

Abstract
Purpose
When integrating molecularly targeted compounds in radiotherapy, synergistic effects of the systemic agent and radiation may extend the limits of patient tolerance, increasing the demand for understanding the pathophysiological mechanisms of treatment toxicity. In this Pelvic Radiation and Vorinostat (PRAVO) study, we investigated mechanisms of adverse effects in response to the histone deacetylase (HDAC) inhibitor vorinostat (suberoylanilide hydroxamic acid, SAHA) when administered as a potential radiosensitiser.
Materials and Methods
This phase I study for advanced gastrointestinal carcinoma was conducted in sequential patient cohorts exposed to escalating doses of vorinostat combined with standard-fractionated palliative radiotherapy to pelvic target volumes. Gene expression microarray analysis of the study patient peripheral blood mononuclear cells (PBMC) was followed by functional validation in cultured cell lines and mice treated with SAHA.
Results
PBMC transcriptional responses to vorinostat, including induction of apoptosis, were confined to the patient cohort reporting dose-limiting intestinal toxicities. At relevant SAHA concentrations, apoptotic features (annexin V staining and caspase 3/7 activation, but not poly-(ADP-ribose)-polymerase cleavage) were observed in cultured intestinal epithelial cells. Moreover, SAHA-treated mice displayed significant weight loss.
Conclusion
The PRAVO study design implemented a strategy to explore treatment toxicity caused by an HDAC inhibitor when combined with radiotherapy and enabled the identification of apoptosis as a potential mechanism responsible for the dose-limiting effects of vorinostat. To the best of our knowledge, this is the first report deciphering mechanisms of normal tissue adverse effects in response to an HDAC inhibitor within a combined-modality treatment regimen.
Keywords: Histone deacetylase inhibitors, Drug-related side effects and adverse reactions, Apoptosis, Radiotherapy, Phase I Clinical Trials
Introduction
Combined-modality therapies comprise the future of cancer medicine, and recent developments in radiobiology have made integration of molecularly targeted therapeutics in clinical radiotherapy increasingly conceivable [1]. One example is the combination of radiation with histone deacetylase (HDAC) inhibitors. Inhibition of HDAC enzymes increases histone acetylation, leading to modification of chromatin structure and changes in gene expression [2,3]. However, the exact mechanisms by which this class of drugs enhances therapeutic DNA-damaging effects, including those from radiation, remain to be understood. Nevertheless, it is assumed that they comprise cell cycle arrest, DNA damage repair and apoptosis [4-6]. Adding to the complexity of HDAC inhibitor mechanisms is the fact that they are not solely targeting histones, but also other signalling proteins [7]. While histone acetylation occurs in both normal and tumour cells, it has been contended that HDAC inhibitors selectively affect the latter [8-10].
Within the context of combined-modality therapy, the enhanced efficacy of radiation combined with a radiosensitising agent may reflect a therapeutic dose at the brink of patient tolerance. Furthermore, evaluating the treatment toxicity that arises from the systemic component (e.g., the radiosensitiser) is challenging, especially if its toxicity profile is indistinguishable from normal tissue effects in the radiotherapy target volume [11]. In this context, sampling of a non-irradiated surrogate tissue may enable investigation of the targeted agent’s mechanism-of-action which is both separate from radiation-induced molecular perturbations and representative of normal tissue responses to the particular compound.
The Pelvic Radiation and Vorinostat (PRAVO) study for symptom palliation in advanced gastrointestinal cancer was first to evaluate the therapeutic use of an HDAC inhibitor in clinical radiotherapy [12]. This phase I study evolved from a preclinical investigation of tumour radiosensitisation by HDAC inhibitors in experimental colorectal carcinoma (CRC) models [13-16]. Sequential patient cohorts were exposed to escalating doses of vorinostat combined with fractionated radiotherapy to pelvic target volumes. Because common side effects of vorinostat single-agent therapy include intestinal toxicities [17], the primary objective of this study was to determine tolerability to vorinostat in combination with pelvic radiation [12,18]. In addition, the peripheral blood mononuclear cells (PBMC) of patients were sampled before and after vorinostat administration to explore molecular markers of vorinostat action through PBMC gene expression analysis. We previously showed that the expression kinetics of several genes encoding DNA damage response factors involved in clinical radiation sensitivity [19] reflected the appropriate timing of vorinostat administration in the fractionated radiotherapy protocol [20].
To better understand mechanisms of normal tissue adverse events, the PBMC gene expression profiles were reanalyzed with respect to vorinostat dose and reported toxicities within the study protocol as assessed by the Common Terminology Criteria for Adverse Events (CTCAE) scoring. Vorinostat-specific dose-limiting toxicity (DLT) in terms of CTCAE grade 3 intestinal adverse events was observed at a dose of 400 mg [12,18], and the present analysis revealed significant alterations in gene expression solely in this patient cohort. Because induction of genes involved in cell death processes was amongst the major PBMC responses, the hypothesis that apoptosis might be a fundamental process in vorinostat-induced toxicity was tested experimentally in vitro using both normal and CRC cell lines, and further functional end-points were examined in an in vivo mouse model. In particular, the use of intestinal epithelial cells was considered essential in the light of the reported intestinal events [12,18].
Materials and Methods
1. Ethics, consent, and permissions
This PRAVO study (ClinicalTrials.gov NCT00455351) was approved by the Institutional Review Board and the Regional Committee for Medical and Health Research Ethics (reference number REK S-06289) and performed in accordance with the Helsinki Declaration. Written informed consent was required for participation. Housing and all procedures involving research animals were developed according to protocols approved by the Animal Care and Use Committee at the Department of Comparative Medicine, Oslo University Hospital (reference number 885-2616-2919-2928-3688), in compliance with the National Committee for Animal Experiments’ guidelines on animal welfare.
