

Abstract
Genetic maps in linkage disequilibrium (LD) units play the same role for association mapping as maps in centimorgans provide at much lower resolution for linkage mapping. Association mapping of genes determining disease susceptibility and other phenotypes is based on the theory of LD, here applied to relations with three phenomena. To test the theory, markers at high density along a 10-Mb continuous segment of chromosome 20q were studied in African-American, Asian, and Caucasian samples. Population structure, whether created by pooling samples from divergent populations or by the mating pattern in a mixed population, is accurately bioassayed from genotype frequencies. The effective bottleneck time for Eurasians is substantially less than for migration out of Africa, reflecting later bottlenecks. The classical dependence of allele frequency on mutation age does not hold for the generally shorter time span of inbreeding and LD. Limitation of the classical theory to mutation age justifies the assumption of constant time in a LD map, except for alleles that were rare at the effective bottleneck time or have arisen since. This assumption is derived from the Malecot model and verified in all samples. Tested measures of relative efficiency, support intervals, and localization error determine the operating characteristics of LD maps that are applicable to every sexually reproducing species, with implications for association mapping, high-resolution linkage maps, evolutionary inference, and identification of recombinogenic sequences.
Gene localization is based on four maps, each with additive distances. Two of these maps are physical, the high-resolution genome map in base pairs (bp) and the low-resolution cytogenetic map in chromosome bands of estimated physical lengths. The other two maps are purely genetic, the linkage map in Morgans or centimorgans (cM) and the map of linkage disequilibrium (LD) in LD units (LDU), which approximates the product of the sex-averaged linkage map and the effective number of generations since a major bottleneck. The primary utility of LD maps is for association mapping of unsequenced determinants of disease susceptibility or other phenotypes, but they also provide unique information about crossing over, selective sweeps, and population history. Linkage maps specifying order were introduced by Sturtevant (1), and the distances were made approximately additive by Haldane (2). Most geneticists are familiar with subsequent refinements and use of linkage, which was introduced to human genetics by Bernstein (3) and subsequently evolved into current maps reliable to a resolution of ≈1 cM or ≈1 Mb (4, 5). On the contrary, LD maps at much higher resolution depend critically on a reliable DNA sequence, first available for the human genome 2 years ago (6). On this framework, various approaches and substitutes for an LD map have been proposed, the evaluation of which depends on aspects of population genetics that we consider here. The data consist of 47 founders in Centre d'Etude du Polymorphisme Humain (CEPH) families, 96 U.K. Caucasians, 97 African Americans, 10 Chinese, and 32 Japanese samples. Single-nucleotide polymorphisms (SNPs) at a density of ≈1 per 2 kb were typed along a 10-Mb continuous segment of chromosome 20q12–13.2. These samples and the genotyping and error checking to which they were submitted are described elsewhere (7). In some analyses, the samples were pooled into two groups, African American and Eurasian, which were analyzed both separately and pooled as “cosmopolitan.” The mixture of theory, data, and analysis is different for population structure, effective bottleneck time, and allele frequency, which have measurably different impacts on LD. We have therefore treated each of these topics in a different section.
LD and Population Structure
Methods. Population structure reflected by inbreeding in contemporary populations is a possible source of error in LD mapping because common methods to infer haplotypes in ostensibly unrelated individuals assume random mating, and therefore markers significantly deviant from Hardy–Weinberg proportions are often rejected as likely errors. Inbreeding, although less than in the past, is not negligible in many populations. For a particular population, let F0 denote the maximal inbreeding coefficient in generation 0, after which a higher immigration rate m and larger effective population size N prevailed. In the next generation, the expected inbreeding was F1 = (1 – m)2 [1/2N + (1 – 1/2N)F0]. At equilibrium under constant m and N, the expected inbreeding will be L = 1/(1 + 4Nm). In generation t, the expected inbreeding is Ft = (1 – L)Me–2mt + L, where M = (F0 – L)e–t/2N/(1 – L) and Ft approaches L if t ≫ 1/(2m + 1/2N). Then, the impact of F0 is lost, M = –Le–t/2N/(1 – L), and Ft approximates L[1 – e–t/2NL], the form it takes when F0 = 0 (8). L is usually small, and the approach to equilibrium is rapid (9, 10). Inbreeding is defined with respect to a particular population and increases as that reference is enlarged. Wright (11) provided the hierarchical model FIT = FIS + (1 – FIS) FST, where FIS is inbreeding relative to a population S and its allele frequencies, FST measures the divergence among such populations, and FIT is inbreeding relative to the collective and its allele frequencies. Then, FST may be estimated as (FIT – FIS)/(1 – FIS), where FIS is the mean among populations that are pooled for FIT.
In a random sample from a particular population, the expected frequency of a heterozygote between alleles or haplotypes with frequencies qi,qj is 2qiqj(1 – F) and the expected frequency of the ith homozygote is qi2(1 – F) + qiF, where F is either FIS or FIT as appropriate (11). For the general case, we used the implementation of Gomes et al. (12) in the BETA suite (http://cedar.genetics.soton.ac.uk/public_html). However, in small samples with an uncommon allele, the frequency of a rare genotype may by chance be 0. This outcome is most serious in the diallelic case, where there are three parameters (minor allele frequency x, sample size n, and inbreeding F). If x exceeds 0, but the frequency of the corresponding homozygote is 0, the estimate of F is –x/(1 – x), and so the information matrix for x and F becomes singular (Table 1). Therefore, F may be estimated if the allele frequency in the population is known without error, and vice versa, but simultaneous estimation has indeterminate error. Omission of these samples greatly overestimates F when x is small. This problem was not recognized by earlier workers.
