiration state 3 and in RCR observed in VCP TG mouse heart was abolished upon deletion of iNOS. There was also a significant decrease in maximum respiration capacity as measured after the addition of the uncoupling agent FCCP in VCP TG/iNOS KO−/− mice compared to VCP TG mice (Fig. 4d). These data indicate that iNOS is necessary for the stimulation of respiration by VCP.
iNOS inhibition prevents VCP-induced increase in mitochondrial respiration in isolated cardiomyocyte in vitro
To further determine whether the enhancement of mitochondrial respiration observed in VCP TG mice originated directly from cardiomyocytes, rat neonatal cardiac myocytes (RNCMs) were transfected with adenoviruses harboring the full-length VCP sequence (Ad-VCP) and compared to the Ad-β-Gal control (Ad-β-Gal). iNOS activity was increased by 2-fold upon the overexpression of VCP versus β-Gal. Addition of iNOS inhibitor 1400 W abolished VCP-mediated activation of iNOS (Fig. 4g). Consistent with the change in VCP TG mice, compared to Ad-β-Gal, RNCMs treated with Ad-VCP exhibited a 2.2-fold increase in oxygen consumption rates (OCR), which represents an index of mitochondrial respiration capacity. This effect was abolished by addition of the iNOS inhibitor, 1400 W (Fig. 4h). Therefore, these results in cultured cardiomyocytes offer further support to the results obtained from genetically-modified mice showing that the effect of VCP on mitochondrial respiration is dependent upon increased iNOS activity.
Over-expression of VCP prevents mPTP opening
In order to further investigate the protective role of VCP in IR, we next examined its potential effect on the mPTP opening, which is a critical mechanism to promote cell damage during IR, particularly at the early stage of reperfusion3. Mitochondria were isolated from the heart of WT, VCP TG and VCP TG/iNOS KO−/− mice. mPTP opening was induced by high concentration of calcium (600 μM CaCl2) and determined by the calcium–overload swelling assay. As showed in Fig. 5a and b, compared to the WT, the mitochondria isolated from the heart of VCP TG mice exhibits a significantly less decrease in absorbance at 540 nm upon the addition of CaCl2, indicating the inhibition of calcium induced mPTP opening. This protection was abolished by the deletion of iNOS in VCP TG/iNOS KO−/− mice (Fig. 5b). Furthermore, addition of cyclosporine A (CsA), a known mPTP inhibitor, prevented the mPTP opening in all the three groups and eliminated the difference between WT and VCP TG (Fig. 5a and b). These data further support that VCP protects the mitochondria from the calcium-load induced mPTP opening.
Figure 5. Effect of VCP overexpression on mPTP opening.

(a) Representative examples of calcium-loaded swelling assay with or without the presence of Cyclosporine (CsA). (b) Quantitative representation of panels a. n = 5 per group *p < 0.05 vs. WT; #p < 0.05 vs. VCP TG. (c) Representative examples of mitochondrial calcein fluorescence after addition of FCCP at the concentration of 1 μmol/L (1) or 10 μmol/L (10) in RNCMs. (d) Representative tracing curve of calcein fluorescence over time in the same groups. (e) Quantitative representation of panels d. n = 11 to 22/group. Data are the mean ± SEM. ^p < 0.05 vs. corresponding control, *p < 0.01 vs. Ad-β-Gal, #p < 0.01 vs. Ad-VCP.
mPTP opening was also determined in isolated cardiomyocytes by a reduction in mitochondrial calcein fluorescence signal after addition of FCCP, which is an uncoupler of the mitochondrial respiratory chain, at the concentration of 1 μmol/L or 10 μmol/L in myocytes treated for 48 hours with Ad-β-Gal and Ad-VCP with or without the iNOS inhibitor 1400 W (Fig. 5c). Calcein fluorescence signaling was traced over time after addition of FCCP (Fig. 5d). Compared to the untreated control, the calcein fluorescence signal in Ad-β-Gal-treated RNCMs declined upon FCCP treatment (p < 0.05), reflecting mPTP opening. RNCMs treated with Ad-VCP preserved the same calcein fluorescence signal as control cells upon FCCP stimulation, indicating that VCP prevented the induced mPTP opening in RNCMs. This prevention conferred by VCP was abolished by the addition of the iNOS inhibitor 1400 W (Fig. 5e). Therefore, increased expression of iNOS by VCP is necessary for the reduction in mPTP pore opening in cardiomyocytes.
