

Abstract
Developing new therapies for stroke is urgently needed, as this disease is the leading cause of death and disability worldwide, and the existing treatment is only available for a small subset of patients. The interruption of blood flow to the brain during ischemic stroke launches multiple immune responses, characterized by infiltration of peripheral immune cells, the activation of brain microglial cells, and the accumulation of immune mediators. Copper is an essential trace element that is required for many critical processes in the brain. Copper homeostasis is disturbed in chronic neurodegenerative diseases and altered in stroke patients, and targeted copper delivery has been shown to be protective against chronic neurodegeneration. This study was undertaken to assess whether the copper bis(thiosemicarbazone) complex, CuII(atsm), is beneficial in acute brain injury, in preclinical mouse models of ischemic stroke. We demonstrate that the copper complex CuII(atsm) protects neurons from excitotoxicity and N2a cells from OGD in vitro, and is protective in permanent and transient ischemia models in mice as measured by functional outcome and lesion size. Copper delivery in the ischemic brains modulates the inflammatory response, specifically affecting the myeloid cells. It reduces CD45 and Iba1 immunoreactivity, and alters the morphology of Iba1 positive cells in the ischemic brain. CuII(atsm) also protects endogenous microglia against ischemic insult and reduces the proportion of invading monocytes. These results demonstrate that the copper complex CuII(atsm) is an inflammation-modulating compound with high therapeutic potential in stroke and is a strong candidate for the development of therapies for acute brain injury.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-016-0504-9) contains supplementary material, which is available to authorized users.
Key Words: CuII(atsm), Stroke, Microglia, Neuron, Neuroinflammation
Introduction
Ischemic stroke, caused by blockage of blood flow in the brain, is a leading cause of death and long-term disability because of limited therapeutic interventions that are available in clinics. Despite significant efforts, drugs that have the potential to limit neuronal damage after the onset of ischemic stroke have not been discovered. The lack of efficient therapeutics for stroke is largely related to the poor translation of preclinical results into clinics. Due to the heterogeneity of stroke types, this disorder is challenging to model in laboratory settings, and results gained from one preclinical model rarely represent stroke in a broad number of patients.
The interruption of blood flow to the brain during ischemic stroke launches multiple immune responses, which are characterized by infiltration of peripheral immune cells, the activation of resident brain immune cells, and the accumulation of immune mediators [1]. Microglia, the resident immune cells of the brain, are a heterogeneous group of cells that have multiple roles in promoting and modulating brain functions under physiological conditions, including phagocytosis of debris or dying cells, neurogenesis and synaptic modulation [2, 3]. Microglia also have the potential to contribute to the neurological outcome of brain injury [4]. During acute brain injury, microglia rapidly undergo dramatic morphological and phenotypic changes. Considering the heterogeneous nature of microglia, it is not surprising that studies assessing the effect of microglia on stroke outcome have yielded controversial results. In any case, neuroinflammation and blood brain barrier disruption are critical steps in the loss of the neurogliavascular network integrity and exacerbation of ischemic damage, and activated microglia are a driving factor in these phenomena [5]. They up-regulate the expression of particular cell surface antigens and produce inflammatory mediators such as tumor necrosis factor (TNF), which can promote neurotoxicity [6]. However, microglia also possess several beneficial functions in stroke, including the promotion of neurogenesis and production of neurotrophic factors [7]. Furthermore, ablation of microglia has been shown to exacerbate ischemic injury in the brain [8].
Copper is an essential trace element that is required for many critical processes in the brain [9]. While copper homeostasis is disturbed in chronic neurodegenerative diseases such as Alzheimer’s disease (AD) [10], little is known about copper homeostasis in acute ischemic stroke. Several years ago, Kodali et al. reported an increase in the plasma copper concentrations of ischemic stroke patients [11]. This observation was supported by a recent paper demonstrating that serum levels of both total copper and copper loosely bound to small molecules are elevated in serum of ischemic stroke patients [12]. These papers provide evidence for copper dyshomeostasis upon ischemic damage in patients. In mouse models, chronic intake of copper has been reported to aggravate ischemic damage, an effect attributed to reduction of angiogenesis [13].
Restoration of brain copper homeostasis is protective against chronic neurodegeneration in animal models of disease [14]. The copper bis(thiosemicarbazones) are stable, lipophilic neutral copper(II) complexes that are capable of crossing both cell membranes and the blood brain barrier [15, 16]. These metal delivery and redistribution agents are neuroprotective in animal models of AD, Parkinson’s disease, and amyotrophic lateral sclerosis [17–20]. Furthermore, we have recently demonstrated that CuII(atsm) reduces brain inflammation caused by peripheral administration of bacterial lipopolysaccharide, and that copper delivery is anti-inflammatory in the chronic neuroinflammatory milieu of AD model mice (Choo et al. submitted). To our knowledge, therapeutic approaches targeted to modulation of copper homeostasis in ischemic stroke, where significant neuroinflammation takes place, have not previously been tested.
