

Abstract
The amino acid γ-aminobutyric acid (GABA) is thought to play a key role in shaping the activity of the GnRH neurons throughout embryonic and postnatal life. However, the physiological roles of direct GABA inputs to GnRH neurons remain unknown. Using a Cre-LoxP strategy, we generated a targeted mouse line, in which all (98 ± 1%) GnRH neurons had the γ2-subunit of the GABAA receptor deleted. Electrophysiological recordings of GABAA-mediated postsynaptic currents from green fluorescent protein-tagged GnRH neurons with the γ2-subunit knocked out (GnRH γ2 KO) showed that the amplitude and frequency of GABAA postsynaptic currents were reduced by 70% (P < 0.01) and 77% (P < 0.05), respectively, and that the response to exogenous GABA was reduced by 90% (P < 0.01). Evaluation of male and female GnRH γ2 KO mice revealed completely normal fecundity, estrous cycles, and puberty onset. Further investigation with gonadectomy and different steroid replacement regimens showed normal basal levels of LH in both sexes, and a normal estradiol-evoked positive feedback mechanism in females. However, the increment in LH after gonadectomy in GnRH γ2 KO female mice was double that of controls (P < 0.05) and also more potently suppressed by 17-β-estradiol (P < 0.05). A similar but nonsignificant trend was observed in GnRH γ2 KO male mice. Together, these findings show that 70–90% reductions in the normal levels of GABAA receptor activity at the GnRH neuron appear to impact upon the estrogen negative feedback mechanism but are, nevertheless, compatible with normal fertility in mice.
The deletion of the great majority of GABAA receptor signaling at the GnRH neuron has no effect on fertility in transgenic mice.
Inputs from γ-aminobutyric acid (GABA)ergic neurons to GnRH neurons are thought to play a major role in determining the output of the GnRH neuronal network and, thus, fertility. Studies in GnRH transgenic mouse lines have demonstrated that GABA exerts a potent and tonic modulatory influence over the excitability of GnRH neurons in situ (1, 2). Single cell recording and gene profiling studies have shown that GnRH neurons express multiple GABAA receptor subunits (3, 4, 5, 6) and are subjected to an on-going barrage of GABA release (3, 7). Although GnRH neurons express the GABAB receptor (8), all fast GABAergic transmission occurs through the GABAA receptor in these cells (3, 7).
A large number of in vivo studies have examined the effects of GABAA receptor manipulation on the secretion of gonadotropins in multiple species. These have suggested key roles for the GABAergic modulation of GnRH neurons in the onset of puberty (9, 10), generation of pulsatile and surge modes of LH secretion (11, 12, 13, 14), and seasonal transitions in fertility (15, 16). One of the difficulties in interpreting these studies, however, is that of determining the location of the neurons modulated by GABA that underlie these effects. The same issue exists for in vivo microinfusion studies that have attempted to modulate GABAA receptor occupancy within the immediate vicinity of the GnRH neuron cell bodies located in the rostral preoptic area (rPOA) (17, 18, 19).
There is little doubt that GABA modulates directly the electrical excitability and intracellular signaling environment of GnRH neurons (3, 20, 21, 22) and that GABAergic neurotransmission plays an important role in the regulation of gonadotrophin secretion (9, 11, 13). However, the physiological roles of direct GABA inputs to GnRH neurons in terms of GnRH secretion at the median eminence remain unknown. One important clue that they may be of significance has come from electrophysiological studies in which the activity of GABAA receptors located on GnRH neurons has been correlated with different physiological states. These studies have shown correlations between the frequency of GnRH neuron GABAA postsynaptic currents (PSCs) and gonadal steroid levels (23, 24, 25, 26, 27), including the onset of the LH surge (28) and nutritional status (29).
In the present study, we have sought to examine the physiological importance of direct GABAergic inputs to GnRH neurons by generating a mutant mouse line in which there is a GnRH neuron-selective reduction in GABAA receptor signaling. The GABAA receptor is a pentamer made up primarily of different combinations of α-, β-, and γ-subunits. Of these, the γ2-subunit is particularly important, because not only it is expressed in the great majority of GABAA receptors within the brain (30), including mouse GnRH neurons (3, 31), but it is also required for the targeting of the GABAA receptor to the postsynaptic membrane (32, 33, 34). Global knockout of the γ2-subunit results in mice that die soon after birth (35). Hence, to assess the physiological roles of direct GABAergic inputs to GnRH neurons, we have used a GnRH-Cre, γ2-subunit flox strategy to generate mice in which all GnRH neurons have reduced GABAA signaling.
Materials and Methods
Experimental animals
All experiments were approved by the University of Otago Animal Welfare and Ethics Committee. Investigations were undertaken using male and female mice maintained under 12-h light, 12-h dark cycle lighting conditions (lights on 0700 h) with food and water available ad libitum. Four mouse lines were used: C57BL/6J;CBA/Ca GnRH-enhanced green fluorescent protein (EGFP) (1), C57BL/6J;CBA/Ca GnRH-Cre (36), 129SvJ/X1 (formerly 129SvJ), GABAA receptor γ2-subunit flox (fγ2) (32), and C57BL/6J ROSA26-Cre reporter (37). All mice were genotyped by PCR using primers detailed in the publications listed above.
Analysis of GnRH neuron γ2-subunit-deleted mice
The GnRH-Cre mouse line used in this study has been reported to target 97 ± 2% of GnRH neurons and, in the adult male and female, Cre expression is restricted to the GnRH neuron population (36). However, several neuronal populations express GnRH1 during embryonic and early postnatal development in the mouse (38), and it is important to identify the locations of Cre recombination throughout development, because this will reflect the extent of γ2-subunit deletion when crossed with the GnRH-Cre line. This was investigated by crossing the GnRH-Cre mouse with the Soriano ROSA26 Cre reporter line and examining four adult GnRH-Cre+/−;ROSA26+/− male and female offspring with Xgal histochemistry using the same method reported previously (38, 39). In brief, mice were anesthetized, perfused with 4% paraformaldehyde, and their brains cut on a freezing microtome in the coronal plane at 30-μm thickness. Two sets of sections were then reacted overnight in 2% Xgal and one mounted on glass slides, whereas the other was processed for dual label GnRH immunocytochemistry exactly as reported previously (40).
A two-generation GnRH-Cre x fγ2 cross was used to produce mutant GnRH-Cre+/−;fγ2/fγ2 mice (GnRH γ2 KO) with, depending on breeding strategy, either fγ2/fγ2 or GnRH-Cre+/− littermates used as controls. Confirmation of Cre-induced recombination of the floxed γ2 locus was undertaken by PCR of brain tissue dissected from the rPOA and cerebellum using primers 2 and 5 as detailed previously (32).