2. Patients and study objectives
The principal eligibility criterion was histologically confirmed pelvic carcinoma scheduled to receive palliative radiation to 30 Gy in 3-Gy daily fractions. Other details regarding eligibility are given in the initial report [12]. This dose-escalation study adopted a phase I conventional 3+3 expansion cohort design in which patients with advanced gastrointestinal carcinoma were enrolled onto four sequential dose levels of vorinostat (Merck & Co., Inc., Whitehouse Station, NJ), starting at a daily dose of 100 mg with dose escalation in increments of 100 mg, given 3 hours (at 9 AM) before the daily radiotherapy fraction (at 12 PM) [20]. The primary objective was to determine the tolerability to vorinostat, defined by the DLT and maximum-tolerated dose, when administered concomitantly with palliative radiation to pelvic target volumes. Amongst secondary objectives was the identification of possible biomarkers of treatment toxicity. The study data describing patient treatment tolerability, tumour histone acetylation following vorinostat administration and radiologic treatment response, as well as the initial exploration of mechanisms of vorinostat activity, have been reported previously [12,18,20].
3. Patient blood sampling and RNA isolation
Peripheral blood was drawn on PAXgene Blood RNA Tubes (Qiagen Norge, Oslo, Norway) and collected at baseline (before commencement of the treatment) and on-treatment (day 3), 2 hours (at 11 AM) and 24 hours (at 9 AM) after the patient received the preceding daily dose of vorinostat (at 9 AM) [20]. A full set of three samples was obtained from 14 of the 16 evaluable study patients. The tubes were stored at –70°C until analysis. Total RNA from PBMC was isolated using a PAXgene Blood RNA Kit (Qiagen) according to the manufacturer’s protocols. RNA concentration and quality were assessed using a NanoDrop 1000 and Agilent 2100 Bioanalyzer (Thermo Fisher Scientific Norway, Oslo, Norway).
4. Gene expression microarray analysis
Gene expression analysis was performed using Illumina Human WG-6 v3 Expression BeadChip arrays with 48,000 probes (Illumina, Inc., San Diego, CA), as previously described [20]. The primary array data are available in the Gene Expression Omnibus data repository under accession number GSE46703. Following quality control and pre-processing of the array data, including log2 transformation, differential gene expression analysis of PBMC samples taken before and after vorinostat administration was conducted using the significance analysis of microarrays algorithm and by applying the setting of paired-comparisons with a false discovery rate of 5% [21]. Cluster analysis of the differentially expressed genes was conducted by employing the Euclidean distance method and using the R software v3.1.1 and the pheatmap package (http://cran.r-project.org/web/packages/pheatmap). Gene ontology analysis of the differentially expressed genes was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7 software [22].
5. Cell cultures
Rat intestinal epithelial cells (IEC-6) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and grown in high glucose (4.5 g/L) Dulbecco’s modified Eagle’s medium (Sigma Aldrich, Schnelldorf, Germany) supplemented with 0.1 U/mL bovine insulin (Sigma Aldrich) and 10% foetal bovine serum (Sigma Aldrich). IEC-6 cells were used between passages 17 and 21. Human BJ fibroblasts were purchased from ATCC and grown in Eagle’s minimum essential medium modified (ATCC) supplemented with 10% serum. The human CRC cell lines HCT-116 and HT29 were originally purchased from ATCC, and the identities of our laboratory’s versions were confirmed by short tandem repeat analysis. These cell lines were kept as previously described [15]. All cell cultures were maintained in a humidified atmosphere under 5% CO2 at 37°C. Treatment with suberoylanilide hydroxamic acid (SAHA; Cayman Chemical, Ann Arbor, MI), the active component of vorinostat, or vehicle (dimethyl sulfoxide) was administered for 24 or 48 hours.
6. Immunoblot analysis
Protein lysates from both floating and adherent cells (cell cultures) were prepared as previously described [23]. Intestines (mice experiments) were homogenised with a FastPrep-24 instrument (MP Biomedicals, http://www.mpbio.com) three times for 30 seconds each using ceramic beads (MP Biomedicals) and RIPA buffer (Sigma Aldrich) to obtain protein lysates. Equal amounts of protein (20 μg for cell culture lysates and 30 μg for tissue extracts) were separated by 4%-12% NuPAGE Bis-Tris gels (Life Technologies, Carlsbad, CA), transferred by electrophoresis to an Immobilon membrane (Millipore Corporation, Billerica, MA), and probed with the following antibodies: anti-acetyl-histone H3, anti–poly-(ADP-ribose) polymerase (PARP), and anti–heat shock protein 70 (Hsp70; Cell Signaling Technology Europe, B.V., Leiden, The Netherlands) and anti–α-tubulin (Calbiochem/EMD Chemicals, Inc., San Diego, CA). The peroxidise activity was visualised using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific Norway). A sufficient amount of lysate from each sample was prepared to run on two gels, and the two blotting membranes were considered identical; thus, proteins of the same size (kDa) were visualised on different membranes. All experiments were performed at least three times.
7. Flow cytometry analysis of cell death
Cells were seeded in flasks and allowed to grow overnight before medium change and treatment with SAHA or vehicle. Both floating and adherent cells were harvested and stained using the Annexin V Alexa Fluor 488 and propidium iodide (PI) kit (Life Technologies). The samples were analyzed with a FACS Diva flow cytometer (Becton Dickinson, http://www.bd.com), and data processing was performed with the commercially available FlowJo v7 software (Tree Star, Inc., Ashland, OR). A total of 10,000 cells were analyzed per sample. Cells were considered as early or late apoptotic if they were annexin-V+/PI– or annexin-V+/PI+, respectively, and necrotic if they were annexin-V–/PI+.