Discussion
As presented in Fig. 6, our results demonstrate that over-expression of VCP in a genetically-modified mouse model in vivo provides cardiac protection in ischemic heart equivalent to that provided by the SWOP16,17; This effect is linked to a preservation of the capacity of mitochondrial respiration and a prevention of mPTP opening under the stress; The mechanistic link between VCP-mediated cardioprotection and preservation of mitochondrial function under the cardiac stress is an increased expression and activity of iNOS. Taken together, these results demonstrate that VCP is a novel agonist of iNOS-mediated mechanisms of cardioprotection against ischemia.
Figure 6. Summary of the findings.
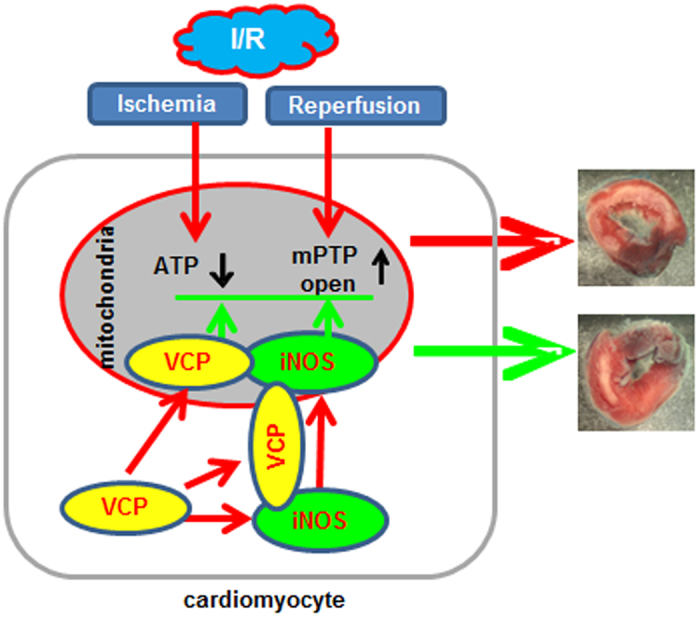
A schematic presentation of VCP action showing the potential mechanism of action of VCP.
Very little information is available about the function of VCP in the heart, at the opposite of other tissues. VCP is a member of the type I AAA (ATPases associated with various cellular activities). By interacting with several sets of adaptor proteins19, VCP is involved in a variety of cellular pathways that are essential for cell homeostasis, such as cell cycle control, transcriptional regulation, apoptosis, protein degradation, and cellular stress response20,21,22,23. Despite this information gained from other tissues, the physiology of VCP in the heart remains largely unknown. We first identified VCP in the heart when we showed that it acts as a novel downstream effector of the cardioprotective signaling mechanism conferred by Hsp2215. Our initial studies in vitro showed that VCP represents the link between Hsp22-mediated activation of Akt and nuclear factor-kappa B (NF-ĸB)-induced expression of iNOS in cardiac myocytes, thereby playing a central role in the mechanisms of cardiac cell survival promoted by Hsp22. We also showed that overexpression of VCP protects cardiomyocytes against apoptosis15. Based upon this evidence obtained in vitro, we tested the physiological relevance of our findings in vivo by generating a cardiac-specific VCP TG mouse.
In the present study, we identified for the first time a specific physiological function of VCP in the mammalian heart, in both normal and stress conditions. Our results show that no change was observed in TG mouse compared with WT in baseline conditions when considering heart mass, ventricular structure, contractile function, and the size and viability of cardiomyocytes. These data demonstrate that chronic stimulation of VCP at this overexpression level has no toxic effects on the heart tissue. Thus, the VCP TG mouse provides an original biological tool for the investigation of the mechanisms underlying VCP-mediated actions on cardiac stress.
A major observation of the present study is that overexpression of VCP protects the heart against IR-induced damage. Acute MI remains one of the leading causes of morbidity, mortality and disability worldwide1. Early and successful reperfusion is the most effective strategy for reducing the IS and for improving the clinical outcome. However, the process of restoring blood flow to the ischemic myocardium can create injury itself, for which no effective therapy is currently available5,24. IPC is well recognized as the most powerful endogenous mechanism of cardioprotection, however the clinical translation of IPC remains limited because of the problematic requirement of pre-emptive ischemic episodes16,17,24,25. Our results defined VCP as a new and powerful mediator of cardiac protection during IR, demonstrating that VCP may represent a novel therapeutic avenue for the prevention of MI.