To address the issue of poor translation of stroke therapeutics into human patients, in this study we tested the copper complex CuII(atsm) in both permanent and transient ischemia mouse models, and characterized the inflammatory responses after delivery of copper in the ischemic brains. We demonstrate that CuII(atsm) is protective in both permanent and transient ischemia models, and that copper delivery in the ischemic brains modulates the inflammatory response, specifically affecting the myeloid cells. These results demonstrate that CuII(atsm) is a compound with high therapeutic potential in stroke and is a strong candidate for the development of therapies for acute brain injury.
Materials and Methods
Cortical Neuron Cultures and Excitotoxicity Assay
Primary cortical neuron cultures were prepared from E15 embryos. Cortices were dissected, after which tissues were dissociated with trypsin (0.0125% for 15 min at 37 °C, Sigma-Aldrich, St. Louis, MO, USA). Neurons were counted and plated on poly-d-lysine (Sigma-Aldrich, St. Louis, MO, USA) coated 48-well plates at a density of 150,000 cells/well in Neurobasal media containing 2% B27, 500 μM l-glutamine and 1% penicillin–streptomycin (all ThermoFisher Scientific, Waltham, MA, USA). 50% of the medium was changed four days after plating and cultures were used for experiments on days 6–7 in vitro.
Treatment of cortical neurons was carried out in 50% fresh Neurobasal medium containing 2% B27, 500 μM l-glutamine and 1% penicillin–streptomycin and 50% media collected from the cells. Neurons were treated with 1 μM CuII(atsm) prepared as above and/or 400 μM glutamate (Sigma-Aldrich, St. Louis, MO, USA) for 24 h prior to measurement of cell viability by the MTT assay.
N2a Cell Cultures and Oxygen-Glucose Deprivation (OGD)
Mouse neuroblastoma 2a cell line (N2a) was cultured on standard cell culture dishes in Dulbecco’s Modified Eagle Medium with GlutaMAX-1 containing D-glucose (4.5 g/L) and sodium pyruvate (0.11 g/L) supplemented with 10% heat inactivated FBS and 1% penicillin-streptomycin (all ThermoFisher Scientific, Waltham, MA, USA). For experiments, cells were plated on 48-well plates (coated similarly as the plates for primary cortical neurons) at the density of 37,500 cells/well. 24 h after plating, media was replaced with DMEM without glucose and sodium pyruvate but supplemented as the culture media (ThermoFisher Scientific, Waltham, MA, USA). The plate was then placed into an incubator with 1% oxygen and 5% carbon dioxide (ProOx C21, Biospherix Ltd, Parish, NY, USA). A control plate was maintained in culture media in a regular incubator. Passages 10–14 were used for the experiments.
MTT Viability Assay
The MTT reduction assay was performed 24 hours after exposure to glutamate or OGD according to Denizot & Lang [21] with the following modifications. Briefly, following removal of the media (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) was added to cells at a concentration of 120 μM, after which the cells were incubated for 2 hours at 37 °C at 5% CO2. Following removal of the media cells were dissolved in DMSO. The absorbances were read at 585 nm with a Wallac Victor2 1420 multilabel counter (PerkinElmer Inc, Waltham, MA, USA). The results were calculated as relative absorbances compared to the control wells. All cell viability assays were repeated three times with six replicates in each treatment group.
Mice
All animal experiments were approved by the National Animal Experiment Board of Finland and followed the Council of Europe Legislation and Regulation for Animal Protection, or by the Florey Institute animal ethics committee (15-019) and were performed in accordance with the National Health and Medical Research Council Australia guidelines. Mice were housed in individual cages in conditions of controlled humidity, temperature, and light conditions. Water and food were provided ad libitum. Three- to seven-month old male Balb/cOlaHsd mice (Harlan Laboratories B.V., An Venrey, Netherlands) were used for the permanent ischemia studies and 3-month old male C57Bl/6Arc mice (Animal Resources Centre, Western Australia) for the transient ischemia studies. Mice were randomized into treatment groups using GraphPad QuickCalcs (GraphPad Software, San Diego, CA, USA), and all the analyses were performed blinded to the experimental groups.