Electrophysiological assessment of GABAA receptors in GnRH neurons
To assess the functional impact of deletion of the γ2-subunit in GnRH neurons, a triple cross was undertaken to generate GnRH-Cre+/−;fγ2/fγ2;GnRH-EGFP+/− mice with fγ2/fγ2;GnRH-EGFP+/− and GnRH-EGFP+/+ used as controls. Whole-cell PSCs were recorded as reported previously (24). In brief, acute coronal brain slices (200-μm thickness) were prepared from adult male and female transgenic mice (6–10 wk old) and whole-cell currents recorded at room temperature (21–23 C) with the visualization of individual neurons undertaken with brief fluorescence followed by differential interference contrast microscopy (BX50WI; Olympus, Tokyo, Japan). Open resistance was in the range 4–8 mΩ, and seal resistance was in the range 1–3 GΩ. Patch pipettes were filled with a solution containing (in mm): 140 KCl, 20 HEPES, 0.5 CaCl2, 5 EGTA, and 5 MgATP (pH 7.2). The electrical activity of GnRH neurons was recorded with a headstage connected to a Multiclamp 700A amplifier (Molecular Devices, Sunnyvale, CA). Current signals were digitized at 10 kHz and filtered at 1 kHz using an analog-digital converter (Digidata 1322A) and pClamp (version 10.1; Molecular Devices).
Initially, the firing pattern of GnRH neurons was recorded for 3 min in the voltage clamp, on-cell mode with a tight gigaohm seal before membrane rupture. The firing pattern of each GnRH neuron was classified into three groups: 1) silent cell, that exhibited no action currents; 2) bursting cell, that had clusters of action currents (bursts) occurring with an interburst interval of more than 4 sec; and 3) irregular cell, all other patterns of firing.
After membrane rupture, GnRH neurons were clamped to −70 mV and allowed to stabilize for 10 min before switching to artificial cerebrospinal fluid (2–3 ml/min) containing 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX) (20 μm), DL-2-amino-5-phosphonopentanois acid (DL-AP5) (20 μm), and tetrodotoxin (TTX) (0.5 μm) to isolate miniature PSCs (mPSCs) originating from GABAA receptor activation. The amplitude, frequency, and decay time of mPSCs was measured from the last 2 min of the 10-min recording period using the Mini Analysis program (version 6.0; Synaptosoft, Inc., Leonia, NJ). A threshold for detection of mPSCs was set at 3-fold root mean square noise. The effect of endogenous GABA was assessed by measuring peak inward current. If access resistance of the whole-cell recording was higher than 30 mΩ or if there were no more than three events detected during 2 min, the cell was excluded from the analysis. Cells were then treated with 50 μm of GABA by addition to the artificial cerebrospinal fluid for 2 min in the presence of CNQX, DL-AP5, and TTX to test the effect of exogenously applied GABA. The data are expressed as mean ± sem with two-tailed Mann-Whitney U tests used to assess significance between groups. One to three GnRH neurons were recorded from each animal, and results combined to provide an average value for each animal that was then used to generate group means, i.e. n number represents animal number.
Assessment of fertility
The fertility of GnRH γ2 KO mice with either fγ2/fγ2 or GnRH-Cre+/− littermates as controls was assessed as detailed previously (36, 41). In brief, this included:
1) Puberty onset by examining for the day of vaginal opening followed by daily vaginal smearing to establish the first day of estrus.
2) Estrous cycles by undertaking daily vaginal smears for a period of 17 d from adult (>50 d old) female mice.
3) Fecundity by mating transgenic adult male mice with wild-type C57BL/6J females for a period of 3 months and noting the numbers of litters produced and the numbers of pups per litter (see Ref. 41).
4) Estrogen positive feedback by ovariectomizing female mice, implanting a 1 μg 17-β-estradiol-containing SILASTIC brand capsule and 6 d later treating the mice with 1 μg/20 g body weight estradiol benzoate. The day after, mice were anesthetized and killed at 1900–1930 h with blood taken for LH RIA and the brain perfused to enable c-Fos-GnRH dual immunocytochemistry to be undertaken (see Ref. 36).
We also assessed negative feedback in both males and females. Negative feedback in adult male mice was assessed by gonadectomy. Males were anesthetized with Halothane, a tail blood sample taken, and bilaterally gonadectomized through the scrotum. Two weeks later mice were killed by cervical dislocation and decapitation with blood collected for LH assay.
Negative feedback in female mice was assessed by gonadectomy and estradiol replacement. Mice were anesthetized with Avertin (200 μl, sc), a tail blood sample was taken and bilaterally ovariectomized. Two weeks later, mice were anesthetized with Avertin, and a further tail blood sample taken. This was followed by a sc injection of 1 μg/20 g body weight 17-β-estradiol (Sigma, South Croydon, Australia) and 3 h later cervical dislocation and decapitation, with blood collected for LH assay. Statistical analysis was undertaken by nonparametric Mann-Whitney tests for two group comparisons and ANOVA followed by Student-Newman-Keuls post hoc tests for three group comaprisons.
Plasma LH concentrations were determined by RIA using the anti-rLH-S-11 antiserum and mLH-RP reference provided by A. F. Parlow (National Hormone And Peptide Program). The intra-and interassay coefficients of variation were 8.3 and 11.2%, respectively.
Results
Cre recombination in the forebrain of the GnRH-Cre line
To verify that the GnRH-Cre line was able to recombine the floxed γ2 allele, PCR across the γ2 locus was undertaken in tissue taken from the rPOA, where the GnRH neurons reside, and from the cerebellum, where there is no Cre expression. PCR products of both approximately 1.8 kb (γ2 locus) and 850 bp (floxed locus) were detected in the rPOA, whereas only the 1.8-kb band was detected in the cerebellum (data not shown).
Histochemistry for Xgal in GnRH-Cre+/−;ROSA26+/− mice resulted in blue dot-like staining within the cytoplasm of cells located in several highly circumscribed regions within the forebrain of both males and females. Outside of the “inverted-Y” Xgal distribution typical of the GnRH neurons in the basal forebrain, Xgal staining was observed in large numbers of cells located in the lateral septum (Fig. 1A), bed nucleus of the stria terminalis, and thalamic nuclei (paraventricular, parafascicular, reunions, and medial geniculate nuclei) (Fig. 1B). A smaller number of scattered Xgal-positive cells was detected in the striatum (Fig. 1A), POA, parafornical nuclei, and zona incerta. The patterns of staining were identical in males and females.