Cardiomyocyte death induced by IR is the major cause of myocardial infarct and loss of cardiac function. VCP has been identified as an apoptosis regulator in other mammalian cells and in yeast26,27,28, Recent studies have identified that loss of VCP activity due to mutations is linked to cell death in different human diseases, including a variety of neurodegenerative diseases, such as Alzheimer’s disease, Pakinson’s disease and amyotrophic lateral sclerosis26,29,30. VCP has also been shown to be associated with cell survival of cancer cells31,32,33,34. We demonstrated previously the protective role of VCP against apoptosis in mammalian cardiomyocytes in vitro15. However, whether this anti-apoptotic effect of VCP protects the heart against ischemia-induced cell death remained unclear. Our data further show that overexpression of VCP by 3.5-fold is sufficient to provide prophylactic cardioprotection against cell death in the heart under ischemic stress.
Another important finding in the present study is a novel role for VCP in mitochondria of the mammalian heart. Previous mechanistic studies on VCP-mediated cell survival focus on its involvement in endoplasmic reticulum (ER) stress-triggered apoptotic pathway35,36. VCP deficiency is associated with decreased mitochondrial membrane potential in human dopaminergic neuroblastoma cell line37,38,39. It has been shown that mutations or deficiency of VCP cause profound mitochondrial dysfunction which results in a significant reduction of ATP synthesis, making neuronal cells more vulnerable to ischemia and cell death37,38,39. However, the potential role of VCP in cardiac mitochondria has not been tested. We first showed that, in a mouse model, VCP exhibits a preferred accumulation in cardiac mitochondria as compared to other sub-cellular fractions, which highlights the possibility of an important mitochondrial function for VCP under stress conditions. This is supported by our observation that VCP TG mice exhibit a significant elevation of the capacity of mitochondrial respiration in the state 3, with no change in states 2 and 4. State 3 is defined as ADP-stimulated respiration. When ADP binds ATP synthase, protons are driven into the matrix from the outside of the inner membrane. The energy released by this proton flux directly drives ATP synthesis. Therefore, an increase in state 3 respiration indicates an increased capacity of ATP synthesis6, while states 2 and 4 are viewed as the steady states of mitochondrial respiration. Therefore, an increase in RCR, which reflects the state 3/state 4 ratio, is an excellent indicator of improved respiratory activity and efficiency6. Additionally, the increase of oxygen consumption was blocked by the inhibitor of cytochrome oxidase, KCN, further supporting the enhancement of mitochondrial respiratory chain by VCP. These data together indicates that overexpression of VCP promotes mitochondrial respiration efficiency, which enhances cellular resistance to oxidative damage, and thus may act as a mechanism of the prevention of cardiomyocyte death during ischemia6.
There is also increasing evidence that mPTP opening plays a central role in mediating both the necrotic and apoptotic components of IR injury particularly at the onset of reperfusion3,12,40,41. Importantly, it has been shown that IPC attenuates IR-induced opening of the mPTP12. Recent studies also demonstrated that the use of mPTP opening inhibitors, such as cyclosporin A, reduces IS in animal models of acute IR injury41. Despite intensive investigation, the molecular identity of the mPTP remains undefined. Our findings showed that overexpression of VCP reduced mPTP opening, indicating that VCP targets an important mitochondrial regulator of cardioprotection against reperfusion injury during IR.
Our next observation is that VCP regulates mitochondrial function in an iNOS-dependent manner. The effect of VCP on mitochondria is related to iNOS for the following reasons: (1) VCP induces the expression of iNOS not only in cardiac myocytes in vitro15 but also in the heart of VCP TG mice in vivo. (2) The cytoprotection conferred by VCP was abolished by the addition of a selective iNOS inhibitor15. (3) iNOS is necessary for Hsp22-related mitochondrial regulation and cytoprotection18. To further investigate if the effect of VCP on mitochondrial function is mediated through the increase of iNOS expression, both pharmacological and genetic approaches were used. We first generated a bigenic mouse in which iNOS was deleted from the VCP TG mouse. This model confirms that enhanced mitochondrial respiration by VCP overexpression in the mouse heart was abolished by the deletion of iNOS. Importantly, VCP exhibited no stimulatory effect on another NOS isoform, such as eNOS. In addition, we also showed in isolated RNCMs that VCP overexpression results in stimulation of mitochondrial respiration, an effect which was eliminated upon treatment with the iNOS inhibitor 1400 W. Although the precise mechanism of VCP on mitochondrial function is largely unknown, our results imply that iNOS is a crucial mediator of such effect. Furthermore, it has been shown previously that mitochondria extracted from iNOS TG mouse hearts subjected to IR have a decreased mPTP opening compared to WT42. We demonstrated that overexpressing VCP decreased the rate of mPTP opening, and that such effect was completely abolished by the deletion or inhibition of iNOS, further indicating that iNOS is the critical mediator of the effect of VCP in the inhibition of mPTP opening.