Table 1. Inbreeding analysis for a diallelic locus: general and special case.
| Expected f
|
∂ Inf/∂X = UX
|
∂ Inf/∂F = UF
|
|||||
|---|---|---|---|---|---|---|---|
| Genotype | Observed | General | n22 = 0 | General | n22 = 0 | General | n22 = 0 |
| 11 | n11 | (1 - x)2 + x(1 - x)F | 1 - 2x | 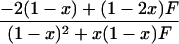 |
 |
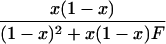 |
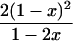 |
| 12 | n12 | 2x(1 - x)(1 - F) | 2x | 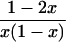 |
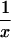 |
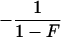 |
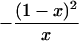 |
| 22 | n22 | x2 + x(1 - x)F | 0 | 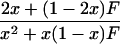 |
0 | 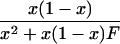 |
0 |
| Total | n | 1 | 1 | ∑fU 0 | 0 | 0 | 0 |
| n22 = 0 | ∑fUxUF = -(1 - x)2 | 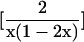 |
∑fU2 | — | 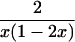 |
— | 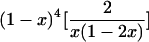 |
A rigorous theory that allows for this and other biases in F would be welcome. In its absence, we analyzed diallelic SNPs, excluding estimates for x < 0.08, where most of the indeterminacies occur. Then, for each value of x ≥ 0.08, we replaced the maximum likelihood score ΣU by ΣU + k/2 with nominal information ΣK, where k is the number of SNPs with a 0 frequency for one of these genotypes. SNPs with 0 frequencies for two genotypes were excluded. The adjustment of F by k/2ΣK approximates F/2n, the bias correction suggested by Robertson and Hill (13). It becomes very small in these samples at about x = 0.15, reaching 0 at about x = 0.28. The smaller the sample, the greater this adjustment. Within a narrow band of x (taken to be 0.02, and, therefore, with midpoints 0.09, 0.11,..., 0.49) the estimate of F from s SNPs is (ΣU + Σk/2)/ΣK with variance v estimated as [Σ(U2/K) – (ΣU + Σk/2)2/ΣK]/(s – 1). To obtain SE and in regression, F is weighted by information estimated as W =ΣK/v if v >1 and by ΣK otherwise. When values of x are pooled, weighted by W, the variance V of F among x classes may be greater and then the SE of F is taken to be 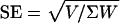 . The quantity (F/SE)2 approximates χ2 but corresponds more precisely when x is small to Fisher's F test, confusing in the context of inbreeding.
. The quantity (F/SE)2 approximates χ2 but corresponds more precisely when x is small to Fisher's F test, confusing in the context of inbreeding.
Results. All five samples and five groups have residual variance V among values of x that is greater than the variance v among SNPs within x values. We attribute this outcome to the autocorrelation of x values among neighboring SNPs, especially within blocks, which inflates the residual variance for LD mapping (14, 15). Allowing for V, there is no evidence against the null hypothesis that FIS is 0 within the four Eurasian groups, whether the scores are pooled (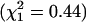 ) or tested separately (
) or tested separately (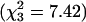 ), although the negative value of FIS in the CEPH sample is barely significant when tested by (F/SE)2 = 4.68 (Table 2). This deviation disappears when estimates of F are pooled with the U.K. sample. The pooled value of FIS = 0.0014 is in close agreement with other regional studies (16). However, when the Chinese and Japanese samples are pooled into a single Asian sample the value of (F/SE)2 = 8.49 is highly significant, which is in agreement with other evidence (17). FIS within the two samples is nonsignificant, and the deviations in the CEPH and Japanese samples are of doubtful significance, given their consistency with complementary samples in the same region. On the contrary, the African Americans give highly significant evidence against the null hypothesis with (F/SE)2 = 35.56, and heterogeneity with the Eurasian groups is highly significant (
), although the negative value of FIS in the CEPH sample is barely significant when tested by (F/SE)2 = 4.68 (Table 2). This deviation disappears when estimates of F are pooled with the U.K. sample. The pooled value of FIS = 0.0014 is in close agreement with other regional studies (16). However, when the Chinese and Japanese samples are pooled into a single Asian sample the value of (F/SE)2 = 8.49 is highly significant, which is in agreement with other evidence (17). FIS within the two samples is nonsignificant, and the deviations in the CEPH and Japanese samples are of doubtful significance, given their consistency with complementary samples in the same region. On the contrary, the African Americans give highly significant evidence against the null hypothesis with (F/SE)2 = 35.56, and heterogeneity with the Eurasian groups is highly significant (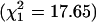 ). This result undoubtedly reflects stratification due to a combination of introgressive hybridization from the parental groups with assortative mating for phenotype and cultural background, a phenomenon that has been studied in northeastern Brazil (18). There has been no comparable research on African Americans, and the selection of our sample is too uncertain to speculate about the relative contributions of assortative mating or isolation by distance to their value of FIS, which is slightly less than the estimate of FIT in Asians, arguing against an important effect of null alleles caused by primer polymorphism in Africans. This is critical evidence because diallelic markers do not distinguish null alleles from inbreeding. Isolate breaking is associated with improved transportation, and its demonstrated effect on inbreeding began a few generations ago with the Industrial Revolution. As expected from this short history, regression of inbreeding on x within samples is nonsignificant (coefficient = –0.004 ± 0.015, P = 0.8).