Fig. 1.
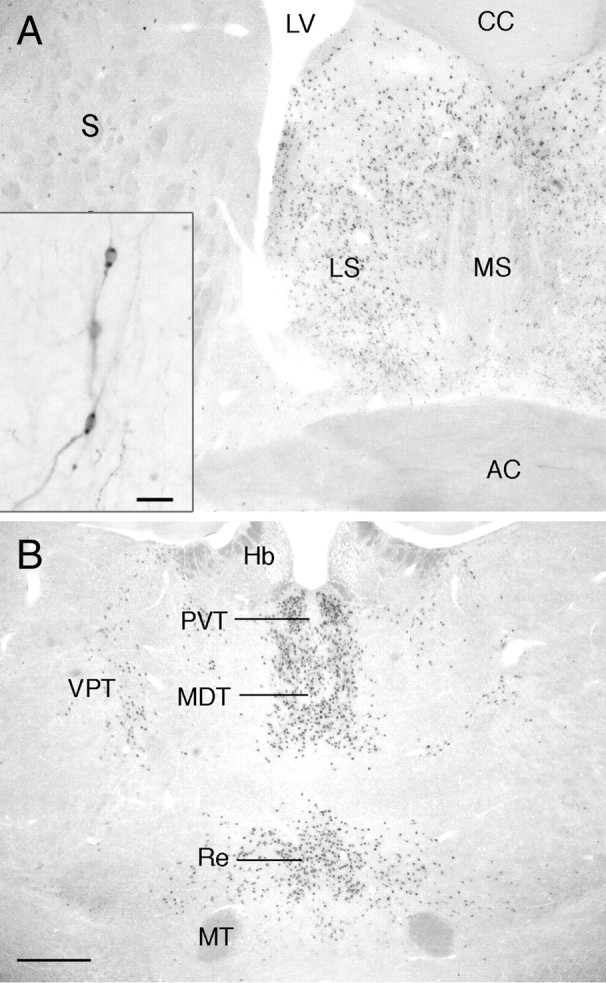
Xgal histochemistry in the brain of an adult female GnRH-Cre;GTRosa26 mouse showing the locations of cells where Cre has been effective in recombining LoxP sites to enable expression of β-galactosidase. A, A low-power view of a coronal section showing dense Xgal expression in the medial septum (MS) and a small number of scattered cells in the striatum (S). Inset shows higher power view of two Xgal-expressing GnRH neurons in the plane of focus from Xgal-GnRH dual labeling. Scale bar, 30 μm. B, Low-power view of the thalamus showing dense Xgal expression in several midline nuclei, including the paraventricular (PVT), mediodorsal (MDT), and reunions (Re) thalamic nuclei, as well as a smaller population of cells in the ventroposterior thalamic nuclei (VPT). AC, Anterior commisure; CC, corpus callosum; Hb, habenular; LV, lateral ventrical; MT, mammilothalamic tract. Scale bar, 200 μm.
Dual label Xgal-GnRH immunocytochemistry demonstrated that 98 ± 1% of GnRH neurons expressed Xgal independent of their anatomical location in the forebrain (n = 3 male and n = 3 female) (Fig. 1A, inset). These observations demonstrate that the GnRH-Cre line induces recombination of the fγ2 locus within GnRH and other neurons in the forebrain. Interestingly, in early studies, we observed that another GnRH-Cre line (42) was not able to recombine the fγ2 locus.
Abnormal GABAA receptor signaling in γ2-subunit-deleted GnRH neurons
Experiments were undertaken on littermate mice forming two groups “γ2 intact” (GnRH-EGFP+/− and GnRH-EGFP+/−;fg2/fg2) and “γ2 deleted” (GnRH-EGFP+/−; GnRH-Cre+/−;fg2/fg2). Mice from all genotypes appeared healthy and did not exhibit any overt behavioral abnormalities. The resting membrane potential and input resistance of GnRH neurons in the two groups were not different: γ2 intact (n = 15), −55 ± 2 mV and 1047 ± 109 mΩ; and γ2 deleted (n = 6), −56 ± 3 mV and 1089 ± 209 mΩ. The analysis of GnRH neuron-firing pattern revealed a nonsignificant (P = 0.1096; Chi-square test) trend toward fewer bursting cells in γ2-deleted mice. Of 28 GnRH neurons recorded from γ2-intact mice, 10 (36%), 6 (21%), and 12 (43%) exhibited irregular, silent, or bursting-type firing patterns, respectively. In contrast, of 16 GnRH neurons from γ2-deleted mice, 8 (50%), 6 (38%), and 2 cells (12%) were irregular, silent, and bursting, respectively.
Exogenous GABA (50 μm) evoked large (159 ± 36 pA) inward currents from GnRH neurons in the γ2-intact group (n = 12; Fig. 2A) but had a markedly (89%) reduced action on γ2-deleted mice (18 ± 7 pA, P = 0.008, Mann-Whitney U test; n = 6) (Fig. 2, A and B). To verify the specificity of the targeted Cre deletion, we also examined the effects of exogenous GABA on randomly selected neurons in the striatum at the same coronal level as the rPOA. In this region, we observed the same response to exogenous GABA in both γ2-intact (n = 6) and γ2-deleted (n = 7) mice (Fig. 2B).
Fig. 2.

Diminished GABAA receptor signaling in GnRH γ2 KO. A, Representative voltage-clamp trace showing the effect of 50 μm GABA on two EGFP-tagged GnRH neurons, one with intact (fg2/fg2) and one with deleted (GnRH-Cre,fg2/fg2) γ2-subunit. B, Comparison of GABA-evoked inward currents in GnRH neurons of γ2-intact (n = 12) and γ2-deleted mice (n = 6) and in unidentified neurons located in the striatum (n = 6 and 7). C and D, Representative traces showing mPSCs from GnRH neurons in which the γ2-subunit is intact (fg2/fg2) or deleted (GnRH-Cre,fg2/fg2). E, Comparison of the shape of averaged mPSC traces from GnRH neurons in which the γ2-subunit is intact (fg2/fg2) or deleted (GnRH-Cre,fg2/fg2). F, Cumulative probability plot of mPSCs amplitude. G–I, Comparison of mPSC amplitude, frequency, and decay time constant in GnRH neurons of γ2-intact (n = 14) and γ2-deleted (n = 6) mice. *, P < 0.05; **, P < 0.01; Mann-Whitney U test.