Although the role of iNOS in cardioprotection has been firmly established, the effect of NO in the mitochondria has been controversial. Indeed, the detrimental effect of NOS on mitochondrial respiration were observed in several conditions related to excessive formation of NO43,44, such as during hypoxia45,46,47 or under treatment with exogenous NO stimulators48. However, our data clearly show that the moderate increase of endogenous iNOS by 2- to 3-fold, which reproduces the extent of overexpression during SWOP, provides cardiac protection during IR injury and promotes mitochondrial respiration. These data are also consistent with our previous observation in a Hsp22 TG mouse model18,49 as well as in ischemic preconditioning50. In addition, different conclusions were made when comparing the increased iNOS expression originating from resident inflammatory cells51 or from circulatory blood cells52 compared to the iNOS generated by cardiac myocytes52. Also, we recently showed that it, instead of the total cellular iNOS content; it is rather the abundance of iNOS in the mitochondria that is crucial for Hsp22-mediated stimulation of oxidative phosphorylation18. Accordingly, the present study shows that iNOS has a preferred location in mitochondria of VCP TG mouse heart, which further supports the concept that stimulation of endogenous iNOS expression and its subsequent translocation to the mitochondria in cardiac myocytes is crucial for its effects on both mitochondrial respiration and cardioprotection. This may also explain the divergent results observed in previous studies in which NO production originated from “non-myocyte” iNOS47,52 or from exogenous NO donors48. The cellular origin and subcellular distribution of iNOS are likely major determinants of its beneficial effect against ischemic injury and mitochondrial respiration. The VCP TG mouse that we generated provides a biological tool for the further study of the mechanisms underlying endogenous iNOS mediated protection.
In summary, the data presented in this study provide the following novel information in the heart in vivo: (1) Cardiac-specific overexpression of VCP in a TG mouse did not change the overall physiological and morphological characteristics of the mouse in baseline conditions. (2) The VCP TG mouse provides a significant reduction in IS during IR injury, which is quantitatively comparable to that observed in IPC. (3) Overexpression of VCP in the heart in vivo leads to an increase in endogenous iNOS expression and activity. (4) Increased iNOS by VCP shows a preferential localization to the mitochondria in TG mouse heart. (5) VCP overexpression leads to an increase in mitochondrial respiratory capacity and an inhibition of the rate of mPTP opening, which are dependent upon iNOS expression and activity. Altogether, these data further highlight a novel cardioprotective mechanism for VCP in vivo. Therefore, pre-emptive activation of VCP and its downstream targets may represent a novel approach to prevent ischemic damage in the heart at risk of suffering from subsequent ischemic stress.
Methods
Animal Models
Generation of the VCP TG mouse
A construct harboring a 2.4 Kb coding sequence of VCP was generated, in which the transgene expression was under the control of the cardiac-specific promoter of the α-myosin heavy chain (αMHC). The plasmid was digested with Bam HI, and introduced by pronuclear microinjection in zygotes of FVB mice, as described previously53. Positive mice were mated with wild type, and their offspring were screened.
Generation of the VCP TG/iNOS KO bigenic mouse
VCP TG mice (FVB) were cross mated with a homozygous iNOS KO−/− (C57BL/6, Jackson Laboratory, stock Number: 002609). After 5 generations of background clearing, F1 generation (VCP TG/iNOS KO+/− and WT/iNOS KO+/−) (FVB) mice were mated together to generate a VCP TG/iNOS KO−/− homozygote (FVB). Litter-matched VCP TG (VCP TG/iNOS+/+) (FVB) and WT (WT/iNOS+/+) (FVB) were used as controls.
Three to four month-old mice, both male and female, were used for this study. Animals were euthanized with 100 mg/kg pentobarbital. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85-23, revised 2011), and with the approval of the IACUC committee at the local institutions (Rutgers, The State University of New Jersey, and Loma Linda University).