). This result undoubtedly reflects stratification due to a combination of introgressive hybridization from the parental groups with assortative mating for phenotype and cultural background, a phenomenon that has been studied in northeastern Brazil (18). There has been no comparable research on African Americans, and the selection of our sample is too uncertain to speculate about the relative contributions of assortative mating or isolation by distance to their value of FIS, which is slightly less than the estimate of FIT in Asians, arguing against an important effect of null alleles caused by primer polymorphism in Africans. This is critical evidence because diallelic markers do not distinguish null alleles from inbreeding. Isolate breaking is associated with improved transportation, and its demonstrated effect on inbreeding began a few generations ago with the Industrial Revolution. As expected from this short history, regression of inbreeding on x within samples is nonsignificant (coefficient = –0.004 ± 0.015, P = 0.8).
Table 2. Inbreeding in different samples.
|
FIS
|
FIT
|
|||||
|---|---|---|---|---|---|---|
| Sample | Mean | SE | (F/SE)2 | Mean | SE | (F/SE)2 |
| Caucasian | 0 | 0.0026 | 0.00 | 0.0032 | 0.0033 | 0.95 |
| Asian | 0.0065 | 0.0038 | 2.97 | 0.0169 | 0.0058 | 8.49 |
| Eurasian | 0.0014 | 0.0021 | 0.44 | 0.0449 | 0.0037 | 147.86 |
| Cosmopolitan | 0.0069 | 0.0018 | 14.37 | 0.0734 | 0.0038 | 376.92 |
| U.K. | 0.0036 | 0.0040 | 0.78 | |||
| CEPH | -0.0060 | 0.0028 | 4.68 | |||
| Chinese | -0.0021 | 0.0068 | 0.09 | |||
| Japanese | 0.0095 | 0.0046 | 4.22 | |||
| African American | 0.0165 | 0.0028 | 35.36 | |||
Generalizing from alleles to haplotypes, which have the same values of F, we conclude that departure from random mating may be neglected for the two Caucasian samples and the Chinese and Japanese samples if their diplotypes are kept separate. However, the African-American, Asian, Eurasian, and cosmopolitan diplotype samples should be disaggregated into more homogeneous samples whose haplotype frequencies may be estimated separately and then pooled if desired. Disaggregation may be by stated ancestry, phenotype, or marker frequencies as appropriate. With this precaution, inbreeding in these populations is not a problem for LD mapping, and there is no significant relationship with allele frequency. Rare genotypes in populations with preferential consanguineous marriage raise a problem unless haplotype frequencies are estimated conditional on F.
LD and the Effective Bottleneck Time
Methods. A physical map of I markers, with distance di in the ith interval between markers i and i + 1, has length Σdi for i = 1,..., I – 1. Usually, di is reported as kb with three decimal places. The corresponding distance in the linkage map is wi Morgans (usually expressed as 100wi cM) with length Σwi = RΣdi, where R = Σwi/Σdi is the ratio of lengths in the linkage and physical maps. An approximate estimate of R over a larger or smaller distance gives the least reliable estimate of wi as diR, by using no information about crossing over in the ith interval. Although there is considerable difference between linkage maps for eggs and sperm, autosomal values are often sex-averaged. These relations are useful to interpolate estimates from a very large sample of meioses in sperm to female and sex-averaged maps (19). At present, meiotic data are available only for a few short sequences in males. Coalescent and LD maps (both sex-averaged by necessity) can also be used to interpolate distances into small intervals of a low-density linkage map, conserving intervals established by linkage. The distance in an LD map is approximately wit, where t (assumed constant for a particular population) is the number of generations since LD began to decline from a bottleneck when effective size was reduced by mortality, migration, selective sweep, or other factors (15, 20). The estimate of t does not depend on association at bottleneck time. As yet, there is no experience with these methods to increase resolution of the linkage map, which is useful for genome scans and refining candidate regions by linkage but useless for association mapping unless t is inferred from an LD map. This result severely limits both meiotic data (t = 1) and coalescent methods that scale recombination, not by t but by effective population size (21).
Time has a great many applications in population genetics, each of different span but relevant to LD or the polymorphisms that define it. Kimura and Ohta (22) derived an expression for the mean age t of a polymorphism with minor allele frequency x in the current population. Their result (following a suggestion by A. Robertson from ref. 2) may be written as t = 4Nγ, where N is effective population size, t is measured in generations, and γ = – [x ln x + (1 – x)ln(1 – x)]. The corresponding time in years is T = gt, where g is the mean generation time in years. No assumption is made about whether the currently rarer allele is younger, but their derivation assumed neutrality, no more than two alleles segregating in any generation, random mating, an effective size great enough so that the distribution of x among loci reaches a steady state, and no error in estimating x. Even under these constraints, the variance of t is very large. Their solution was suggested earlier by Watterson (23) for the mean time until extinction of a polymorphism, which under these assumptions is identical to its mean age (24). Most estimates of nominal N range from 10,000 to 20,000, with g between 20 and 25 years (25). By using the term thousand years ago (kya) (17), we tentatively assume Ng = 250 kya, which agrees with other evidence (17, 26). Most alleles with frequencies of <0.02 have arisen since migration out of Africa, whereas many alleles with frequencies >0.05 antedate our species (Table 1). Unfortunately, there is no precise and independent estimate of t to make a more rigorous test of the model. Estimates on the evolutionary scale from coalescent theory have been disappointingly variable (27).
The problems become more serious when recombination between two markers is introduced. The time required to go halfway to equilibrium in a closed population of effective size N and recombination rate θ per generation depends on the mutation rate μ. Assuming that μ is negligible compared with θ and 1/2N, that n = 10,000, and there is no selection, the halfway time if g = 25 years per generation is predicted to be T = 25(ln 2)/(θ + 1/2N) years (28). Genes separated by 1 cM have T = 2 kya, whereas genes separated by 0.0001 cM (≈0.1 kb) have T = 340 kya (Table 2). Over such time spans, a steady state undisturbed by population bottlenecks is unimaginable. Bottlenecks are central to the evolutionary ideas of Wright (ref. 29, p. 215): “Every deme at any given time has a history of passage through a great many bottlenecks of small numbers on being traced back from place to place, and because a few momentarily flourishing demes may be the source from which many new colonies are founded, large areas or even the whole species may, in the course of time, trace to a single deme that has passed through many bottlenecks.” Conquest, slavery, and admixture of populations with different fertilities, especially with persistent stratification because of nonrandom mating, are three mechanisms that can reduce effective size without necessarily reducing census size.