Ischemia Surgeries and Treatment of Mice with CuII(atsm)
Transient acute focal cerebral ischemia was induced by the intraluminal middle cerebral artery occlusion (MCAO) as described previously [22, 23]. Mice were deeply anaesthetized with 5% isoflurane in 30% O2/70% N2O using the Anesthesia system (Mediquip, Australia). The level of isoflurane was then reduced and maintained at 1%. A 4-mm distal nylon monofilament (30 mm in length, 0.16 mm in diameter, Amber, Japan) segment was coated with 0.21–0.22 mm diameter silicone (Henkel, Australia). MCAO was performed by insertion of the monofilament via the common carotid artery into the left internal carotid artery, advanced 9–10 mm past the carotid bifurcation until a slight resistance was felt. The filament was left in place for 60 min, and then withdrawn for reperfusion. In the sham-operated animals, the occluding filament was inserted only 5 mm above the carotid bifurcation. Mice were excluded from further studies if excessive bleeding occurred during surgery, if the operation time exceeded 90 min, if the mouse failed to recover from anesthesia within 15 min, or if hemorrhage was found in the brain slices or at the base of the circle of Willis during postmortem examination.
For permanent occlusion of the MCA, mice were deeply anesthetized with 5% isoflurane in 30% O2/70% N2O and the anesthesia was maintained at 2% during the surgery. The left MCA was permanently occluded as previously described [24]. Briefly, first the temporal bone was first exposed and a 1-mm-diameter hole was drilled to expose the artery. The dura was removed, after which the artery was lifted and occluded by a thermocoagulator (Aaron Medical Industries Inc., Clearwater, FL, USA). MCA occlusion was confirmed by cutting the artery, after which the temporal muscle was replaced and the wound was sutured. The body temperatures of animals were maintained at 37 ± 0.5 °C throughout the procedures using a heat-pad. The temperature and respiratory rates were monitored during the surgery. The sham mice went through all the same procedures except the occlusion of the MCA. The exclusion criteria were predetermined; mice with hemorrhages visible in magnetic resonance imaging (MRI), bleeding during the surgery, and unsuccessful induction of ischemia were excluded.
CuII(atsm) was prepared according to published procedures [25]. The mice were treated with CuII(atsm) dissolved in standard suspension vehicle (SSV) solution containing 0.9% (w/v) NaCl, 0.5% (w/v) sodium carboxymethylcellulose, 0.5% (v/v) benzyl alcohol, 0.4% (v/v) Tween 80, or SSV alone by oral gavage. For the transient MCAO mice were treated daily at a dose of 15 mg/kg while for the permanent MCAO the mice were treated daily at a dose of 60 mg/kg. Mice were sacrificed at 1 or 3 days post injury (dpi) and tissues were collected as described below.
Post-Surgery Evaluation of Outcome
After 24 h of transient MCAO induction, neurological deficits of three mice in each treatment group were evaluated by a simple scale (five-point scale) as described previously [26]: 0, no observable deficit; 1, right forelimb flexion; 2, decreased resistance to left lateral push (and right forelimb flexion) without circling; 3, same behavior as grade 2, with circling to right; 4, severe rotation progressing into barreling, loss of walking or righting reflex. Two investigators blinded to the experimental groups performed the neurological assessment post-surgery.
At 1 dpi the locomotor activity of the mice subjected to permanent MCAO was assessed by the latency-to-move test as previously described [27]. The mice (8–12 in each treatment group) were placed on a flat surface and the time to move one body length (7 cm) was recorded by an investigator blinded to the treatment groups. Each mouse received two trials.
Measurement of Lesion Volume
TTC staining was used to quantify the lesion size following transient ischemia using three animals in each treatment group. 24 hours after the induction of MCAO the mice were euthanized with an overdose of sodium pentobarbitone (Lethabarb, 100 mg/kg). The brain was removed rapidly and frozen at −20 °C for 20 min. Coronal slices were made at 2 mm intervals from the frontal poles, and sections were immersed in 0.5% 2,3,5-tripenyltetrazolium chloride (TTC, Sigma-Aldrich, St. Louis, MO, USA) in phosphate buffered saline (PBS) at 37 °C for 20 min. The presence or absence of infarction was determined by examining TTC-stained sections for the areas on the side of infarction that did not stain with TTC. The brain slices were fixed in 4% paraformaldehyde at 4 °C until imaging. Serial sections were photographed using a digital camera and the area of infarct was quantified with Image J (1.49 m, NIH) by an investigator blinded to the experimental groups. The area of infarct, the area of ipsilateral hemisphere, and the area of the contralateral hemisphere were measured for each section by a blinded operator. The volume was calculated by summing the representative areas in all sections and multiplying by the slice thickness, then correcting for edema, as previously described [28]: Corrected Infarct Volume (CIV) = contralateral hemisphere volume – (ipsilateral hemisphere volume – infarct volume).