Culture of RNCMs
RNCMs were prepared from Sprague-Dawley rat pups (Charles River Laboratories, Wilmington, MA) as described previously15,18,49,54. Recombinant adenoviruses (Ad) harboring the full-length VCP sequence (Ad-VCP) or β-Gal control (Ad-β-Gal) was generated with the AdEasy XL adenoviral vector system (Agilent, Santa Clara, CA). Myocytes were infected with Ad-VCP and Ad-β-Gal for 48 hours after 24 hours of serum-free starvation. Inhibition of iNOS was initiated 24 hours before the collection of the cells upon addition of 100 μmol/L 1400 W (Sigma-Aldrich, St. Louis, MO)15,18.
Echocardiography
Cardiac function and LV structure were measured in the VCP TG mice and WT littermates by two-dimensional echocardiography as described previously49. After determination of body weight, mice were anesthetized with 2% isoflurane (JD Medical, AZ). Echocardiography was performed using a Logiq E vet with a 13-MHz probe (12L-RS). Heart rate, LVEDD and LVESD, wall thickness, and contractility (EF and FS) were measured.
Surgical procedure
IR was performed on both VCP TG and WT mice and the infarct area and the area at risk were measured as described previously55. Briefly, mice were anesthetized with ketamine 65 mg/kg, xylazine 1.2 mg/kg, and acepromazine 2.17 mg/kg intraperitoneally and ventilated via tracheal intubation connected to a rodent ventilator with a tidal volume of 0.2 ml and a respiratory rate of 110 breaths per minute. The heart was exposed through a thoracotomy and the left anterior descending (LAD) artery was located and was occluded for a period of 45 min to induce ischemia followed by 24 hours of reperfusion. At the end of each experiment, the LAD was re-ligated to determine the area at risk by low-pressure retrograde injection of 0.5 mg/ml Alcian blue through the aorta. The infarct size was measured following incubation in 1% Triphenyl Tetrazolium Chloride (TTC) at 37 °C for 15 min. Image-Pro software was used to measure and analyze of the infarct area and the area at risk from each section.
Histopathology
Hearts form both VCP TG and WT were collected and weighed. The LV/BW, LV/TL and LW/TL ratios were measured. Heart samples were fixed in 10% formalin and cut into 7-μm-thick sections. Collagen accumulation, myocyte cross-sectional area and myocyte apoptosis were measured as described previously49.
qPCR and Western blotting
mRNA was extracted from mouse heart tissues and qPCR was performed as described previously49,54. Protein extraction and subcellular fractions were performed as described previously49,54. Targeted proteins including VCP, eNOS and iNOS were detected by western blotting as previously described15,18. The primary antibodies used for western blotting were anti-VCP (catalog no. 2648S, rabbit, dilution of 1:1000, Cell Signaling Technology), anti-iNOS (catalog no. ab136918, rabbit, dilution of 1:1000, Abcam), and anti-eNOS (catalog no. Sc-654, rabbit, dilution of 1:1000, Santa Cruz Biotechnology). Anti-GAPDH (Catalog no. ab9484, mouse, dilution of 1:10,000, Abcam) and anti- voltage-dependent anion-selective channel protein 1(VDAC) (Catalog no. 4866, rabbit, dilution of 1:5,000, Cell Signaling Technology) were used as loading controls.
iNOS activity assay
iNOS activity was measured with the Nitric Oxide (NO) Assay Kit (Oxford Biomedical Research, Oxford, UK) as described previously18.
Mitochondrial isolation
Mitochondria were isolated from mouse heart as described previously15,18,49. Briefly, sub-cellular fractions were prepared by differential centrifugation of tissue homogenized manually in a Dounce homogenizer using hypotonic buffer. After an initial spin at 100 g (5 minutes) to discard the cellular debris and unbroken cells, the nuclear fraction was pelleted at low-speed centrifugation (500 g, 10 minutes). The supernatant was further centrifuged (10,000 g, 10 minutes) to pellet the mitochondrial fraction. The resulting supernatant was ultra-centrifuged (100,000 g, 90 minutes) to obtain the cytosolic fraction (supernatant) and a microsomal fraction (pellet). Pellets were washed 3 times with 1x phosphate-buffered saline and re-suspended in RIPA buffer (150 mM NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0))15,18,49. The purification of the isolated mitochondria from heart tissue was verified by western blotting, using glyceraldehyde 3-phosphate dehydrogenase (GAPDH, cytosol), histone 1 (nucleus), and voltage-dependent anion channel (VDAC, mitochondria) as described previously15,18,49.