Results. A general theory for multiple bottlenecks has eluded population geneticists, but the special case of a pair of diallelic markers and a single bottleneck at which founders had association ρ0 gives the recurrence ρt = (1 – μ)(1 – θ) [1/2Nt–1 + (1 – 1/2Nt–1)ρt–1] with solution ρt = (1 – L)Me–θt + L, where ρt is the association probability in the tth generation after the bottleneck, M = (ρ0 – L)e–(μ+1/2N)t/(1 – L), where N is the harmonic mean of the Nj for j = 1,..., t, and L is the asymptote as Me–θt approaches 0 (20). This representation is called a Malecot model because the recurrence uses methods introduced by Malecot (30, 31) and leads to a form he derived for isolation by distance and other problems with different parameters. For example, substituting μ = 0, θ = m, and ρ = F gives the inbreeding formula derived in the preceding section. Only small values of θ contribute to LD, as shown in Table 3, and so θ and distance Σwi in Morgans are interchangeable. Likewise, recombination in a small interval is proportional to distance, and so θt may also be expressed as tΣwi =Σεidi, where εi = twi/di. One LDU corresponds to Σεidi = 1, which is proportional to both chromosome distance and time. For association mapping, εidi is much more useful than twi, because di is known and εi may be estimated directly, whereas t is unknown and a linkage map is at far too low resolution to estimate wi for an LD map (21). Applications have been made to association mapping of rare major genes (32) and oligogenes (33), population differences (14), construction of LD maps (15), and proof that ρ fits pairwise LD better than other metrics (20), and that conservation of haplotype diversity by selection of single SNPs does not retain power (34). Applications before invention of LD maps (15) used the kb map and assumed that εi is constant (ε). Because that is not true, the kb map does not fit LD nearly as well as an LD map, but Σεidi/Σdi ≈ ε.
Table 3. Halfway time T kya to LD equilibrium.
| Recombination θ | cM | Nominal kb | T |
|---|---|---|---|
| 10-6 | 0.0001 | 0.1 | 340 |
| 10-5 | 0.001 | 1 | 289 |
| 10-4 | 0.01 | 10 | 116 |
| 10-3 | 0.1 | 100 | 17 |
| 10-2 | 1 | 1,000 | 2 |
Extrapolating from chromosomes 6, 21, and 22, samples from large Eurasian populations suggest ≈59,000 LDU (F.M. de la Vega, unpublished work) in an autosomal, euchromatic genome of 34.36 Morgans, implying 59,000/34.36 = 1,717 generations or ≈43 kya to the hypothesized bottleneck. This is less than half the time to migration out of Africa, suggesting that lesser bottlenecks have subsequently contributed to LD, in accordance with Wright's insight (29). It is therefore appropriate to call the LDU/Morgan ratio the effective bottleneck time, by analogy with the effective population size. LD maps and genome length in LD, yet to be determined precisely, are the relevant parameters for association mapping. However, tentative inferences can be made about evolutionary time, even if t in the Malecot model corresponds to the effective bottleneck time for multiple bottlenecks of different magnitude. If migration out of Africa is assigned to 100 kya, a major bottleneck in Homo sapiens (perhaps but not necessarily speciation) can be dated to ≈100 times the ratio of LD map length in Africans and Eurasians, or ≈174 kya (14), in good agreement with the first fossil evidence of our species dated to 157 ± 3 kya (26), and in support of the hypothesis that multiple bottlenecks, although not explicit in the Malecot model, are accurately reflected by it.
LD and Allele Frequency
Methods. Some authors have expressed concern that multiple bottlenecks might create a relation between allele frequency and the Malecot parameters that mimics the prediction for mutation of single markers (7, 35). Suppose that a mutation increases in a local population (deme) by direct selection, LD with a selected marker, or drift. The small effective size of a deme favors rapid change in gene frequency. Subsequent expansion of that deme may by chance or selection give a mean age much smaller than the predictions in Table 4, which are for a larger population, without considering their high variance. Under a more complex scenario, many alleles with frequencies of >0.02 could have arisen subsequent to migration out of Africa or any other major bottleneck. This result would make t increase with x, and to that extent violate the assumptions about LD of both coalescent and Malecot models. The strength and limitations of these models cannot be evaluated without access to the computer programs that implement them, of which a critical one does not estimate time and is presently unavailable (21). Another coalescent approach has a location error and support interval twice as great as the Malecot model (36, 37). To orient a comprehensive comparison, we investigate LD in pairs of SNPs, classified by minor allele frequency x. The obvious ways of doing this select on both SNPs, for example, by rejecting all SNPs with x less than some value. The association probability ρ satisfies conditions on both members of a pair. If the haplotype counts in their 2 × 2 table are  with a + b + c + d = n, the conditions are ad – bc ≥ 0 and b ≤ c, implying that x = (a + b)/n cannot be selected without imposing a lower limit to c and thereby failing to model random sampling of the second SNP with allele frequencies (a + c)/n and (b + d)/n. In LD mapping, each SNP is paired with a second SNP without regard to its frequency. To approach such randomness under the constraint of classification by x, we assigned each pair on the basis of the minor allele frequencies, either drawing one at random or assigning both to their respective class or classes, taken as 0–0.01, 0.01–0.02, etc. For both sampling schemes and for each class, the mean minor allele frequency for the other SNP was close to the sample mean (0.23–0.25) with no trend, which is consistent with random selection. As usual, each value of ρ was weighted by its nominal information Kρ to give χ21 = ρ2Kρ under the null hypothesis and a composite likelihood with residual variance