MRI imaging was utilized to determine the lesion volume following permanent MCAO. A vertical 9.4 T Oxford NMR 400 magnet (Oxford Instrument PLC, Abington, UK) was used for visualizing the lesion as previously described [24]. The mice were anesthetized with 5% isoflurane in 30% O2/70% N2O and multislice T2-weighted images (repetition time 3000 ms, echo time 40 ms, matrix size 128 × 256, field of view 19.2 × 19.2 mm2, slice thickness 0.8 mm and number of slices 12) were obtained. The images were analyzed using in-house made software Aedes in the Matlab environment (Math-works, Natick, MA, USA). The infarction volume was calculated as previously described by using the following formula: Infarct volume = (volume of left hemisphere − (volume of right hemisphere − measured infarct volume)) / volume of left hemisphere [29].
Immunohistochemistry
Mice were euthanized at 1 (7–8 mice/treatment group) or 3 dpi (5–8 mice/treatment group) for tissue collection. Mice were anesthetized with 250 mg/kg Avertin and perfused transcardially with heparinized (2500 IU/L) saline. The brains were removed and post-fixed in 4% paraformaldehyde for 20 h, after which they were cryoprotected for 48 h in 30% sucrose solution, frozen in liquid nitrogen and cut into 20-μm thick sections with a cryostat (Leica Microsystems, Wetzlar, Germany). Six sections at an interval of 400 μm were selected for immunohistological staining.
The brain sections were incubated overnight at room temperature with primary antibodies (CD45, 1:100 dilution, Bio-Rad, Hercules, CA, USA; CD68, 1:2000 dilution, Bio-Rad, Hercules, CA, USA; GFAP, 1:500 dilution, ABR Affinity BioReagents, Golden, CO, USA; Iba-1, 1:250 dilution, Wako Chemicals, Tokyo, Japan; Ly6-G neutrophil, 1:100 dilution, BioLegend, San Diego, CA, USA; phospho-p38, 1:100 dilution, Cell Signaling Technology, Inc., Danvers, MA, USA). All stainings except the Ly6-G neutrophil stain required antigen retrieval in 10 mM aqueous solution of sodium citrate dihydrate (pH 6, preheated to 92 °C) before application of primary antibodies. Secondary antibodies were applied on sections after three washes in 0.05% Tween20 in PBS. Fluorescent Alexa 568-conjugated secondary antibody (1:200 dilution, Abcam, Cambridge, UK) was used with CD45 and CD68, and 488-conjugated secondary antibody with Iba-1, Ly6G and GFAP. With the phospho-p38 staining, biotinylated secondary antibody (1:200 dilution, Vector Laboratories, Burlingame, CA, USA) was used instead of a fluorescent one, followed by reaction with avidin-biotin complex reagent (1:200 dilution, Vector Laboratories, Burlingame, CA, USA) according to manufacturer’s instructions. The bound immunoreactivity was then visualized by development with nickel-enhanced 3,3’-diaminobenzidine.
For quantification of CD68, GFAP, Iba-1 immunoreactivities in the peri-ischemic area, a 718 × 532 μm cortical area adjacent to the infarct border was imaged using 10x magnification on an AX70 microscope (Olympus corporation, Tokyo, Japan) with an adjacent digital camera (Color View 12 or F-View, Soft imaging system, Muenster, Germany) running AnalySis software (Soft Imaging System). For CD45 and neutrophil stainings, images of equal size were taken from the lesion area, where the majority of the immunoreactivity was observed. Quantification of the immunoreactivities was performed blinded to the study groups using ImagePro Plus software (Media Cybernetics, Rockville, MD, USA) at a predefined range and presented as relative immunoreactive area.
Measurement of Cell Morphology
The morphological reconstitution of Iba1 positive cells at 1 dpi was carried out by Matlab v2013b as described [30, 31]. Several characteristics of the segmented cell processes and cell bodies were analyzed, including area, perimeter and diameter. The images for morphological assessment were taken by a Zeiss LSM 700 confocal microscope (Zeiss Inc., Maple Grove, USA) with an attached digital camera (Color View 12 or F-View; Soft Imaging System, Munster, Germany) running Zen 2012 Image analysis Software (Zeiss inc., Maple Grove, USA).