Oxygen consumption assay
Oxygen consumption of isolated mitochondria was measured at 30 °C with a Clark-type electrode fitted to a water-jacketed reaction chamber of 0.5 mL. Mitochondria (0.3–0.4 mg/mL) were isolated from mouse heart and incubated in a respiration buffer containing 100 mmol/L KCl, 50 mmol/L sucrose, 10 mmol/L HEPES, 5 mmol/L KH2PO4, pH 7.4. The following parameters of mitochondrial respiration were evaluated49,56: 1. Substrate-dependent respiration rate (state 2 and state 4 with 5 mmol/L pyruvate or 10 mmol/l glutamate/5 mmol/L malate) in the absence of exogenous ADP; 2. ADP-stimulated respiration rate (state 3) in presence of 250 mmol/L ADP; 3. Oxygen-consumption with the addition of following reagents: 5 mmol/L succinate in the absence or in the presence of rotenone (2 μmol/L); cytochrome C (4 μmol/L); KCN (50 μmol/L) and oligomycin (10 μmol/L); 4. Respiration uncoupling in presence of 0.2 mmol/L FCCP.
Oxygen consumption rates (OCR) were also measured in intact RNCMs with a Clark-type electrode as described previously18. Briefly, RNCMs infected with Ad-β-Gal and Ad-VCP were collected and suspended in extracellular buffer as described18,57. Glucose was used as a substrate. OCR was determined after addition of 6 μmol/L oligomycin or 5 μmol/ FCCP. The ratio of FCCP-stimulated to oligomycin-inhibited OCR in the myocytes was calculated18.
Mitochondrial swelling assays
Mitochondria were isolated as described above. mPTP opening was induced by calcium loading via addition of 600 μM of CaCl2. The mitochondrial swelling was monitored by the decrease in light-scattering at 540 nm in a spectrophotometer (Thermo Scientific Multiskan GO) every 6 second for 10 minutes in the presence or absence of 10 μM Cyclosporin A58.
mPTP opening assay in intact cardiomyocytes
mPTP opening was determined by analyzing the mitochondrial calcein leak as previously described59,60. Briefly, RMCMs were loaded with 1 μmol/L calcein-AM and 1 mmol/L CoCl2 for 30 min at room temperature, and calcein fluorescence quenching was measured. mPTP opening was evaluated after addition of 1 μmol/L or 10 μmol/L FCCP, which uncouples the mitochondrial respiratory chain, and depolarizes mitochondrial membrane potential. Opening of mPTP promotes mitochondrial calcein quenching. During the whole experiments, the loaded cells were excited at 470 nm and the emitted light was collected at 510 nm. The mPTP opening was indicated by a reduction in mitochondrial calcein fluorescence signal.
Statistical analysis
Results are the mean ± SEM for the number of samples indicated in the figure legends. A one-way ANOVA was used, and a Student-Newman-Keuls post hoc correction was applied for multi-group comparisons. A value of p < 0.05 was considered significant.
Additional Information
How to cite this article: Lizano, P. et al. The valosin-containing protein is a novel mediator of mitochondrial respiration and cell survival in the heart in vivo. Sci. Rep. 7, 46324; doi: 10.1038/srep46324 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
This work is partially supported by a grant 1R01 HL115195-01 from NIH (H.Q.) and a grant 0835182N from American Heart Association (H.Q.).
Footnotes
The authors declare no competing financial interests.
Author Contributions H.Q., P.L., E.R., S.S. and C.D. conceived and designed the study, analyzed the data and wrote the manuscript; P.L. and N.Z. designed, performed and analyzed the experiments shown in Figures 1 and 2 and Table 1. S.S., E.R. and G.Q. designed, performed and analyzed the experiments shown in Figure 4. S.S., E.R., H.W. and L.X. designed, performed and analyzed the experiments shown in Figure 5. N.Z., S.S. and T.H. designed, performed and analyzed the experiments shown in Figure 3. All authors discussed and analyzed the results and reviewed and approved the final version of the manuscript.