To assess endogenous GABA signaling at GnRH neuron GABAA receptors, we next examined the characteristics of mPSCs in the presence of CNQX, DL-AP5, and TTX in γ2-intact (n = 14) and γ2-deleted (n = 6) mice. Frequent (0.34 ± 0.08 Hz) mPSCs with a mean amplitude of 30.3 ± 3.2 pA and decay time of 16.3 ± 2.1 msec were observed in all GnRH neurons forming the γ2-intact group (Fig. 2C). In comparison, a reduction in the number and size of mPSCs was observed in γ2-deleted mice (Fig. 2D). Significant decreases were found in the amplitude (70% reduction; 8.8 ± 1.9 pA, P < 0.01) (Fig. 2, E–G), frequency (77% reduction; 0.08 ± 0.01 Hz, P < 0.05) (Fig. 2H), and decay time (54% reduction; 7.6 ± 2.1 msec, P < 0.05) (Fig. 2, E and I) of GnRH neurons mPSCs in γ2-deleted mice. All mPSCs were abolished by the selective GABAA channel blocker gabazine (10 μm) (data not shown).
Together, these findings indicate that the Cre-LoxP strategy used here has effectively and selectively targeted the GnRH neurons with exogenous GABAA receptor-evoked currents reduced by 90% and endogenous synaptic currents reduced by 70%.
Normal fertility in GnRH γ2 KO male mice
The fecundity of male GnRH γ2 KO mice (n = 5) was normal (Fig. 3A). Compared with littermate control mice (n = 5), the numbers of litters produced and numbers of pups per litter were not different (Fig. 3A).
Fig. 3.
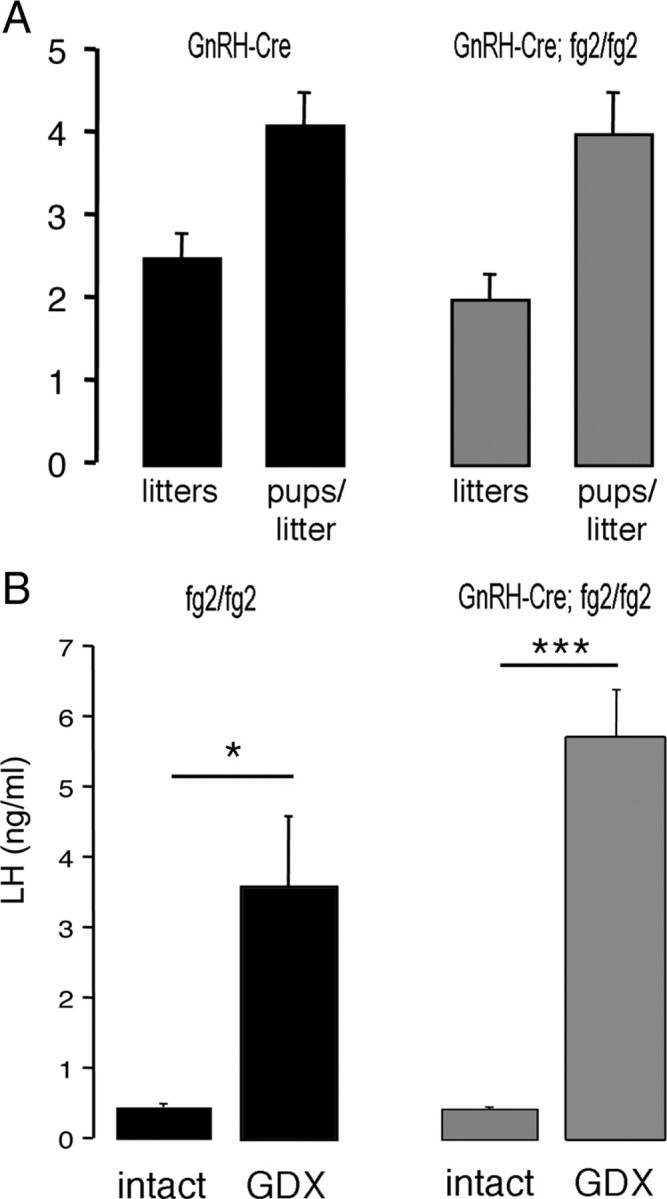
Male fertility and negative feedback in GnRH γ2 KO mice. A, Histograms showing the mean + sem numbers of litters and pups per litter born to breeding pairs in which wild-type females were mated with either GnRH γ2 KO (GnRH-Cre, fg2/fg2; n = 5) or control (GnRH-Cre, n = 5) mice. B, Histograms showing mean + sem. LH levels of intact and goandectomized (GDX) GnRH γ2 KO (n = 5) and control (fg2/fg2; n = 5) mice. *, P < 0.05; ***, P < 0.001.
Because GABA has been implicated in the negative feedback pathway suppressing LH secretion in rats (43), we examined basal LH levels and their response to gonadectomy in GnRH γ2 KO mice. Basal levels of LH were not different (P > 0.05) between control (fg2/fg2) and GnRH γ2 KO mice (0.5 ± 0.2 vs. 0.7 ± 0.3 ng/ml, respectively; n = 5 each group). Both genotypes displayed robust, significant increases in LH secretion 2 wk after gonadectomy (Fig. 3B). Although a trend for a larger increment in LH was observed in GnRH γ2 KO mice compared with controls (3.5 ± 1.3 vs. 5.0 ± 0.7ng/ml, respectively; n = 5 each group), this was not significantly different.
Normal puberty and cyclicity in GnRH γ2 KO female mice
The onset of puberty was assessed in GnRH γ2 KO and control (fg2/fg2) mice. No difference in the day of vaginal opening was found between GnRH γ2 KO (d 30.6 ± 0.5; n = 10) and control (fg2/fg2) (d 30.0 ± 0.4; n = 5) mice (Fig. 4A). Similarly, no differences were detected between the day of first estrus in GnRH γ2 KO and control mice (Fig. 4B).
Fig. 4.