8. Analysis of caspase 3/7 activity
Cells were seeded in 96-well plates and allowed to grow overnight before changing the medium and treating the samples with varying concentrations of SAHA or vehicle. Caspase 3/7 activity was measured using a Caspase-Glo 3/7 Assay kit (Promega, Madison, WI), and chemiluminescence readings were obtained with Fujifilm FLA-5100 (Fujifilm Corporation, Tokyo, Japan).
9. Animal experiments
Locally bred immunocompetent BALB/c mice, 6-8 weeks old, were allowed to acclimatise for approximately one week before the start of the study. Food and water were supplied ad libitum. SAHA (Selleckchem, Houston, TX) was dissolved in hydroxypropyl-β-cyclodextrin solution (Sigma Aldrich) at a 1:5 molar ratio. Thirteen mice, five in each treatment group and three in the control group, were administered with either SAHA or vehicle once daily for five consecutive days via oral gavage. The weight of each individual mouse was measured every 24 hours and compared to the starting weight. On day 5 of treatment and 3 hours after the last drug or vehicle administration, the mice were sacrificed and organs were collected for further analysis.
10. Immunohistochemistry
Intestinal sections were opened longitudinally, rinsed in saline and fixed in formalin. Sections were then rolled in a “Swiss roll” fashion (luminal side outwards) and embedded in paraffin [24] before standard staining with hematoxylin and eosin and examination by a specialist bowel pathologist. A TACS 2 TdT-Fluor In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD) was used according to the manufacturer’s instructions to detect apoptotic cells based on terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL). Tissue slides were viewed using an IX81 inverted fluorescencent microscope (Olympus, http://www.olympus-lifescience.com ). Sections from each group were examined and representative images of intestinal sections were taken for each mouse.
11. Statistical analysis
Differences between treatment groups were identified by t tests, with p < 0.05 considered statistically significant.
Results
1. The PRAVO study: vorinostat toxicity
Of 16 eligible study patients, three reported study-specific DLTs, one at the 300 mg dose level of vorinostat and two at 400 mg. These DLTs were all CTCAE grade 3 intestinal and related adverse events [12]. PBMC samples were not available from two cases at the 300 mg dose level, including the patient that experienced DLT [20]. However, in this case, the DLT was determined as an adverse radiation-induced effect rather than a toxic effect caused by vorinostat [18], so no information about the molecular mechanisms of vorinostat-specific toxicity from PBMC was lost from further analysis. In contrast, all of the DLT events reported at the 400 mg dose level were interpreted as true vorinostat-induced side effects [18]. Since the first study patient who was enrolled (at the 100 mg dose level) experienced no higher than CTCAE grade 1 side effects during the complete follow-up period [12], enrolment onto the next dose level (200 mg) proceeded directly, in accordance with common manner of conduct of studies with this design. Gene expression data from the single patient at the lowest dose level were omitted from further analysis. Table 1 shows the study population that remained for investigations in the present report.
Table 1.
Study patients in the present report
| Vorinostat dose (mg daily) | Age (yr) | Sex | Dose-limiting toxicity |
|---|---|---|---|
| 200 | 49 | Female | - |
| 200 | 64 | Female | - |
| 200 | 66 | Female | - |
| 300 | 66 | Female | - |
| 300 | 77 | Male | - |
| 300 | 81 | Female | - |
| 300 | 82 | Male | - |
| 400 | 45 | Female | - |
| 400 | 55 | Male | - |
| 400 | 62 | Male | - |
| 400 | 75 | Female | Diarrhoea, fatigue, hypokalemia |
| 400 | 83 | Female | Diarrhoea, anorexia, hyponatremia |
| 400 | 85 | Female | - |
2. Transcriptional responses to vorinostat
We previously analyzed gene expression array data from study patients’ PBMC sampled at baseline and on-treatment two and 24 hours after the patients had received the daily dose of vorinostat. Since vorinostat-induced tumour histone acetylation was observed at all dose levels (100-400 mg daily) [12], array data from each of the time points, regardless of the actual vorinostat dose, were analyzed to identify generic molecular indicators of HDAC inhibitor activity [20].
However, we investigated responses at each of the individual dose levels in the current study. Differential expression analysis revealed that profound transcriptional responses were only seen in patients who received the highest dose of vorinostat (i.e., the dose level at which DLTs were reported), and that these gene expression alterations were transient and observed only two hours after the patients had received the drug (i.e., they were restored to baseline at the 24-hour sampling point). Specifically, the number of genes significantly altered two hours after vorinostat administration was 1,294 in the 400 mg cohort, but only five and six in the 200 mg and 300 mg cohorts, respectively. Table 2 lists transcripts that were regulated by 2.5-fold or more, where HSPA1B, encoding a major member of the Hsp70 protein family that has been suggested to promote immunogenic cell death following therapeutic cell stresses [25], showed the greatest induction in the patient cohort. Fig. 1 illustrates clustering of patients and differentially regulated genes in the highest dose cohort. Gene ontology analysis of the differentially expressed genes showed significant over-representation of a designated repertoire of biological processes (Table 3). In general terms, induced genes were over-represented in chromatin organisation and cell death categories, with the latter including pro-apoptotic factors, whereas repressed genes were over-represented in gene transcription and RNA processing categories.
Table 2.