with a + b + c + d = n, the conditions are ad – bc ≥ 0 and b ≤ c, implying that x = (a + b)/n cannot be selected without imposing a lower limit to c and thereby failing to model random sampling of the second SNP with allele frequencies (a + c)/n and (b + d)/n. In LD mapping, each SNP is paired with a second SNP without regard to its frequency. To approach such randomness under the constraint of classification by x, we assigned each pair on the basis of the minor allele frequencies, either drawing one at random or assigning both to their respective class or classes, taken as 0–0.01, 0.01–0.02, etc. For both sampling schemes and for each class, the mean minor allele frequency for the other SNP was close to the sample mean (0.23–0.25) with no trend, which is consistent with random selection. As usual, each value of ρ was weighted by its nominal information Kρ to give χ21 = ρ2Kρ under the null hypothesis and a composite likelihood with residual variance 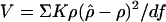 , where
, where 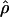 is an estimate with predicted value ρ under the Malecot model (20), V is minimized for ε and M with L predicted (15), and degrees of freedom (df) is the difference between the number of pairs and the number of parameters estimated. A map in LDU fits association data much better than either a high-resolution physical map or a linkage map that cannot reflect the LD pattern at higher resolution (14, 38). However, when ρ is partitioned by minor allele frequency x, the density is greatly reduced and the LD map becomes unreliable. For this unusual problem, we therefore fit the Malecot model to the physical map, pooling the very small Chinese sample with Japanese as “Asian.”
is an estimate with predicted value ρ under the Malecot model (20), V is minimized for ε and M with L predicted (15), and degrees of freedom (df) is the difference between the number of pairs and the number of parameters estimated. A map in LDU fits association data much better than either a high-resolution physical map or a linkage map that cannot reflect the LD pattern at higher resolution (14, 38). However, when ρ is partitioned by minor allele frequency x, the density is greatly reduced and the LD map becomes unreliable. For this unusual problem, we therefore fit the Malecot model to the physical map, pooling the very small Chinese sample with Japanese as “Asian.”
Table 4. Mean age T kya for a polymorphism with current minor allele frequency x and Ng = 250 kya.
| x | T | Concurrent event |
|---|---|---|
| 0.50 | 693 | Homo habilis → Homo erectus |
| 0.05 | 199 | Homo erectus → Homo sapiens |
| 0.02 | 98 | Out of Africa |
The public version of our program ldmap has several improvements over earlier versions applied to maps that were small or at low resolution (15, 38). To reduce computing time, the marker pairs are selected not to exceed a specified kb length and number of intervening markers, defaulted to 500 and 100, respectively. These analyses were performed by the ldmap program at http://cedar.genetics.soton.ac.uk/public/html. The estimates of y = ε or –ln M were fitted to regression models with 2 df for estimated parameters α and β. The four models were linear on x (y = α + βx), linear on the γ parameter of Kimura and Ohta (22) (y = α + βγ), increasing exponential on x (y = α[1 – e–βx], β > 0), and decreasing exponential on x (y = α[1 + e–βx], β > 0). The linear models increase if β > 0 and decrease otherwise, the latter being contrary to expectation, like β < 0 for the exponential models. Significance is tested by Fisher's F1, r–2 as (r – 2) (SS0/SS1 – 1), where SS0, SS1 are the residual sum of squares for the constant model and the alternative, respectively, and r is the number of minor allele classes.
Results. The residual variance for estimates of ε follows the same pattern in every sample. The worst fit is for the model of constant ε, and the best fit is for an exponential increasing model (Table 5). The ratio of residual variances for worst and best models is a measure of relative efficiency, which is least for African Americans. The γ model of Kimura and Ohta (22) fits poorly, as expected from its foundation on mutational age rather than time after a bottleneck. Significance tests against the null hypothesis of a constant model reveal the same pattern, with the greatest difference for African Americans (Fig. 1). The exponent β for the U.K. sample is significantly greater than for the rest (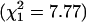 ), but residual variation in β is nonsignificant (
), but residual variation in β is nonsignificant (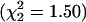 ).
).
Table 5. Residual variance of 105V with four models.
| Linear | Linear | Exponential x
|
|||||
|---|---|---|---|---|---|---|---|
| Dependent variable | Sample | Relative efficiency, worst/best | Constant | x | γ | Increasing | Decreasing |
| ε | African American | 0.32 | 2.75 | 2.13 | 1.58 | 0.89 | 2.30* |
| Asian | 0.54 | 2.04 | 1.80 | 1.49 | 1.10 | 1.90* | |
| U.K. | 0.60 | 1.24 | 1.13 | 0.98 | 0.74 | 1.10* | |
| CEPH | 0.54 | 1.76 | 1.57 | 1.37 | 0.95 | 1.60* | |
| —InM | African American | 0.81 | 1,294 | 1,114* | 1,053* | 1,257 | 1,050 |
| Asian | 0.95 | 661 | 657* | 643* | 665 | 634 | |
| U.K. | 0.97 | 419 | 428* | 423* | 414 | 427 | |
| CEPH | 0.96 | 502 | 495* | 480* | 494 | 480 | |
β < 0.
Fig. 1.

Best-fitting two-parameter model for ε (increasing exponential) in four samples. AA, African American.