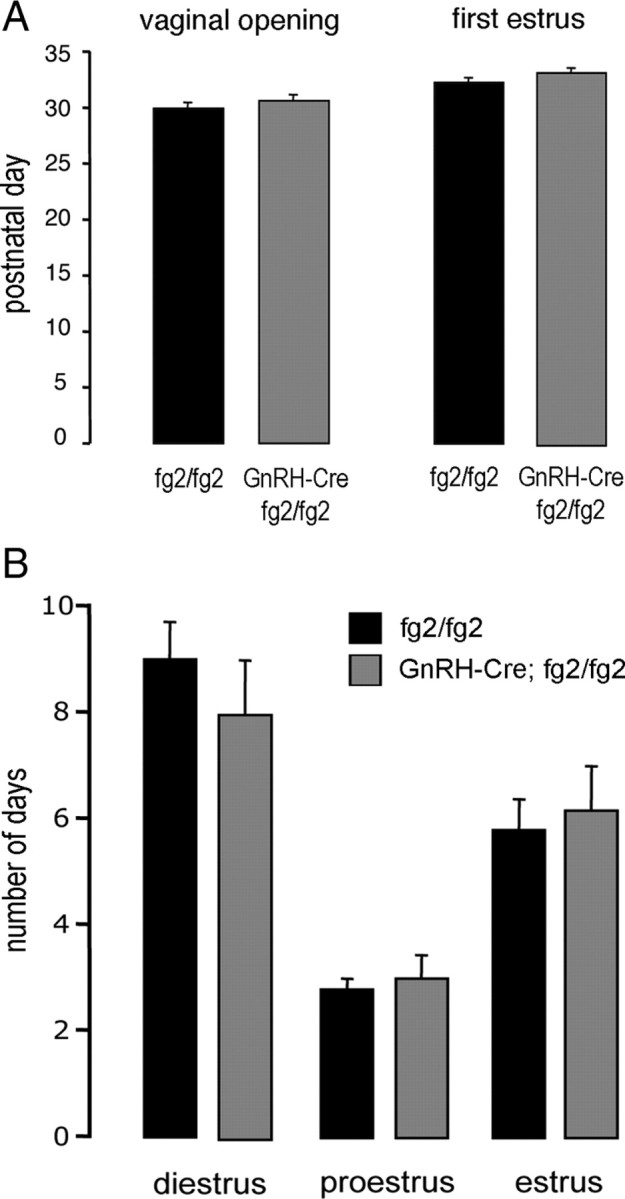
Female puberty and estrous cyclicity in GnRH γ2 KO mice. A, Histograms showing the mean + sem postnatal day of vaginal opening and first estrus for GnRH γ2 KO (n = 10) and control (fg2/fg2, n = 5) mice. B, Histograms showing the mean + sem number of days GnRH γ2 KO (n = 6) and control (fg2/fg2, n = 6) female mice were in diestrus, proestrus, and estrus over a 17 d vaginal smearing period.
The estrous cycles of GnRH γ2 KO mice (n = 6) were not different to those of controls (n = 5) with the numbers of complete cycles observed over the 17-d smearing period being 2.5 ± 0.2 and 2.3 ± 0.2, respectively, and the percentage of time spent in diestrus, proestrus, and estrus being the same in both genotypes (Fig. 4B). Because estrous cyclicity was normal, we did not formally test the fecundity of mutant mice, although GnRH γ2 KO female mice were found to be fertile in the breeding program.
Abnormal estrogen feedback in GnRH γ2 KO female mice
Negative feedback
Basal levels of LH were the same in GnRH γ2 KO (n = 10) and control (fg2/fg2 or wild type; n = 10) mice (Fig. 5A). Two weeks after ovariectomy, LH levels were significantly increased in both genotypes (P < 0.001 mutants and controls). Assessment of the ovariectomy-induced increment in LH from basal levels showed that LH levels in GnRH γ2 KO mice (1.5 ± 0.3 ng/ml) were approximately double that of controls (0.8 ± 0.1, P < 0.05) (Fig. 5A). Treatment with 17-β-estradiol significantly (P < 0.05) decreased LH concentrations 3 h later in both control and GnRH γ2 KO mice (Fig. 5A).
Fig. 5.

Estrogen positive and negative feedback in female GnRH γ2 KO mice. A, Negative feedback. Histograms showing mean + sem. LH levels in mice before (intact) and 2 wk after ovariectomy (OVX) and then 3 h after treatment with 17-β-estradiol (E2) in GnRH γ2 KO (n = 10) and control (fg2/fg2 or wild type, n = 10) mice. The increment from basal in LH after OVX is shown to the right. *, P < 0.05; **, P < 0.01; ***, P < 0.001. B–D, Estrogen positive feedback. Histogram showing (B) the mean + sem number of GnRH-immunoreactive neurons detected per section at the level of the rPOA, (C) mean + sem % GnRH neurons expressing c-Fos, and (D) the mean + sem surge levels of LH in GnRH γ2 KO (n = 5) and control (fg2/fg2; n = 6) mice.
Positive feedback
Female mice, ovariectomized and treated with estradiol that were killed at 1900–1930 h, the time of lights out, exhibited surge-like levels of LH being 12.0 ± 4.5 and 8.3 ± 2.7 ng/ml, respectively, in GnRH γ2 KO and control (fg2/fg2) mice that were not different from one another (Fig. 5D). As shown previously (36, 41), dual-label immunocytochemistry for GnRH and c-Fos resulted in the typical inverted-Y scattered distribution of the GnRH neurons stained brown and the heterogeneous black nuclear staining for c-Fos. The numbers and topography of GnRH neurons detected within the brain were the same in GnRH γ2 KO and control mice (Fig. 5B), with 40–50% of GnRH neurons in the rPOA exhibiting c-Fos immunoreactivity (Fig. 5C) in both genotypes.
Discussion
We report here that the reproductive axis is essentially normal in male and female mice, in which GnRH neurons have the great majority of their GABAA receptor activity abolished. We show that our Cre-LoxP strategy targets virtually all (98%) GnRH neurons and results in a 70–90% reduction in the amplitude of their responses to exogenous and endogenous GABA. We also find that the frequency of GABAA receptor-dependent PSCs is reduced by 77%. A change in PSC frequency is typically interpreted as a change in presynaptic GABA release, but under the present conditions, where postsynaptic GABAA receptor functioning is robustly suppressed, this reduced frequency likely represents failure of the postsynaptic membrane to respond to GABA.
As predicted by the multiple populations of cells known to express GnRH1 during embryonic development (38), we find that the GnRH-Cre mouse model used here also results in the expression of Cre outside the GnRH neuronal phenotype. This is typical for all GnRH-Cre transgenic mice generated to date (44, 45), including the knock-in DuLac line (42) that, interestingly, did not recombine the fγ2 locus (Campbell, R., and A. Herbison, unpublished data). We note that the expression of Xgal reported here is much wider than for Cre itself (36), because the expression of Xgal represents any cell that has expressed Cre at any time during development, whereas Cre represents only cells actively transcribing the transgene around the time of death. The Xgal in multiple limbic and thalamic brain regions indicates that GABAA receptor activity would have been reduced in cells within all of these regions in our γ2-deleted mice. However, neurons in these brain regions are not believed to be involved in the GnRH neuronal network and clearly had no impact on fertility or generated any other obvious phenotype. We note, however, that we did not formally assess mating behavior (45).