Significantly altered genes in peripheral blood mononuclear cells 2 hours after vorinostat administration
| Gene | Fold-changea) | q-valueb) |
|---|---|---|
| HSPA1B | 4.2 | 4.1 |
| PHF13 | 3.8 | 1.6 |
| PTTG1 | 3.5 | 4.1 |
| ING2 | 3.5 | 3.3 |
| C17ORF91 | 3.2 | 1.6 |
| TNFRSF12A | 3.1 | 2.8 |
| SPATA2L | 3.1 | 7.7 |
| PTTG3P | 3.0 | 4.7 |
| PTTG1 | 3.0 | 9.4 |
| ADRB2 | 2.7 | 4.7 |
| ZC3H12A | 2.7 | 0 |
| MSH6 | 2.6 | 5.9 |
| C16ORF61 | 2.6 | 3.7 |
| GADD45B | 2.6 | 4.1 |
| CKS2 | 2.5 | 4.1 |
| ZNF791 | 2.5 | 2.8 |
| SGK | 0.30 | 0 |
| DEFA3 | 0.30 | 6.7 |
| DEFA1B | 0.30 | 7.7 |
| DEFA1 | 0.30 | 5.9 |
| LOC653600 | 0.30 | 7.7 |
| ZC3HC1 | 0.30 | 0 |
| GIMAP8 | 0.30 | 0 |
| MS4A7 | 0.40 | 1.5 |
| MYC | 0.40 | 1.6 |
| IL27RA | 0.40 | 0.72 |
| PDK4 | 0.40 | 0.72 |
| MYC | 0.40 | 2.8 |
| CYP1B1 | 0.40 | 2.7 |
Fold-change from baseline expression,
q-value represents an adjusted p-value, the lowest false discovery rate at which the gene is deemed to have changed significantly.
Fig. 1.
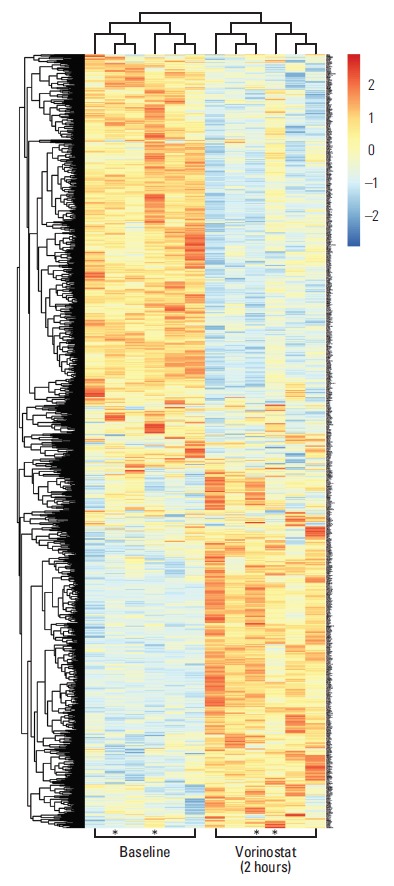
Heat-map presentation of differentially expressed transcripts in peripheral blood mononuclear cells sampled at baseline (before commencement of study treatment) and 2 hours after administration of vorinostat (400 mg), with colours representing high (red) to low (blue) levels of log2-transformed data and asterisks indicating the two patients who experienced intestinal dose-limiting toxicities.
Table 3.
Enriched biological processes in peripheral blood mononuclear cells 2 hours after vorinostat administration (400 mg)
| Gene ontology (GO) term | p-value | Gene |
|---|---|---|
| Induced process | ||
| GO:0051276 chromosome organisation |
1.5×10-5 | UTP3, HIST2H2AA3, ING2, RBM4, CBX4, CBX3, TERF2IP, PTTG1, HIST2H4A, CBX6, BRPF1, TSPYL2, SMARCD2, H2AFZ, HIST3H2A, BCOR, KDM5B, BUB3, PINX1, MLL, MSH3, HIST1H1C, TAF5, PTTG3P, NASP, H2AFJ, DAPK3, C20ORF20, JMJD6, BCORL1, RRS1, H3F3B, SETD8, RBM14 |
| GO:0008219 cell death |
3.1×10-5 | MICB, PPARD, HRAS, TNF, TNFRSF12A, PMAIP1, MTP18, PDCD1, GLRX2, SETX, RRAGC, BAG5, BAK1, HTRA2, TARDBP, DAD1, ZC3H12A, PSENEN, BMF, CASP2, TRAF4, MLL, SRA1, TBRG4, POLB, DAPK3, ELMO2, PLEKHF1, TNFRSF10C, TNFRSF10B, AKTIP, JMJD6, TDP1, BAX, SLC5A8, APAF1, HSPD1, GADD45B, CLN8, PPP1R15A, UBA52, MAP3K11 |
| GO:0016265 death |
3.6×10-5 | MICB, PPARD, HRAS, TNF, TNFRSF12A, PMAIP1, MTP18, PDCD1, GLRX2, SETX, RRAGC, BAG5, BAK1, HTRA2, TARDBP, DAD1, ZC3H12A, PSENEN, BMF, CASP2, TRAF4, MLL, SRA1, TBRG4, POLB, DAPK3, ELMO2, PLEKHF1, TNFRSF10C, TNFRSF10B, AKTIP, JMJD6, TDP1, BAX, SLC5A8, APAF1, HSPD1, GADD45B, CLN8, PPP1R15A, UBA52, MAP3K11 |
| GO:0006325 chromatin organisation |
1.2×10-4 | UTP3, HIST2H2AA3, ING2, RBM4, CBX4, CBX3, HIST2H4A, CBX6, BRPF1, TSPYL2, SMARCD2, H2AFZ, HIST3H2A, BCOR, KDM5B, MLL, HIST1H1C, TAF5, NASP, H2AFJ, DAPK3, C20ORF20, BCORL1, JMJD6, H3F3B, SETD8, RBM14 |
| GO:0006915 apoptosis |
1.8×10-4 | HRAS, PPARD, TNF, TNFRSF12A, PMAIP1, MTP18, PDCD1, GLRX2, RRAGC, BAG5, BAK1, HTRA2, DAD1, ZC3H12A, PSENEN, BMF, CASP2, TRAF4, MLL, SRA1, TBRG4, DAPK3, ELMO2, PLEKHF1, TNFRSF10C, TNFRSF10B, AKTIP, JMJD6, BAX, SLC5A8, APAF1, HSPD1, GADD45B, PPP1R15A, UBA52 |
| Repressed process | ||
| GO:0006396 RNA processing |
6.7×10-8 | RNASEN, RNASEL, CRNKL1, GAR1, DICER1, DUSP11, POLR2C, XAB2, EXOSC10, INTS9, SFRS6, RBM4B, SFRS4, DHX38, AGGF1, QTRTD1, TRMT5, RBM10, RBM28, GEMIN4, DUS3L, RBM22, PPP2R1A, RBM23, CSTF2, C1ORF25, DDX1, HEATR1, CDC5L, SF3A1, TRMT61A, RBMX, URM1, PHAX, PPIE, PPP1R8, GTF2F1, CPSF3, CWC22, UTP14A, TRUB2, RPP40, RBM16, DUS2L, KIAA1429 |