References
- Mozaffarian D. et al. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 131, e29–322 (2015). [DOI] [PubMed] [Google Scholar]
- Lesnefsky E. J., Moghaddas S., Tandler B., Kerner J. & Hoppel C. L. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol 33, 1065–1089 (2001). [DOI] [PubMed] [Google Scholar]
- Ong S. B., Samangouei P., Kalkhoran S. B. & Hausenloy D. J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 78, 23–34 (2015). [DOI] [PubMed] [Google Scholar]
- Hollander J. M., Thapa D. & Shepherd D. L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 307, H1–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Rodriguez A., Abreu-Gonzalez P. & Reiter R. J. Cardioprotection and pharmacological therapies in acute myocardial infarction: Challenges in the current era. World J Cardiol 6, 100–106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestanello J. A. et al. Mitochondrial function during ischemic preconditioning. Surgery 131, 172–178 (2002). [DOI] [PubMed] [Google Scholar]
- Wojtovich A. P., Nadtochiy S. M., Brookes P. S. & Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol 47, 1–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell L. S. & Brookes P. S. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal 10, 579–599 (2008). [DOI] [PubMed] [Google Scholar]
- Opie L. H. & Sack M. N. Metabolic plasticity and the promotion of cardiac protection in ischemia and ischemic preconditioning. J Mol Cell Cardiol 34, 1077–1089 (2002). [DOI] [PubMed] [Google Scholar]
- Kadenbach B., Ramzan R., Moosdorf R. & Vogt S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion 11, 700–706 (2011). [DOI] [PubMed] [Google Scholar]
- Suleiman M. S., Halestrap A. P. & Griffiths E. J. Mitochondria: a target for myocardial protection. Pharmacol Ther 89, 29–46 (2001). [DOI] [PubMed] [Google Scholar]
- Ong S. B., Dongworth R. K., Cabrera-Fuentes H. A. & Hausenloy D. J. Role of the MPTP in conditioning the heart - translatability and mechanism. Br J Pharmacol 172, 2074–2084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. & Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78, 100–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A. P. & Richardson A. P. The mitochondrial permeability transition: a current (2015). [DOI] [PubMed] [Google Scholar]
- Lizano P. et al. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc Res 99, 685–693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol 292, H19–27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R. et al. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res 81, 1094–1107 (1997). [DOI] [PubMed] [Google Scholar]
- Rashed E. et al. Heat Shock Protein 22 (Hsp22) Regulates Oxidative Phosphorylation upon Its Mitochondrial Translocation with the Inducible Nitric Oxide Synthase in Mammalian Heart. PloS one 10, e0119537 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol 156, 29–40, doi:S1047-8477 (2006). [DOI] [PubMed] [Google Scholar]
- Egerton M. et al. VCP, the mammalian homolog of cdc48, is tyrosine phosphorylated in response to T cell antigen receptor activation. EMBO J 11, 3533–3540 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasure I. T., Black M. M. & Keen J. H. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature 365, 459–462 (1993). [DOI] [PubMed] [Google Scholar]
- Rabinovich E., Kerem A., Frohlich K. U., Diamant N. & Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22, 626–634 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. & Latterich M. The AAA team: related ATPases with diverse functions. Trends Cell Biol 8, 65–71 (1998). [PubMed] [Google Scholar]
- Ibanez B., Heusch G., Ovize M. & Van de Werf F. Evolving Therapies for Myocardial Ischemia/Reperfusion Injury. J Am Coll Cardiol 65, 1454–1471 (2015). [DOI] [PubMed] [Google Scholar]
- Khan A. R. et al. Cardioprotective role of ischemic postconditioning in acute myocardial infarction: a systematic review and meta-analysis. Am Heart J 168, 512-521 e514 (2014). [DOI] [PubMed] [Google Scholar]
- Braun R. J. & Zischka H. Mechanisms of Cdc48/VCP-mediated cell death: from yeast apoptosis to human disease. Biochim Biophys Acta 1783, 1418–1435 (2008). [DOI] [PubMed] [Google Scholar]
- Shah P. P. & Beverly L. J. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 6, 17725–17737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun R. J. et al. Crucial mitochondrial impairment upon CDC48 mutation in apoptotic yeast. J Biol Chem 281, 25757–25767 (2006). [DOI] [PubMed] [Google Scholar]
- Ishigaki S. et al. Physical and functional interaction between Dorfin and Valosin-containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J Biol Chem 279, 51376–51385 (2004). [DOI] [PubMed] [Google Scholar]
- Mizuno Y., Hori S., Kakizuka A. & Okamoto K. Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci Lett 343, 77–80 (2003). [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. et al. Elevated expression of valosin-containing protein (p97) is associated with poor prognosis of prostate cancer. Clin Cancer Res 10, 3007–3012 (2004). [DOI] [PubMed] [Google Scholar]