We observed only one abnormal phenotype in GnRH γ2 KO mice. Although puberty onset, fertility, estrous cycling, estrogen positive feedback, and basal LH levels were all normal in female GnRH γ2 KO mice, their negative feedback mechanism appeared changed when investigated further with ovariectomy and estrogen replacement. We found that the increment from basal LH levels after ovariectomy in GnRH γ2 KO mice was twice that of control mice and that 17-β-estradiol replacement failed suppressed LH levels after 3 h in intact and mutant mice. These intriguing observations suggest that the estrogen negative feedback mechanism is in some way different in GnRH γ2 KO mice with a more robust suppression of LH secretion. A similar trend was seen for the postgonadectomy rise in LH secretion in GnRH γ2 KO male mice but did not reach statistical significance. Another nonsignificant trend noted in these mice was that relatively few GnRH neurons exhibited burst firing. Interestingly, a recent study that deleted the γ2-subunit selectively from cerebellar Purkinje neurons also noted abnormalities in firing patterning (46), indicating the potential importance of γ2-containing GABAA receptors in generating patterned neural activity.
Prior studies examining the GABAA receptor activity of GnRH neurons have noted both positive and negative correlations between the magnitude of GABAA receptor signaling, GnRH neuron firing rates, and LH secretion (23, 24, 25, 26, 27, 28, 29), suggesting a causal relationship. For example, chronic fasting induces a 50% reduction in GnRH neuron GABAA receptor mPSCs, and this correlates with a significant reduction in circulating LH levels (29). Thus, at face value, a 70% reduction in GnRH neuron GABAA receptor mPSCs would be expected to result in a major disruption of gonadotropin secretion. However, this is clearly not the case. There would appear to be at least two potential explanations for this conundrum: 1) GABAA receptor signaling at the GnRH neurons is not critical in shaping the behavior of the GnRH neuron, and 2) GABAA receptor signaling at the GnRH neuron is important but not critical and can be compensated.
At present, it is hard to differentiate between these two possibilities. Although in vivo experiments have clearly demonstrated rapid effects of GABAA receptor modulation on LH secretion (11, 13, 47), it is not known if these effects occur at the GnRH neuron or elsewhere within the brain. Equally, there is no guarantee that PSC correlative studies undertaken to date have been examining causative factors. For example, the 50% reduction in GABAA PSCs with fasting may represent only one of several altered inputs to GnRH neurons and may not itself be responsible for the suppressed LH release. Thus, one possibility that cannot be excluded as yet is that direct GABAA receptor signaling is not necessary for shaping the electrical activity of GnRH neurons to physiological stimuli.
The alternative explanation is that GABAA receptor signaling is either sufficient at a much lower level (i.e. 25%) and/or its loss can be compensated for in a knockout mouse line such as this. In this regard, it is important to highlight that the γ2-subunit will be deleted from GnRH neurons during embryogenesis, when Cre driven by this transgene begins to be expressed in differentiated GnRH neurons (38). As such, all GnRH neurons in GnRH γ2 KO mice will have migrated into the brain and integrated into the GnRH neuronal network without γ2-dependent GABAA receptors. As an aside, we note here again that the correct migration of GnRH neurons does not require the γ2-subunit in mice (31). The deletion of the γ2-subunit early in development would likely provide ample time for multiple levels of compensation to occur, including the use of other subunits to generate GABAA receptors, the up-regulation of GABAB receptors, alterations in intrinsic conductances, or replacing GABA’s influence with other neurotransmitters.
Although a very substantial reduction in GABAA receptor functioning occurs, it is not ablated completely. The identity of the remaining GABAA receptors is not known, and may represent receptors not normally expressing the γ2-subunit, including pentamers made from only α- and β-subunits, or receptors in which the loss of γ2 has been compensated for by the incorporation of other subunits. There is evidence in GnRH neurons for low levels of expression of the γ1-, δ-, and ε-subunits (4, 27, 48), each of which can replace the γ2-subunit in the GABAA receptor pentamer (30). However, evidence from the whole brain of neonatal γ2 knockout mice suggests that little if any subunit compensation occurs at this time point (35). Although GnRH neurons express GABAB receptors (8), and GABAB receptors in the brain are involved in regulating GnRH neurons (49), the residual GABA PSCs observed in γ2-deleted mice are totally dependent upon GABAA receptors. Another level of “GABA compensation” that is known to occur in GABAA receptor subunit-deleted mice is that of up-regulating tonic GABA signaling through the modulation of GABA transporters (50). If this occurs in GnRH γ2 KO, it is subtle, because the loss of response here to both exogenous and endogenous GABA indicates that both extrasynaptic (tonic) and synaptic GABAA receptors are mostly abolished in GnRH neurons of GnRH γ2 KO mice. Another plausible scenario is that the loss of GABAA influence in the GnRH neuron is compensated for through the modulation of other ion channels. In this regard, it is notable that deletion of the GABAA receptor α6- and δ-subunits from cerebellar granule cells results in the up-regulation of a likely potassium-leak conductance that maintains their excitability (51).
Finally, there is a possibility that other neurotransmitters may have taken over roles of GABAergic inputs, but the identity of these inputs is unknown. In this regard, it is interesting to note the altered negative feedback dynamics in γ2-deleted mice. Although a case has been made for GABAergic inputs mediating negative feedback in both males and females, this is by no means an exclusive role and in vivo studies support the involvement of several other neurotransmitters in this mechanism (see Ref. 11). Thus, it may be that the more potent suppression of LH revealed by ovariectomy in GnRH γ2 KO mice represents the use of a different combination of neuronal inputs that are, nevertheless, able to maintain normal feedback and normal fertility. It seems clear that the GnRH neurons themselves have substantial in-built redundancy in terms of maintaining fertility (41), and this may extend to rearrangement of their synaptic environment.
In summary, using a Cre-LoxP strategy, we show here that normal GABAA receptor activity at the GnRH neuron is largely dispensable for normal reproductive activity in the mouse. Reducing the GABA response of GnRH neurons by 70–90% had no effect on male fertility or female puberty onset and cyclicity, although the negative feedback mechanism appeared to be operating differently. These findings suggest that either γ2-subunit-deleted GnRH neurons can “rebalance” their electrical excitability through unknown compensatory mechanisms or that normal levels of GABAA receptor-mediated transmission at the GnRH neuron are not important in shaping GnRH secretion at the median eminence.