| GO:0006350 transcription |
3.3×10-7 | CNOT2, MED24, APOBEC3G, GTF2IRD2B, EPC1, EPC2, OLIG1, ZNF593, ZNF45, ZHX2, ZNF37A, ZNF193, MGA, DEAF1, ZNF613, TH1L, KEAP1, XAB2, SMARCB1, ZNF329, LEO1, ZNF426, RNF14, SETDB1, TAF4, IKZF3, IKZF1, RFX5, MAFB, KLF11, SUV39H1, KAT5, HOXB2, ZNF211, ZNF317, GTF2F1, DNMT1, KLF2, ZNF85, TAF1C, CRCP, CTCF, ZEB2, DMAP1, RFXANK, ZKSCAN4, C10ORF137, ZNF350, ASH2L, CCDC101, E4F1, MKL1, MYC, ELP3, MTERF, GMEB2, GMEB1, ZNF543, ZNF142, CDK7, MCM2, CDC5L, ZNF689, ZNF688, ZNF140, PCGF1, ZFP37, MED6, ZNF341, ZNF134, NCOA5, NCOA6, ZNF274, ING3, NMI, ZNF559, ZNF805, NFYC, HCFC1, ZNF35, ZNF366, ZNF653, POLR2C, CCDC59, ZNF32, ZNF708, ECD, ZNF473, GTF3C6, THAP1, GTF3C2, ERCC3, GTF3C1, TRIP11, ZNF605, GTF3C3, ZNF565, CEBPB, HSFX1, L3MBTL2, ZNF260, ZNF23, IRF9, PHF17, ADNP2, RPAP1, UBTF, ZNF362, SP2, PPP1R8, RBM16, VPS25 |
| GO:0016071 mRNA metabolic process |
2.7×10-6 | RNASEL, C17ORF71, CRNKL1, XAB2, POLR2C, ZFP36L1, EXOSC10, SFRS6, DCPS, RBM4B, SFRS4, DHX38, RBM10, RBM28, GEMIN4, RBM22, RBM23, CSTF2, DDX1, ZHX2, CDC5L, SF3A1, RBMX, PHAX, PPIE, PPP1R8, DCP1A, GTF2F1, CPSF3, CWC22, RBM16, KIAA1429 |
| GO:0006351 transcription, DNA-dependent |
1.8×10-5 | DEAF1, TAF1C, NMI, CRCP, MED24, HCFC1, POLR2C, ASH2L, ECD, GTF3C6, ERCC3, GTF3C2, MYC, TRIP11, GTF3C1, GTF3C3, TAF4, MTERF, CEBPB, GMEB2, KLF11, CDK7, IRF9, GTF2F1, NCOA6, RBM16 |
| GO:0032774 RNA biosynthetic process |
2.2×10-5 | DEAF1, TAF1C, NMI, CRCP, MED24, HCFC1, POLR2C, ASH2L, ECD, GTF3C6, ERCC3, GTF3C2, MYC, TRIP11, GTF3C1, GTF3C3, TAF4, MTERF, CEBPB, GMEB2, KLF11, CDK7, IRF9, GTF2F1, NCOA6, RBM16 |
3. SAHA-induced apoptosis
Because induction of HSPA1B and other genes involved in cell death processes was amongst the major PBMC responses at the 400 mg dose level of vorinostat, we investigated whether similar endpoints might be observed in relevant experimental models. To accomplish this, rat IEC-6 intestinal epithelial cells, human BJ fibroblasts and two human CRC cell lines (HCT-116 and HT29) were exposed to SAHA for 24 and 48 hours. Acetylation of histone H3 was observed with a concentration of 2.5 μM, closely corresponding to the peak plasma concentration (approximately 600 ng/mL) observed after oral intake of vorinostat (molecular weight 264 g/mol) [26], as well as with the supra-pharmacologic concentration of 20 μM for comparison (Fig. 2). Interestingly, 2.5 μM SAHA for 24 hours caused a significant increase in the percentage of apoptotic phenotypes (annexin-V+/PI–, which is regarded as reversible, and annexin-V+/PI+, which is considered irreversible). This was observed in both CRC cell lines and normal intestinal epithelial cells. In contrast, the BJ fibroblasts showed no enhancement of these features (Fig. 3). Moreover, assessment of the caspase 3/7 activity as an apoptotic read-out revealed significant induction in the CRC cell lines and the normal intestinal epithelial cells following treatment with 2.5 μM and higher concentrations of SAHA for 24 hours, while induction of caspase 3/7 activity was only seen at 20 μM in BJ cultures (Fig. 4). Furthermore, as shown in Fig. 2, there was no sign of PARP cleavage in BJ cells; however, in the CRC cell lines, this late-stage apoptotic endpoint was elicited in response to both 2.5 μM and 20 μM SAHA, which was also the case in the IEC-6 cells treated with the higher of the two concentrations.