Cytokine Measurement
Cytokine concentrations were measured in the brain tissues of mice (5–6/treatment group) subjected to permanent MCAO. The mice were sacrificed as described above and following dissection of the peri-ischemic brain area, the samples snap frozen in liquid nitrogen were first homogenized in 20 mM Tris–HCl (pH 7.4), 8.56% sucrose, 0.5 mM EDTA, 0.5 mM EGTA and protease inhibitor cocktail (Complete, Roche Applied Science). The protein levels of IL-6, IL-10, MCP-1, IFN-γ, TNF and IL-12p70 were then measured using the mouse anti-inflammatory cytokine bead array (CBA) kit (BD Biosciences, San Jose, CA, USA), running the samples on a FACS Calibur flow cytometer (BD Biosciences). The results were analyzed using FCAP Array 2.0 software (Soft Flow Hungary Ltd, Pecs, Hungary). Total protein concentrations of the brain samples were determined by Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc, Hercules, CA, USA) and the results of the CBA assay were normalized to the total protein concentrations.
Measurement of Brain Copper Content
Inductively coupled mass spectrometry (ICP-MS) was used to measure copper levels in peri-ischemic brain areas as reported previously [21]. Briefly, cell pellets collected for copper analysis (4–6/treatment group) were digested overnight in concentrated nitric acid (Aristar, BDH, Kilsyth, VIC, Australia), after which samples were heated for 20 min at 90 °C. The volume of each sample was reduced to approximately 40–50 μl then 1 ml of 1% (v/v) nitric acid diluent was added to the samples. Measurements were made using an Agilent ICPMS 7700x series ICP-MS instrument. Results were expressed as micromole per liter concentrations of copper (μmol/l).
Brain Cell Isolation and Flow Cytometry
Mice (4–6/treatment group) were transcardially perfused at 3 dpi with heparnized saline as above. Brains without cerebellum were isolated and put in HBSS on ice. The tissue was minced on ice with forceps in petri dishes in digest buffer containing 0.5 mg/ml collagenase type 4 (Worthington, Lakewood, NJ, USA) DNAse 1, 25 U/ml, and RPMI-1640 (both from Sigma-Aldrich) then incubated at 37 °C, 5% CO2 for 20 min. Samples were put directly back on ice, triturated, and sequentially filtered through 70 μm and 40 μm cell strainers (Falcon, Corning, NY, USA). Homogenates were centrifuged 450 x g for 5 min and resuspended in Miltenyi Myelin Debris Removal Beads II (Miltenyi, Cologne, Germany). Samples were incubated for 15 min at 4 °C then washed with cold MACS buffer (PBS and 0.5% BSA, both from Sigma) and centrifuged at 400 x g for 10 min. Pellets were resuspended in cold MACS buffer and applied to magnetic LD columns (Miltenyi) with 50 μm CellTrics pre-filters (Sysmex, Norderstedt, Germany). The flow-through was collected on ice, counted, then spun 400×g for 10 min and resuspended in RPMI-1620.
Cell surface staining was done on 200,000–300,000 cells per mouse in round bottom polypropylene 96-well plates (Corning). Cells were washed with PBS, stained with Zombie NIR fixable viability dye (BioLegend, San Diego, CA, USA) for 15 min at RT, then blocked with CD16/32 (clone 24G2, BD Biosciences, San Jose, CA, USA) and stained with the following antibodies: CD45 PerCP-Cy5.5 (clone 30 F11, eBioscience, San Diego, CA, USA), CD11b PECy7 (clone M1/70, eBioscience), Ly6G FITC (clone 1A8, BioLegend), F4/80 PE (clone A3-1, AbD Serotec, Oxford, UK), and Ly6C APC (clone AL-21, BD Biosciences) for 30 min at 4 °C. Cells were washed twice in PBS and fixed in 0.5% Ultra Pure formaldehyde (ThermoFisher Scientific, Waltham, MA, USA). Samples were acquired on BD FACSAria III equipped with 488- and 633-nm lasers with standard configuration. Data were analyzed using FCSExpress v5 (DeNovo Software Glendale, CA, USA).
Statistics
Statistical analyses were performed in GraphPad Prism software 5.03 (GraphPad Software, La Jolla, CA, USA) using either unpaired two-tailed t-test, one-way ANOVA or two-way ANOVA as appropriate. The statistical test used and n-numbers are indicated in each figure legend. p-values < 0.05 were considered statistically significant. All data are presented as mean ± standard deviation (SD).