The quantity –lnM shows a different and more complicated pattern. The increasing exponential model is superior to the constant model in only one sample and inferior to the decreasing exponential model in three samples of four (Table 5). These small differences are reflected in significance tests, with African Americans giving the strongest evidence for a decreasing exponential model (Table 6). The exponential rate of change is smaller than for ε, making the γ model appear to fit almost as well as the decreasing exponential. However, in every case the estimate of β is negative, contrary to the mutational model, which we may therefore reject unconditionally as an explanation for the relation of LD to allele frequency and, therefore, time. Significance tests are equivocal compared with ε, but again the African Americans give the strongest evidence against constancy of the dependent variable and in favor of a more ancient bottleneck than Eurasians (Fig. 2).
Table 6. Significance tests F1,df on four regression models vs. constant model.
| Linear | Linear | Exponential x
|
|||
|---|---|---|---|---|---|
| Dependent variable | Sample | x | γ | Increasing | Decreasing |
| ε | African American | 15.34 | 37.27 | 103.66 | 11.27* |
| Asian | 7.44 | 18.53 | 41.40 | 5.71* | |
| U.K. | 6.02 | 13.82 | 34.34 | 4.96* | |
| CEPH | 6.75 | 14.64 | 41.75 | 5.60* | |
| —InM | African American | 8.93* | 12.21* | 2.43 | 12.61 |
| Asian | 1.29* | 2.31* | 0.67 | 2.99 | |
| U.K. | 0.01* | 0.53* | 1.58 | 0.01 | |
| CEPH | 1.74* | 3.24* | 1.77 | 3.26 | |
β < 0.
Fig. 2.

Best-fitting two-parameter model for –lnM (decreasing exponential) in four samples. AA, African American.
Discussion
Slatkin (39) wrote that “Kimura and Ohta's (22)... paper on allele age led to a rich theoretical literature but, until recently, few applications. The reason is that, in a population of constant size, the distribution of ages is so broad that little information about age is provided by allele frequency.” This generalization is certainly true, reflecting the preoccupation of theoretical genetics with tests of neutral mutation theory for a large number of nucleotides over evolutionary time. Age estimation for a single allele gives errors “too high for these methods to be reliably used in practice” (27). However, errors are controlled when a great many diallelic polymorphisms within narrow bands of allele frequencies are considered. By using this approach, we have shown that the theory of Kimura and Ohta (22) does not describe LD, contrary to the conclusions of several authors (7, 35). This outcome is hardly surprising because the theory deals with time since the first mutation that has not yet become extinct, whereas LD (like inbreeding) depends on the much shorter time since a founder population. If and when a test of the model is made on mutation age, their assumptions (exact population frequency, neutrality, equilibrium, lack of population subdivision, and constant population size) may prove too stringent for real populations.
Although the theory of Kimura and Ohta (22) clearly does not fit LD, small allele frequencies are associated with low ε, indicative of low frequency before migration out of Africa or later by mutation, followed by slow dispersal from one or more local populations (demes). On the contrary, small allele frequencies have high –lnM, with expectation approximately t/2N if the initial association ρ0 approached 1 (20). This finding suggests long persistence in a population of small effective size N, preceding expansion to other populations as described for many isolates (40).
It is characteristic of these and most other studies on population structure that the samples, were poorly specified. The Coriell Institute (Camden, NJ), which is the custodian and distributor of anonymous DNA samples, describes them in terms like “self-declared Caucasians who are unrelated.” The populations and grandparental origins are unidentified and in principle could include participants from Iceland to Bangladesh and from Lapps to Moroccans, and it is inconceivable that all n(n – 1)/2 pairs have been questioned about relationship (violating anonymity) or would be well informed about it. African Americans have a complex and undescribed structure. The CEPH sample is a mixture of Mormon volunteers from Utah. The Asian sample pools Chinese and Japanese, creating significant stratification and not distinguishing north and south populations with different histories and allele frequencies (17). Failure to make the samples representative of a defined population is a less serious problem than that most of the world is not sampled. How to handle this variation in location databases and genetic analysis is one of the disputed problems of the scientific community that includes hapmap (41). Cosmopolitan maps that include several populations offer an efficient solution because they may be scaled by the Malecot model to the density and LD of a particular sample (14). This procedure recovers nearly all information in the sample, and the small remainder can be recovered by simultaneous estimation of the εi, beginning with the values in the cosmopolitan map instead of a scaled physical map that corresponds much less well to LD. This principle may also be used to compare models of different complexity (for example, with L predicted and estimated), of low and high density, or from less and more credible genetic models.
Neglecting approaches not applied since the physical map was nominally finished, three alternatives to LD maps have been proposed, all by using haplotypes. One is non-Bayesian and uses logistic regression based on a similarity dendrogram to select the most significant set of s SNPs when s varies from 4 to 10 and the haplotypes are defined on overlapping windows (42). The best result is assumed to minimize the Bonferroni-corrected P value, and the causal SNP is estimated to lie at the midpoint of that window on the kb map. Best results were reported in a window of size 6, both in simulation and for the CFTR locus that was mapped by restriction fragment length polymorphisms 15 years ago (43). Localization of these markers on the finished kb map was not attempted, and the example was tentative enough not to be mentioned in the abstract, but it reminds us that association mapping is possible without an LD map if only the most extreme outcome is chosen, but discarding all information from other markers and dispensing with a support interval. In short, their evidence favors small haplotypes over single SNPs for association mapping but does not permit comparison with composite likelihood, coalescent theory, Bayesian methods, and alternatives to a kb map.