Fig. 2.

Immunoblot analysis of histone H3 acetylation (Ac-H3) and poly-(ADP-ribose) polymerase (PARP) cleavage in human colorectal carcinoma cells (HCT-116 and HT29), human BJ fibroblasts, and rat intestinal epithelial cells (IEC-6) following suberoylanilide hydroxamic acid (SAHA) treatment; Amido Black total protein staining as loading controls.
Fig. 3.

Flow cytometry histograms (A, B, D, E, G, H, J, K), with bi-exponentially transformed annexin-V/propidium iodide (PI) data, displaying early apoptosis (lower right rectangles), late apoptosis (upper right rectangles), and necrosis (upper left rectangles) in human colorectal carcinoma cells (A-F), human fibroblasts (G-I), and rat intestinal epithelial cells (J-L) treated with suberoylanilide hydroxamic acid (SAHA, 2.5 μM for 24 hours), with corresponding quantifications (average values and standard error of the mean; *p < 0.05, **p < 0.01, ***p < 0.001) of at least three experiments (C, F, I, L).
Fig. 4.
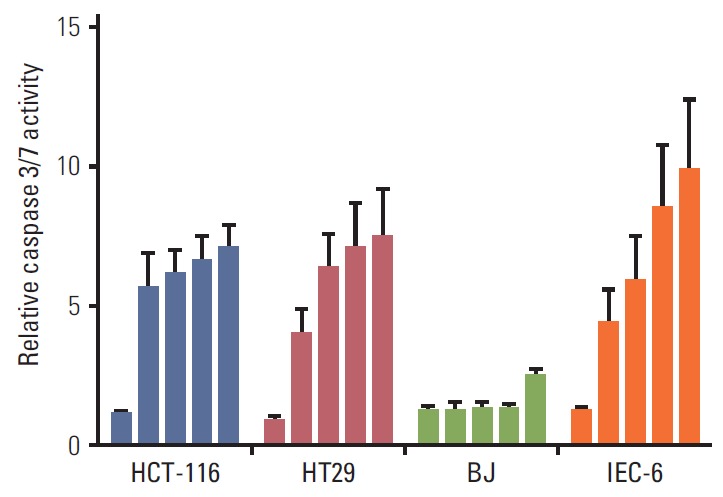
Caspase 3/7 activity in human colorectal carcinoma cells (HCT-116 and HT29), human BJ fibroblasts, and rat intestinal epithelial cells (IEC-6) following exposure to suberoylanilide hydroxamic acid (0.1, 2.5, 5.0, 10, and 20 μM; columns from left to right) for 24 hours. Results were normalised to the respective controls (given the values of 1) and displayed as average values and standard error of the mean of at least three experiments with three technical replicates.
4. In vivo SAHA effects
Following daily SAHA (100 or 200 mg/kg) or vehicle administration for 5 days, intestinal sections were collected 3 hours after the last treatment and evaluated for possible organ effects. All of the 13 mice used in this experimental setup accomplished the planned course. As shown in Fig. 5, SAHA-induced histone acetylation was observed in the intestinal tissue sections. In addition, there was a decline in intestinal expression of the full-length PARP protein, but no evidence of cleaved PARP in one of two investigated mice from the 200 mg/kg group, as well as a slight increase in intestinal Hsp70 expression in both of the investigated mice from the 100 mg/kg group. Moreover, compared with the control group, mice in the 200 mg/kg group showed a significant decrease in body weight with nadir at the start of treatment day 3. However, blinded histopathologic evaluation of tissue rolls of intestinal sections that were sampled 3 hours after drug administration on day 5 revealed no signs of tissue damage, such as epithelial ulceration, erosion, or apoptosis. Finally, TUNEL staining of the sections did not reveal evident changes in DNA fragmentation, which, if present, would have reflected a fulminant apoptotic response in the epithelial lining of the intestinal crypts or villi.
Fig. 5.
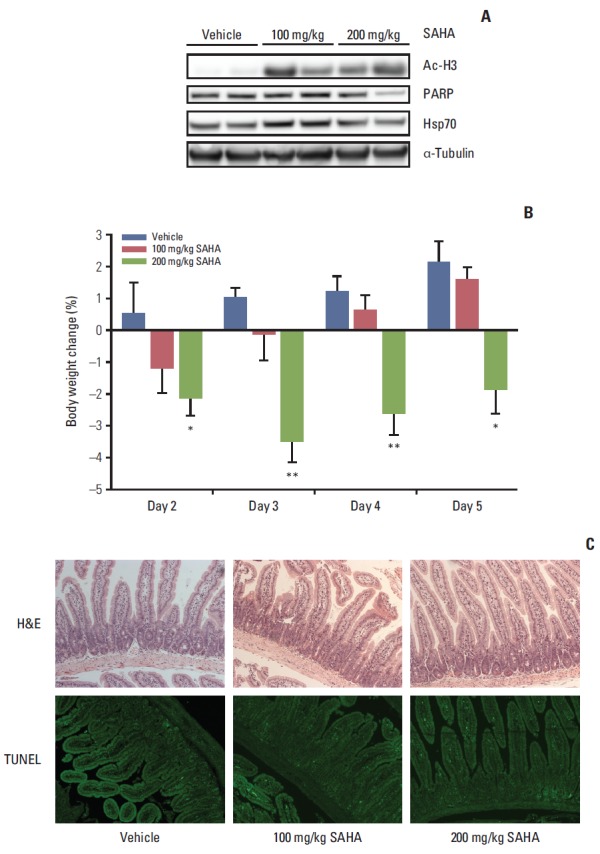
In vivo intestinal effects of suberoylanilide hydroxamic acid (SAHA) in mice treated for five consecutive days and sacrificed on day 5, 3 hours after the oral administration, in terms of histone H3 acetylation (Ac-H3), poly-(ADP-ribose) polymerase (PARP), and heat shock protein 70 (Hsp70) protein expression, verified by immunoblot analysis with α-tubulin expression as a loading control (shown for two mice in each treatment group) (A), body weight change relative to the start of experiment on day 1 (group average values with standard error of the mean; *p < 0.05, **p < 0.01) (B) and histology of tissue rolls of intestinal sections stained with hematoxylin and eosin (H&E) or terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) (all images shown are at ×10 magnification and representative of each treatment group) (C).