Results
The Copper Complex CuII(atsm) is Protective in In Vitro Models of Excitotoxicity and OGD
Excitotoxicity is a primary mechanism of neuronal injury following stroke. To determine the neuroprotective potential of CuII(atsm) against excitotoxic insult, primary cortical neurons were treated with CuII(atsm) in the presence of glutamate. Treating neurons with glutamate for 24 h resulted in approximately 40% reduction in cell viability as measured by the MTT assay (Fig. 1a). Co-treatment with CuII(atsm) significantly improved the viability of glutamate-treated neurons, although the treatment did not entirely rescue the cells from excitotoxicity. At the tested concentrations, neuronal viability was not affected by CuII(atsm) treatment alone. In a more relevant in vitro model of stroke, 24 h OGD caused approximately 50% cell death in N2a cultures (Fig. 1b). Pre-treatment with 0,1 μM CuII(atsm) had a slight but significant protective effect in this assay.
Fig. 1.

CuII(atsm) is protective in in vitro models of stroke. a Primary cortical neurons were treated with CuII(atsm) in the presence of 400 μM glutamate for 24 h, after which neuronal viability was measured by MTT reduction. b N2a cells were exposed to OGD for 24 h with or without 1 h CuII(atsm) pretreatment followed by MTT measurement for cell viability. A control plate underwent similar assay, except the cells were incubated in regular culture conditions. Data show means ± standard deviation (SD) from one representative experiment. One-way ANOVA or paired t-test, *p < 0.05, ***p < 0.001. n = 6 in all groups
CuII(atsm) Reduces Ischemic Injury and Functional Impairment in a Transient Model of Cerebral Ischemia
CuII(atsm) was tested firstly in the transient middle cerebral artery occlusion (MCAO) mouse model of cerebral ischemia. We found that CuII(atsm) treatment (15 mg/kg, by oral gavage) both prior to MCAO, and at the start of reperfusion reduced infarct sizes 24 h later (Fig. 2a-b). Transient MCAO-induced functional impairment indexed by neuroscore was also prevented by pre-treatment of CuII(atsm) (Fig. 2c) 24 h later.
Fig. 2.

CuII(atsm) is protective in transient ischemia. a Representative images of TTC stained brain sections at 1 dpi. b Quantitative analysis of infarct volume at 24 h after tMCAO. Mean ± standard deviation (SD). One-way ANOVA with Tukey post hoc test, *p < 0.05, **p < 0.01 compared to vehicle, n = 3-4/treatment group. c Transient MCAO-induced functional impairment indexed by neuroscore 1 dpi. Mean ± SD, one-way ANOVA with Tukey post hoc test, ***p < 0.001. n = 3/treatment group
CuII(atsm) Confers a Beneficial Effect on the Outcome of Ischemic Stroke in a Permanent Model of Cerebral Ischemia
CuII(atsm) was administered to mice by oral gavage immediately after permanent middle cerebral artery occlusion and the lesion volume was measured 24 h later by MRI. CuII(atsm) administered at a dose of 60 mg/kg caused a significant reduction in the lesion volume (Fig. 3a-b). The locomotor activity of the ischemic mice was assessed using the latency-to-move test, which is reported to be affected for several days after ischemia [32]. Ischemic mice showed a significant reduction in the latency to move at 1 dpi, which was corrected to the levels of sham mice by CuII(atsm) treatment (Fig. 3c). To confirm that the observed beneficial effects were related to copper delivery by CuII(atsm), the ischemic brain tissues were analyzed for copper content by ICP-MS. When compared to vehicle-treated mice, the concentration of copper in the peri-ischemic area of the CuII(atsm) treated mice was 57% higher (Fig. 3d), indicating efficient delivery of copper by the complex.
Fig. 3.

CuII(atsm) is protective in permanent ischemia and increases the brain copper content. a Representative MRI images of ischemic brains at 1 dpi. b Quantitative analysis of infarct volume at 24 h after pMCAO. Unpaired 2-tailed t-test, *p < 0.05. n = 10 in vehicle group, n = 8 in CuII(atsm) group. c The latency-to-move was measured at 1 day after ischemia in sham operated and in mice subjected to pMCAO. Each mouse was tested twice and the data shows the average of two trials for each mouse. Two-way ANOVA, *p < 0.05, **p < 0.01. n = 12 in the sham vehicle group, n = 12 in the sham CuII(atsm) group, n = 8 in the ischemic vehicle group, n = 8 in the ischemic CuII(atsm) group. d The copper concentrations in the peri-ischemic brain areas were measured by ICP-MS at 3 dpi and are shown normalized to mass of the tissue ± standard deviation (SD). Unpaired 2-tailed t-test, *p < 0.05. n = 6 in vehicle group, n = 4 in CuII(atsm) group
CuII(atsm) Reduces CD45 Levels in the Ischemic Core
Previous studies have suggested that copper delivery may have anti-inflammatory effects in neurodegenerative diseases [33]. To assess the effect of copper delivery by CuII(atsm) on neuroinflammation in the context of ischemic stroke, we first assessed astrocytic and myeloid cell activation in the ischemic brains by immunohistochemistry. Astrocytic activation as assessed by GFAP immunohistochemistry was not affected by CuII(atsm) treatment in the peri-infarct area (data not shown). However, analyses of brains subjected to permanent ischemia revealed significant CuII(atsm) induced alterations to the levels of CD45. CD45 is a marker expressed highly by monocytes and to a lower extent by microglia, in which expression is up-regulated after injury [34]. Copper delivery with CuII(atsm) reduced the amount of CD45 immunoreactivity in the ischemic core both at 1 (Fig. 4a-b) and 3 dpi (Fig. 4c-d).