- Yamamoto S. et al. Expression level of valosin-containing protein (p97) is correlated with progression and prognosis of non-small-cell lung carcinoma. Ann Surg Oncol 11, 697–704 (2004). [DOI] [PubMed] [Google Scholar]
- Yamamoto S. et al. Elevated expression of valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence. J Clin Oncol 21, 447–452 (2003). [DOI] [PubMed] [Google Scholar]
- Yamamoto S. et al. Increased expression of valosin-containing protein (p97) is correlated with disease recurrence in follicular thyroid cancer. Ann Surg Oncol 12, 925–934, doi: 10.1245/ASO.2005.07.002 (2005). [DOI] [PubMed] [Google Scholar]
- Lim P. J. et al. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol 187, 201–217 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch E. et al. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 4 (2002). [DOI] [PubMed] [Google Scholar]
- Fang L. et al. Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front Cell Neurosci 9, 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Peng G., Wang Y., Fang S. & Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol Biol Cell 22, 291–300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome F. et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 341 (Pt 2), 233–249 (1999). [PMC free article] [PubMed] [Google Scholar]
- Orzi F., Castri P. & Fornai F. Apomorphine as a neuroprotective drug: a study in MPTP-treated mice and potential relevance to ischemia. Funct Neurol 16, 153–158 (2001). [PubMed] [Google Scholar]
- West M. B. et al. Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial permeability transition. Circulation 118, 1970–1978 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandy P. & Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol 138, 532–543 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungrue I. N. et al. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Invest 109, 735–743 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poderoso J. J. et al. Nitric oxide regulates oxygen uptake and hydrogen peroxide release by the isolated beating rat heart. The American journal of physiology 274, C112–119 (1998). [DOI] [PubMed] [Google Scholar]
- Borutaite V., Moncada S. & Brown G. C. Nitric oxide from inducible nitric oxide synthase sensitizes the inflamed aorta to hypoxic damage via respiratory inhibition. Shock 23, 319–323 (2005). [DOI] [PubMed] [Google Scholar]
- Li W., Jue T., Edwards J., Wang X. & Hintze T. H. Changes in NO bioavailability regulate cardiac O2 consumption: control by intramitochondrial SOD2 and intracellular myoglobin. Am J Physiol Heart Circ Physiol 286, H47–54 (2004). [DOI] [PubMed] [Google Scholar]
- Roberts B. W., Mitchell J., Kilgannon J. H., Chansky M. E. & Trzeciak S. Nitric oxide donor agents for the treatment of ischemia/reperfusion injury in human subjects: a systematic review. Shock 39, 229–239 (2013). [DOI] [PubMed] [Google Scholar]
- Qiu H. et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 124, 406–415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Ischemic preconditioning upregulates inducible nitric oxide synthase in cardiac myocyte. J Mol Cell Cardiol 34, 5–15 (2002). [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H., Zhu L. & Beckman J. S. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys 298, 446–451 (1992). [DOI] [PubMed] [Google Scholar]
- Guo Y. et al. Identification of inducible nitric oxide synthase in peripheral blood cells as a mediator of myocardial ischemia/reperfusion injury. Basic Res Cardiol 107, 253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depre C. et al. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ Res 91, 1007–1014 (2002). [DOI] [PubMed] [Google Scholar]
- Qiu H. et al. Characterization of a novel cardiac isoform of the cell cycle-related kinase that is regulated during heart failure. J Biol Chem 283, 22157–22165 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depre C. et al. H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ Res 98, 280–288 (2006). [DOI] [PubMed] [Google Scholar]
- Lanza I. R. & Nair K. S. Functional assessment of isolated mitochondria in vitro. Methods in enzymology 457, 349–372 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. D., Gaspers L. D., Wang G. & Thomas A. P. Uncoupling protein-2 modulates myocardial excitation-contraction coupling. Circ Res 106, 730–738 (2010). [DOI] [PubMed] [Google Scholar]
- Hafner A. V. et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z. et al. Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PloS one 8, e80574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad T. et al. Mechanisms involved in postconditioning protection of cardiomyocytes against acute reperfusion injury. J Mol Cell Cardiol 58, 209–216 (2013). [DOI] [PubMed] [Google Scholar]