Discussion
The PRAVO study for symptom palliation in advanced bowel cancer was conducted to determine tolerability to the combination of vorinostat with radiation to pelvic target volumes. Because of overlapping and therefore possible synergistic toxicities by the two therapeutic components, the study design implemented a strategy to identify the mechanisms of vorinostat action separately to explore the contribution of this modality within the overall toxicity profile dominated by radiation effects. Using gene expression array analysis of surrogate tissue (PBMC) from study patients, the induction of genes implicated in cell death processes was observed among the top five transcriptional responses at the vorinostat dose level causing CTCAE grade 3 intestinal adverse events. Early apoptotic features at a relevant drug concentration were confirmed as functional endpoints in normal intestinal epithelial cells grown in culture, while treated mice showed significant loss of body weight. These experimental data collectively indicate that intestinal epithelial apoptosis may be one fundamental mechanism in the reported DLTs from vorinostat.
Vorinostat-induced transcriptional responses in PBMC involved genes related to chromatin organisation, cell death, gene transcription, and RNA processing, reflecting known molecular signatures of HDAC inhibitors. For example, some of the genes induced in cell death categories included proapoptotic factors that are implicated in HDAC inhibitor-induced apoptosis [9,27]. The observation that such transcriptional responses were induced in PBMC led to the hypothesis that vorinostat might cause cell death-related phenotypic effects in both normal and cancer cells. Normal intestinal epithelial cells may represent a relevant experimental model for testing acute intestinal-related toxicities, and we used this model together with normal fibroblasts and CRC cell lines to validate the array findings. In all cell line models, histone acetylation was observed following incubation with the pharmacologically relevant SAHA concentration of 2.5 μM [26], clearly indicating that the compound had inhibited its targets at this concentration. Various functional readout assays indicated that apoptotic indicators were observed in the CRC cell lines and the normal intestinal epithelial cells. This was in contrast to the fibroblasts, which showed no appearance of these apoptotic phenotypes, concordant with previously reported data [9]. However, a short-coming of utilising fibroblasts is that the model is more relevant for studying late, as opposed to early adverse effects of radiation, which might be the case for other agents as well.
Nevertheless, the results of this study support the notion that vorinostat may induce apoptosis in both normal and malignant epithelial cells of the intestinal mucosal lining. However, upon treatment of mice with SAHA, even though intestinal histone acetylation was observed together with a hard clinical endpoint (weight loss), no morphological or functional signs of apoptotic damage were detected in the epithelial lining of the intestinal crypts or villi. It is worth noting that detection of apoptosis in the intestinal tract could be hindered by the fact that intestinal epithelial cells are shed into the luminal space at the end of their life cycle and not processed by detectable phagocytosis as in other tissues [28]. Alternatively, the intestinal apoptotic response may not have reached an irreversible and complete stage. Notably, the transcriptional response of PBMC following administration of 400 mg vorinostat was transient at 2 hours, but not retrieved as a long-term effect (i.e., at 24 hours). Additionally, in contrast to the CRC cell lines, late-stage apoptotic PARP cleavage was not observed in the IEC-6 cells in response to incubation with the pharmacologically relevant SAHA concentration of 2.5 μM. Similarly, no PARP cleavage, but a decline in the full-length PARP protein and a small increase in the Hsp70 protein associated with therapy-induced immunogenic cell death [25] were observed in intestinal tissue from SAHA-treated mice. The observed endpoints in study patients [12,18] and SAHA-treated mice likely reflected reversible normal tissue responses to the treatment [11], which might have been caused by transient and repeat early apoptotic effects in the intestinal epithelium. Recently, SAHA was shown to repress intestinal crypt cell proliferation in experimental models [29], which may be a central manifestation of toxicity to the drug. Finally, the clinical toxicities must also be interpreted in a wider mechanistic context.
Acute radiation-induced adverse effects are commonly manifested within a short time of the treatment completion, typically in tissues with a hierarchical proliferative structure such as the mucosal linings of the intestinal tract [30]. Because of the additional complexity of biological responses to injuries in combined-modality treatments, correlative biomarker studies should preferably be conducted within the context of prospective assessments of side effects [11]. Nevertheless, biomarker development studies could be challenging given the multiplicity of molecular responses to radiation, which justifies the application of multiplex technologies and matched computational tools. Ideally, when searching for biomarkers of treatment toxicity, the normal tissue that best manifests the actual clinical phenotype should be analyzed for causative mechanisms of treatment severity at the individual level. However, since that normal tissue may not always be readily available, a surrogate tissue is commonly accepted as being more feasible for correlative mechanistic analysis at the study population level. Any candidate biomarkers could then be phenotypically validated using relevant preclinical assessment tools, as illustrated in the present study.
Nevertheless, there are limitations to analysing PBMC as target tissue for the systemic agent in the context of concomitant radiotherapy. For example, the possibility of additional systemic effects of the local radiation must be considered. However, in this study, exposed PBMC was sampled both at 9 AM and 11 AM on day 3 [20]. If radiation exposure (daily at 12 PM) [20] had caused molecular perturbations in PBMC, both on-treatment sampling points would likely have displayed differentially expressed genes (compared to baseline) that reflected the two radiation fractions already given. This was not the case as only PBMC collected at 11 AM, 2 hours after vorinostat administration (and solely at the 400 mg dose level), was different from baseline.