Fig. 4.

CuII(atsm) reduces CD45 immunoreactivity in ischemic mice. a Typical example images of CD45 immunoreactivity in the ischemic lesion in vehicle-treated and CuII(atsm) treated mouse brains at 1 day after pMCAO. Scale bar = 20 μm. b Quantitative analysis of CD45 immunoreactivity in the lesion at 1 dpi. Data are shown as means ± standard deviation (SD). Unpaired 2-tailed t-test, *p < 0.05. n = 7 in both groups. c Typical example images of CD45 immunoreactivity in the ischemic lesion in vehicle-treated and CuII(atsm)-treated mouse brains at 3 dpi. Scale bar = 20 μm. d Quantitative analysis of CD45 immunoreactivity in the lesion at 3 dpi. Data are shown as means ± SD. Unpaired 2-tailed t-test, *p < 0.05. n = 5 in both groups
CuII(atsm) Reduces Iba1 Immunoreactivity and Alters the Morphology of Iba1-Positive Cells in the Ischemic Brain
To extend our analyses of myeloid cell involvement in CuII(atsm)-mediated protection from ischemic injury we undertook a panel of immunohistological stainings in order to determine which cell types are affected by the treatment. While the immunoreactivities for CD68 (a marker of active phagocytosis) and Arginase-1 (a marker of alternatively activated cells) remained unaltered (data not shown), Iba1 levels in the peri-ischemic brains were altered by copper delivery. Iba1 is expressed by microglia and monocytes and its expression is reported to increase during ischemia [35]. It is considered one of the useful proteins for distinguishing microglia through immunostaining especially in stroke studies [35]. The Iba1 staining revealed a significant reduction of immunoreactivity by the CuII(atsm) treatment at 3 dpi in mice subjected to pMCAO (Fig. 5a-b).
Fig. 5.

CuII(atsm) reduces Iba1 immunoreactivity and alters the morphology of Iba1-positive cells in ischemic mice. a Typical example images of Iba1 immunoreactivity in the peri-ischemic brain area in vehicle-treated and CuII(atsm)-treated mouse brains at 3 dpi. Scale bar = 20 μm. b Quantitative analysis of Iba1 immunoreactivity in the peri-ischemic brain area at 3 dpi. Data are shown as means ± standard deviation (SD). Unpaired 2-tailed t-test, *p < 0.05. n = 5 in both groups. c Typical example images of the algorithm used to determine the morphology of Iba1 positive cells. Scale bar = 50 μm. d Quantification of the cellular area of the Iba1 positive cells in the peri-ischemic brain areas of mice treated with CuII(atsm) at 1 dpi. Data are shown as means ± SD. Unpaired 2-tailed t-test, *p < 0.05. n = 8 in both groups
While measuring immunoreactivity following histological staining provides information about changes in the cells, it does not give detailed information about whether the change is related to the presence of more or less cells, larger or smaller cells or more or less intensively stained cells. Moreover, as microglial morphology is tightly coupled to their function we next assessed the effect of copper delivery on the structure of the Iba1-positive cells. The morphology of the Iba1 positive cells was analyzed as reported [30, 31]. The digital reconstruction (Fig. 5c) revealed that the morphology of Iba1-positive cells was altered by copper delivery in ischemic mice. CuII(atsm) did not affect cellular branching or processes (data not shown). However, despite the reduction in immunoreactivity (Fig. 5a-b) the treatment significantly increased the cellular area of the Iba1-positive cells (Fig. 5d).