The other two alternatives to LD maps based on composite likelihood are at once coalescent, Bayesian, and haplotypic (21, 36). An excellent presentation of coalescent theory concludes that it leads to “estimates of the recombination rate from polymorphism data [that] are extremely unreliable,” with many references to support this conclusion (44). Each population is implausibly assumed to be at equilibrium, differing only in effective population size. It is therefore necessary to scale coalescents either to the sex-averaged linkage map at low resolution or to LD at much higher resolution, making it dependent on a map in LDU. The better a coalescent approximates a linkage model, the less well it represents the selective sweeps, bottlenecks, and stochastic events that characterize LD maps. Because of excessive smoothing that combines blocks with small steps, the coalescent map (21) of part of the HLA region for which a high-density linkage map is available (19) gives nonzero estimates of recombination in long blocks, where no recombination was observed, in contrast with the Malecot map published 2 years earlier (38). This fact is not a proof that coalescence is worse than an LD map, but a reminder that there is no evidence that is as good for representing either linkage or LD, or, more importantly, for localizing disease genes, the purpose for which LD maps were designed and have been shown to function well. Bayesian statistics applied to coalescent models raise other problems. The “prior probabilities” are based on evidence in the sample and are therefore not prior, making the number of degrees of freedom unclear and residual variance ambiguous. It is difficult to compare results with non-Bayesian LD maps based on a defined set of parameters estimated without preconceptions. Objective criteria for this comparison must be sought, although in other situations the Bayesian vs. non-Bayesian conflict remains dogmatic.
In contrast with these largely unexplored methods, the Malecot model predicts recombination and time as the sole determinants of an LD map, which is therefore expected to be proportional to the linkage map and provides an estimate of time that scales the LD map to linkage. Equilibrium is not assumed, and the parameters of mutation rate and effective size do not determine the LD map. Many evolutionary factors disturb this relationship, including the reduced time we have corroborated for alleles that were rare at the effective bottleneck time or have arisen since. Use of composite likelihood makes it easy to compute its relative efficiency on a given set of data in terms of residual variance. Several studies have determined their operating characteristics for association mapping, which would be enhanced with haplotype analysis that estimates an LD location without imposing a genealogy and recognizes that “haplotype map” is an oxymoron. Haplotypes fall into arbitrary haplosets that may be used to annotate a physical map but lack the indispensable additivity that defines a linear map. The proportion of haplotypes that have recombined at a given step over thousands of generations can be as little as 0.02 and is rarely >0.4 (45). Because there is no natural haploset, the length and content of haplotypes is completely arbitrary, to be chosen in a way that optimizes association mapping.
In conclusion, the utility of efforts to improve construction of LD maps or find a more efficient substitute may be measured in five ways: (i) correspondence with the sex-averaged linkage map; (ii) residual variance of alternative LD maps; (iii) constancy of effective bottleneck time over chromosomes with sufficient marker density; (iv) capability to identify systematic departures from the scaled linkage map due to selection and other evolutionary events; and (v) power for association mapping. At present, LD maps based on the Malecot model are unique in providing all these data and therefore are a benchmark against which alternatives may be measured. Whatever the final solution, LD maps and their application to localization of genes for disease susceptibility have progressed in the 2 years since they were introduced. Although many questions remain, it is no longer necessary to respond to misunderstanding as Benjamin Franklin did for one of his inventions: “What is the use of a newborn child?”
Acknowledgments
We thank James F. Crow for helpful comments, including a suggestion of the term “effective bottleneck time.” This work was supported by United Kingdom Medical Research Council Grant GM42947.
Author contributions: W.Z., A.C., J.G., W.J.T., S.H., P.D., D.R.B., and N.E.M. performed research.
Abbreviations: LD, linkage disequilibrium; LDU, LD unit; kya, thousand years ago; SNP, single-nucleotide polymorphism; cM, centimorgan; CEPH, Centre d'Etude du Polymorphisme Humain.
References
- 1.Sturtevant, A. H. (1913) J. Exp. Zool. 14, 43–59. [Google Scholar]
- 2.Haldane, J. B. S. (1919) J. Genet. 8, 299–309. [Google Scholar]
- 3.Bernstein, F. (1931) Z. Indukt. Abstammungs. Vererbungsl. 57, 113–138. [Google Scholar]
- 4.Collins, A., Frezal, J., Teague, J. & Morton, N. E. (1996) Proc. Natl. Acad. Sci. USA 93, 14771–14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong, A., Gudbjartsson, D. F., Sainz, J., Jonsdottir, G. M., Gudjonsson, S. A., Richardsson, B., Sigurdardottir, S., Barnard, J., Hallbeck, B., Masson, G., et al. (2002) Nat. Genet. 31, 241–247. [DOI] [PubMed] [Google Scholar]
- 6.Abramowicz, M. (2003) Adv. Genet. 50, 231.-261, 507–510. [DOI] [PubMed] [Google Scholar]
- 7.Ke, X., Hunt, S., Tapper, W., Lawrence, R., Stavrides, G., Ghori, J., Whittaker, P., Collins, A., Morris, A. P., Bentley, D., et al. (2004) Hum. Mol. Genet. 13, 577–588. [DOI] [PubMed] [Google Scholar]
- 8.Morton, N. E., Harris, D. E., Yee, S. & Lew, R. (1971) Am. J. Hum. Genet. 23, 339–360. [PMC free article] [PubMed] [Google Scholar]
- 9.Imaizumi, Y., Morton, N. E. & Harris, D. E. (1970) Genetics 66, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morton, N. E. (1982) in Current Developments in Anthropological Genetics, eds. Crawford, M. H. & Mielke, J. H., (Plenum, New York), Vol. 2, pp. 449–466. [Google Scholar]
- 11.Wright, S. (1943) Genetics 28, 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomes, I., Collins, A., Lonjou, C., Thomas, N. S., Wilkinson, J., Watson, M. & Morton, N. E. (1999) Ann. Hum. Genet. 63, 535–538. [DOI] [PubMed] [Google Scholar]
- 13.Robertson, A. & Hill, W. G. (1984) Genetics 107, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lonjou, C., Zhang, W., Collins, A., Tapper, W. J., Elahi, E., Maniatis, N. & Morton, N. E. (2003) Proc. Natl. Acad. Sci. USA 100, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maniatis, N., Collins, A., Xu, C.-F., McCarthy, L. C., Hewett, D. R., Tapper, W., Ennis, S., Ke, X. & Morton, N. E. (2002) Proc. Natl. Acad. Sci. USA 99, 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morton, N. E. (1992) Proc. Natl. Acad. Sci. USA 89, 2556–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavalli-Sforza, L. L., Menozzi, P. & Piazza, A., (1994) The History and Geography of Human Genes (Princeton Univ. Press, Princeton).