Conclusion
The PRAVO study design enabled the identification of vorinostat-induced transcriptional responses, with apoptosis among the major processes, in surrogate tissue from patients at the dose that caused intestinal DLTs. Moreover, in preclinical models, apoptotic phenotypes in cultured intestinal epithelial cells and weight loss in mice following administration of the compound were observed as functional endpoints, which may point to one major pathophysiological mechanism for the intestinal toxicities of vorinostat. This combined clinical safety and translational approach for studying systemic normal tissue responses together with the organ-confined adverse effects in response to radiation may form a template for future early-phase clinical trials assessing molecularly targeted therapeutics combined with radiotherapy.
Acknowledgments
The South-Eastern Norway Regional Health Authority (grant No. 2014012–ACREDIT to A.H.R.). E.K. and K.R.R. are Postdoctoral Research Fellows supported by the South-Eastern Norway Regional Health Authority (grants No. 2013101 and 2012002 to A.H.R.) and K.R. is a Postdoctoral Research Fellow supported by the Norwegian Cancer Society (grant No. 2180105 to A.H.R). I.S.B. is a Medical Student Research Fellow at the University of Oslo (by funding to K.R.R.). The funding sources played no role in the study design, collection, analysis and interpretation of data, writing of the report or the decision to submit for publication.
Footnotes
This study was supported by Merck & Co., Inc.
References
- 1.Ree AH, Hollywood D. Design and conduct of early-phase radiotherapy trials with targeted therapeutics: lessons from the PRAVO experience. Radiother Oncol. 2013;108:3–16. doi: 10.1016/j.radonc.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–83. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 4.Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB. HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol. 2009;20:1293–302. doi: 10.1093/annonc/mdn792. [DOI] [PubMed] [Google Scholar]
- 5.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 6.Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108:748–54. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 8.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumorselective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 9.Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, et al. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013;4:e519. doi: 10.1038/cddis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ree AH, Meltzer S, Flatmark K, Dueland S, Kalanxhi E. Biomarkers of treatment toxicity in combined-modality cancer therapies with radiation and systemic drugs: study design, multiplex methods, molecular networks. Int J Mol Sci. 2014;15:22835–56. doi: 10.3390/ijms151222835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–64. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 13.Flatmark K, Nome RV, Folkvord S, Bratland A, Rasmussen H, Ellefsen MS, et al. Radiosensitization of colorectal carcinoma cell lines by histone deacetylase inhibition. Radiat Oncol. 2006;1:25. doi: 10.1186/1748-717X-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ree AH, Folkvord S, Flatmark K. HDAC2 deficiency and histone acetylation. Nat Genet. 2008;40:812–3. doi: 10.1038/ng0708-812. [DOI] [PubMed] [Google Scholar]
- 15.Folkvord S, Ree AH, Furre T, Halvorsen T, Flatmark K. Radiosensitization by SAHA in experimental colorectal carcinoma models-in vivo effects and relevance of histone acetylation status. Int J Radiat Oncol Biol Phys. 2009;74:546–52. doi: 10.1016/j.ijrobp.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 16.Saelen MG, Ree AH, Kristian A, Fleten KG, Furre T, Hektoen HH, et al. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat Oncol. 2012;7:165. doi: 10.1186/1748-717X-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 18.Bratland A, Dueland S, Hollywood D, Flatmark K, Ree AH. Gastrointestinal toxicity of vorinostat: reanalysis of phase 1 study results with emphasis on dose-volume effects of pelvic radiotherapy. Radiat Oncol. 2011;6:33. doi: 10.1186/1748-717X-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–53. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 20.Ree AH, Saelen MG, Kalanxhi E, Ostensen IH, Schee K, Roe K, et al. Biomarkers of histone deacetylase inhibitor activity in a phase 1 combined-modality study with radiotherapy. PLoS One. 2014;9:e89750. doi: 10.1371/journal.pone.0089750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Tahiri A, Roe K, Ree AH, de Wijn R, Risberg K, Busch C, et al. Differential inhibition of ex-vivo tumor kinase activity by vemurafenib in BRAF(V600E) and BRAF wild-type metastatic malignant melanoma. PLoS One. 2013;8:e72692. doi: 10.1371/journal.pone.0072692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moolenbeek C, Ruitenberg EJ. The "Swiss roll": a simple technique for histological studies of the rodent intestine. Lab Anim. 1981;15:57–9. doi: 10.1258/002367781780958577. [DOI] [PubMed] [Google Scholar]
- 25.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 26.Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, et al. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin Cancer Res. 2011;17:3956–68. doi: 10.1158/1078-0432.CCR-10-3412. [DOI] [PubMed] [Google Scholar]
- 28.Grossmann J, Walther K, Artinger M, Rummele P, Woenckhaus M, Scholmerich J. Induction of apoptosis before shedding of human intestinal epithelial cells. Am J Gastroenterol. 2002;97:1421–8. doi: 10.1111/j.1572-0241.2002.05787.x. [DOI] [PubMed] [Google Scholar]
- 29.Roostaee A, Guezguez A, Beausejour M, Simoneau A, Vachon PH, Levy E, et al. Histone deacetylase inhibition impairs normal intestinal cell proliferation and promotes specific gene expression. J Cell Biochem. 2015;116:2695–708. doi: 10.1002/jcb.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kavanagh BD, Pan CC, Dawson LA, Das SK, Li XA, Ten Haken RK, et al. Radiation dose-volume effects in the stomach and small bowel. Int J Radiat Oncol Biol Phys. 2010;76(3 Suppl):S101–7. doi: 10.1016/j.ijrobp.2009.05.071. [DOI] [PubMed] [Google Scholar]