Acknowledgments
We thank Dr. Parlow for RIA reagents, Ms. Zhi-Yi Ong for technical assistance, and Dr. X. Liu, S. Constantin and Dr. K. Iremonger for review of an initial draft of the manuscript.
Footnotes
This work was supported by the New Zealand Health Research Council.
Disclosure Summary: The authors have nothing to disclose.
First Published Online June 23, 2010
Abbreviations: CNQX, 6-Cyano-7-nitroquinoxaline-2,3-dione; DL-AP5, DL-2-amino-5-phosphonopentanois acid; EGFP, enhanced green fluorescent protein; GABA, γ-aminobutyric acid; GnRH γ2 KO, GnRH-Cre+/−;fγ2/fγ2 mice; mPSC, miniature PSC; PSC, postsynaptic current; rPOA, rostral preoptic area; TTX, tetrodotoxin.
References
- 1.Han SK, Todman MG, Herbison AE 2004. Endogenous GABA release inhibits the firing of adult gonadotropin-releasing hormone neurons. Endocrinology 145:495–499 [DOI] [PubMed] [Google Scholar]
- 2.Moenter SM, DeFazio RA 2005. Endogenous γ-aminobutyric acid can excite gonadotropin-releasing hormone neurons. Endocrinology 146:5374–5379 [DOI] [PubMed] [Google Scholar]
- 3.Sim JA, Skynner MJ, Pape JR, Herbison AE 2000. Late postnatal reorganization of GABAA receptor signalling in native GnRH neurons. Eur J Neurosci 12:3497–3504 [DOI] [PubMed] [Google Scholar]
- 4.Pape JR, Skynner MJ, Sim JA, Herbison AE 2001. Profiling γ-aminobutyric acid (GABA(A)) receptor subunit mRNA expression in postnatal gonadotropin-releasing hormone (GnRH) neurons of the male mouse with single cell RT-PCR. Neuroendocrinology 74:300–308 [DOI] [PubMed] [Google Scholar]
- 5.Jung H, Shannon EM, Fritschy JM, Ojeda SR 1998. Several GABAA receptor subunits are expressed in LHRH neurons of juvenile female rats. Brain Research 780:218–229 [DOI] [PubMed] [Google Scholar]
- 6.Yin C, Ishii H, Tanaka N, Sakuma Y, Kato M 2008. Activation of A-type γ-amino butyric acid receptors excites gonadotrophin-releasing hormone neurones isolated from adult rats. J Neuroendocrinol 20:566–575 [DOI] [PubMed] [Google Scholar]
- 7.Sullivan SD, DeFazio RA, Moenter SM 2003. Metabolic regulation of fertility through presynaptic and postsynaptic signaling to gonadotropin-releasing hormone neurons. J Neurosci 23:8578–8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ 2009. γ-Aminobutyric acid B receptor mediated inhibition of gonadotropin-releasing hormone neurons is suppressed by kisspeptin-G protein-coupled receptor 54 signaling. Endocrinology 150:2388–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terasawa E, Fernandez DL 2001. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev 22:111–151 [DOI] [PubMed] [Google Scholar]
- 10.Clarkson J, Herbison AE 2006. Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Mol Cell Endocrinol 254–255:32–38 [DOI] [PubMed]
- 11.Herbison AE 2006. Physiology of the GnRH neuronal network. In: Neill JD, ed. Knobil and Neill’s physiology of reproduction. 3rd ed. San Diego, CA: Academic Press; 1415–1482
- 12.Mitsushima D, Tin-Tin-Win-Shwe, Kimura F 2003. Sexual dimorphism in the GABAergic control of gonadotropin release in intact rats. Neurosci Res 46:399–405 [DOI] [PubMed] [Google Scholar]
- 13.Jackson GL, Kuehl D 2002. γ-Aminobutyric acid (GABA) regulation of GnRH secretion in sheep. Reprod Suppl 59:15–24 [PubMed] [Google Scholar]
- 14.Clarke IJ, Scott CJ 1993. Studies on the neuronal systems involved in the oestrogen-negative feedback effect on gonadotrophin releasing hormone neurons in the ewe. Hum Reprod 8(Suppl 2):2–6 [DOI] [PubMed] [Google Scholar]
- 15.Scott CJ, Clarke IJ 1993. Evidence that changes in the function of the subtypes of the receptors for γ-aminobutyric acid may be involved in the seasonal changes in the negative-feedback effects of oestrogen on gonadotropin-releasing hormone secretion and plasma luteinizing hormone levels in the ewe. Endocrinology 133:2904–2912 [DOI] [PubMed] [Google Scholar]
- 16.Jansen HT, Cutter C, Hardy S, Lehman MN, Goodman RL 2003. Seasonal plasticity within the gonadotropin-releasing hormone (GnRH) system of the ewe: changes in identified GnRH inputs and glial association. Endocrinology 144:3663–3676 [DOI] [PubMed] [Google Scholar]
- 17.Jarry H, Leonhardt S, Wuttke W 1991. γ-Aminobutyric acid neurons in the preoptic/anterior hypothalamic area synchronize the phasic activity of the gonadotropin-releasing hormone pulse generator in ovariectomized rats. Neuroendocrinology 53:261–267 [DOI] [PubMed] [Google Scholar]
- 18.Herbison AE, Chapman C, Dyer RG 1991. Role of medial preoptic GABA neurones in regulating luteinizing secretion in the ovariectomised rat. Exp Brain Res 87:345–352 [DOI] [PubMed] [Google Scholar]
- 19.Herbison AE, Dyer RG 1991. Effect on luteinizing hormone secretion of GABA receptor modulation in the medial preoptic area at the time of proestrous luteinizing hormone surge. Neuroendocrinology 53:317–320 [DOI] [PubMed] [Google Scholar]
- 20.DeFazio RA, Heger S, Ojeda SR, Moenter SM 2002. Activation of A-type γ-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol 16:2872–2891 [DOI] [PubMed] [Google Scholar]
- 21.Constantin S, Jasoni CL, Wadas B, Herbison AE 2010. γ-Aminobutyric acid and glutamate differentially regulate intracellular calcium concentrations in mouse gonadotropin-releasing hormone neurons. Endocrinology 151:262–270 [DOI] [PubMed] [Google Scholar]
- 22.Watanabe M, Sakuma Y, Kato M 2009. GABAA receptors mediate excitation in adult rat GnRH neurons. Biol Reprod 81:327–332 [DOI] [PubMed] [Google Scholar]
- 23.Jones BL, Whiting PJ, Henderson LP 2006. Mechanisms of anabolic androgenic steroid inhibition of mammalian epsilon-subunit-containing GABAA receptors. J Physiol 573:571–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romanò N, Lee K, Abrahám IM, Jasoni CL, Herbison AE 2008. Nonclassical estrogen modulation of presynaptic GABA terminals modulates calcium dynamics in gonadotropin-releasing hormone neurons. Endocrinology 149:5335–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan SD, Moenter SM 2005. GABAergic integration of progesterone and androgen feedback to gonadotropin-releasing hormone neurons. Biol Reprod 72:33–41 [DOI] [PubMed] [Google Scholar]