- 18.Krieger, H., Morton, N. E., Mi, M. P., Azevedo, E., Freire-Maia, A. & Yasuda, N. (1965) Ann. Hum. Genet. 29, 113–125. [DOI] [PubMed] [Google Scholar]
- 19.Jeffreys, A. J., Kauppi, L. & Neumann, R. (2001) Nat. Genet. 29, 217–222. [DOI] [PubMed] [Google Scholar]
- 20.Morton, N. E., Zhang, W., Taillon-Miller, P., Ennis, S., Kwok, P.-Y. & Collins, A. (2001) Proc. Natl. Acad. Sci. USA 98, 5217–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McVean, G. A., Myers, S. R., Hunt, S., Deloukas, P., Bentley, D. R. & Donnelly, P. (2004) Science 304, 581–584. [DOI] [PubMed] [Google Scholar]
- 22.Kimura, M. & Ohta, T. (1973) Genetics 75, 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watterson, G. A. (1962) Ann. Math. Statist. 33, 939–957, and correction (1963) 34, 352. [Google Scholar]
- 24.Watterson, G. A. & Guess, H. A. (1977) Theor. Popul. Biol. 11, 141–160. [DOI] [PubMed] [Google Scholar]
- 25.Harpending, H. C., Batzer, M. A., Gurven, M., Jorde, L. B., Rogers, A. R. & Sherry, S. T. (1998) Proc. Natl. Acad. Sci. USA 95, 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark, J. D., Beyene, Y., WoldeGabriel, G., Hart, W. K., Renne, P. R., Gilbert, H., Defleur, A., Suwa, G., Katoh, S., Ludwig, K. R., et al. (2003) Nature 423, 747–752. [DOI] [PubMed] [Google Scholar]
- 27.Basu, A. & Majumder, P. P. (2003) J. Genet. 82, 7–12. [DOI] [PubMed] [Google Scholar]
- 28.Hill, W. G. & Robertson, A. (1966) Genet. Res. 8, 269–294. [PubMed] [Google Scholar]
- 29.Wright, S. (1969) Evolution and the Genetics of Populations (Univ. of Chicago Press, Chicago), Vol. 2.
- 30.Malecot, G. (1948) Les Mathématiques de l'Hérédité (Masson & Cie, Paris).
- 31.Malecot, G. (1973) in Genetic Structure of Populations, ed. Morton, N. E. (Univ. Press of Hawaii, Honolulu), pp. 72–75.
- 32.Collins, A. & Morton, N. E. (1998) Proc. Natl. Acad. Sci. USA 95, 1741–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maniatis, N., Collins, A., Gibson, J., Zhang, W., Tapper, W. & Morton, N. E. (2004) Am. J. Hum. Genet. 74, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, W., Collins, A. & Morton, N. E. (2004) Hum. Genet. 115, 157–164. [DOI] [PubMed] [Google Scholar]
- 35.Abecasis, G. R., Noguchi, E., Heinzmann, A., Traherne, J. A., Bhattacharyya, S., Leaves, N. I., Anderson, G. G., Zhang, Y., Lench, N. J., Carey, A., et al. (2001) Am. J. Hum. Genet. 68, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris, A. P., Whittaker, J. C., Xu, C.-F., Hosking, L. K. & Balding, D. J. (2003) Proc. Natl. Acad. Sci. USA 100, 13442–13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maniatis, N., Morton, N. E., Gibson, J., Xu, C.-F., Hosking, L. K. & Collins, A. Hum. Mol. Genet., in press. [DOI] [PubMed]
- 38.Zhang, W., Collins, A., Maniatis, N., Tapper, W. & Morton, N. E. (2002) Proc. Natl. Acad. Sci. USA 99, 17004–17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slatkin, M. (2002) in Modern Developments in Theoretical Population Genetics, eds. Slatkin, M. & Veuille, M. (Oxford Univ. Press, Oxford), pp. 233–260.
- 40.Thompson, E. A. & Neel, J. V. (1997) Am. J. Hum. Genet. 60, 197–204. [PMC free article] [PubMed] [Google Scholar]
- 41.Couzin, J. (2004) Science 304, 671–673. [DOI] [PubMed] [Google Scholar]
- 42.Durrant, C., Zandervan, K. T., Cardon, L. R., Hunt, S., Deloukas, P. & Morris, A. P. (2004) Am. J. Hum. Genet. 75, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerem, B., Rommers, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., Buchwald, M. & Tsui, L. C. (1989) Science 245, 1073–1080. [DOI] [PubMed] [Google Scholar]
- 44.Nordborg, M. (2001) in Handbook of Statistical Genetics, eds. Balding, D. J., Bishop, M. & Cannings, C. (Wiley, Chichester, U.K.), pp. 179–208.
- 45.Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J. & Lander, E. S. (2001) Nat. Genet. 29, 229–232. [DOI] [PubMed] [Google Scholar]