- 26.Pielecka-Fortuna J, Moenter SM 2010. Kisspeptin increases γ-aminobutyric acidergic and glutamatergic transmission directly to gonadotropin-releasing hormone neurons in an estradiol-dependent manner. Endocrinology 151:291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penatti CA, Davis MC, Porter DM, Henderson LP 2010. Altered GABAA receptor-mediated synaptic transmission disrupts the firing of gonadotropin-releasing hormone neurons in male mice under conditions that mimic steroid abuse. J Neurosci 30:6497–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christian CA, Moenter SM 2007. Estradiol induces diurnal shifts in GABA transmission to gonadotropin-releasing hormone neurons to provide a neural signal for ovulation. J Neurosci 27:1913–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan SD, Moenter SM 2004. γ-Aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology 145:1194–1202 [DOI] [PubMed] [Google Scholar]
- 30.Olsen RW, Sieghart W 2008. International union of pharmacology. LXX. Subtypes of γ-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 60:243–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simonian SX, Skynner MJ, Sieghart W, Essrich C, Luscher B, Herbison AE 2000. Role of the GABAA receptor γ2 subunit in the development of gonadotropin-releasing hormone neurons in vivo. Eur J Neurosci 12:3488–3496 [DOI] [PubMed] [Google Scholar]
- 32.Schweizer C, Balsiger S, Bluethmann H, Mansuy IM, Fritschy JM, Mohler H, Lüscher B 2003. The γ2 subunit of GABA(A) receptors is required for maintenance of receptors at mature synapses. Mol Cell Neurosci 24:442–450 [DOI] [PubMed] [Google Scholar]
- 33.Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B 1998. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nature Neuroscience 1:563–571 [DOI] [PubMed] [Google Scholar]
- 34.Alldred MJ, Mulder-Rosi J, Lingenfelter SE, Chen G, Lüscher B 2005. Distinct γ2 subunit domains mediate clustering and synaptic function of postsynaptic GABAA receptors and gephyrin. J Neurosci 25:594–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Günther U, Bencon J, Benke D, Fritschy JM, Reyes G, Knoflach F, Crestani F, Aguzzi A, Arigoni M, Lang Y, Bluethmann H, Mohler H, Luscher B 1995. Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of γ-aminobutyric acid type A receptors. Proc Natl Acad Sci USA 92:7749–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE 2006. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soriano P 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21:70–71 [DOI] [PubMed] [Google Scholar]
- 38.Skynner MJ, Slater R, Sim JA, Allen ND, Herbison AE 1999. Promoter transgenics reveal multiple gonadotropin-releasing hormone-1-expressing cell populations of different embryological origin in mouse brain. J Neurosci 19:5955–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pape JR, Skynner MJ, Allen ND, Herbison AE 1999. Transgenics identify distal 5′- and 3′ sequences specifying gonadotropin-releasing hormone expression in adult mice. Mol Endocrinol 13:2203–2211 [DOI] [PubMed] [Google Scholar]
- 40.Thanky NR, Slater R, Herbison AE 2003. Sex differences in estrogen-dependent transcription of gonadotropin-releasing hormone (GnRH) gene revealed in GnRH transgenic mice. Endocrinology 144:3351–3358 [DOI] [PubMed] [Google Scholar]
- 41.Herbison AE, Porteous R, Pape JR, Mora JM, Hurst PR 2008. Gonadotropin-releasing hormone neuron requirements for puberty, ovulation, and fertility. Endocrinology 149:597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon H, Enquist LW, Dulac C 2005. Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 123:669–682 [DOI] [PubMed] [Google Scholar]
- 43.Grattan DR, Rocca MS, Sagrillo CA, McCarthy MM, Selmanoff M 1996. Antiandrogen microimplants into the rostral medial preoptic area decrease γ-aminobutyric acidergeic neuronal activity and increase luteinizing hormone secretion in the intact male rat. Endocrinology 137:4167–4173 [DOI] [PubMed] [Google Scholar]
- 44.Wolfe A, Divall S, Singh SP, Nikrodhanond AA, Baria AT, Le WW, Hoffman GE, Radovick S 2008. Temporal and spatial regulation of CRE recombinase expression in gonadotrophin-releasing hormone neurones in the mouse. J Neuroendocrinol 20:909–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimshek DR, Bus T, Grinevich V, Single FN, Mack V, Sprengel R, Spergel DJ, Seeburg PH 2006. Impaired reproductive behavior by lack of GluR-B containing AMPA receptors but not of NMDA receptors in hypothalamic and septal neurons. Mol Endocrinol 20:219–231 [DOI] [PubMed] [Google Scholar]
- 46.Wulff P, Schonewille M, Renzi M, Viltono L, Sassoè-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI 2009. Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci 12:1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terasawa E 2001. Luteinizing hormone-releasing hormone (LHRH) neurons: mechanism of pulsatile LHRH release. Vitam Horm 63:91–129 [DOI] [PubMed] [Google Scholar]
- 48.Moragues N, Ciofi P, Lafon P, Tramu G, Garret M 2003. GABAA receptor epsilon subunit expression in identified peptidergic neurons of the rat hypothalamus. Brain Res 967:285–289 [DOI] [PubMed] [Google Scholar]
- 49.Catalano PN, Di Giorgio N, Bonaventura MM, Bettler B, Libertun C, Lux-Lantos VA 2010. Lack of functional GABA(B) receptors alters GnRH physiology and sexual dimorphic expression of GnRH and GAD-67 in the brain. Am J Physiol Endocrinol Metab 298:E683–E696 [DOI] [PubMed]
- 50.Ortinski PI, Turner JR, Barberis A, Motamedi G, Yasuda RP, Wolfe BB, Kellar KJ, Vicini S 2006. Deletion of the GABA(A) receptor α1 subunit increases tonic GABA(A) receptor current: a role for GABA uptake transporters. J Neurosci 26:9323–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M 2001. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409:88–92 [DOI] [PubMed] [Google Scholar]