CuII(atsm) Increases the Proportion of of CD45low CD11b+ Ly6G- F4/80+ Cells in the Ischemic Brain
To create a multiparameter characterization of the cell types affected by CuII(atsm) during ischemia we isolated immune cells from the brain and applied flow cytometry for several cellular markers. The gating strategies are shown in Fig. 6a. The cells were discriminated based on the expression of CD45, CD11b, F4/80, Ly6C and Ly6G (Fig. 6a-b). In comparison to sham mice, ischemia induced a 59% increase in CD45+CD11b- lymphocytes (p = 0.001), and a 97% increase in Ly6G+ neutrophils (p < 0.001) (data not shown). Ischemia also induced a 95% increase in CD45high CD11b+ Ly6G- myeloid cells (p < 0.001), a 53% increase in the Ly6Chigh CD45low cells (p = 0.007), a 41% increase in Ly6Chigh CD45high cells, and a 70% increase in CD45high F4/80+ cells (p < 0.001) (data not shown).
Fig. 6.
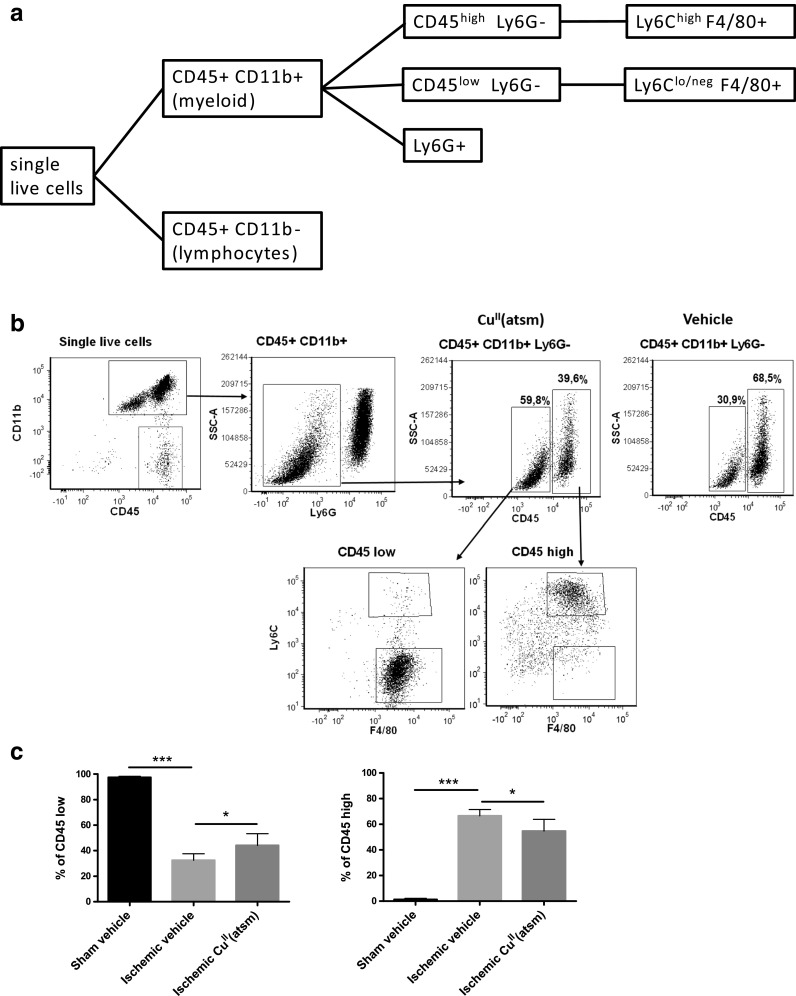
CuII(atsm) alters the ratios of myeloid cell in the ischemic hemisphere at 3 dpi. a Flow cytometric characterization strategy to identify cell populations. b Representative scatter plots showing gating process to ultimately distinguish CD45lowCD11b+ Ly6G- and CD45highCD11b+ Ly6G- cell populations and their subpopulations of Ly6C and F4/80. Vehicle CD45 subpopulations displayed similar cell distributions. c Quantified ratios of CD45high and CD45low CD11b+ Ly6G- microglia in sham operated animals, and in mice that underwent pMCAO with either vehicle or CuII(atsm) treatment. Mean ± standard deviation (SD), 1-way ANOVA, *p < 0.05, ***p < 0.001. n = 6 in stroke groups
The numbers of CD45low CD11b+ Ly6G- cells were reduced by 86% (p < 0.001) in ischemic brains (data not shown). CuII(atsm) treatment had a significant effect on this cell population, especially the cells that were Ly6Glow and F4/80+ (Fig. 6b). When compared to vehicle-treated ischemic mice, the percentages of resident CD45low CD11b+ Ly6G- cells were increased by 20%, while invading CD45high CD11b+ Ly6G- cells were reduced by 20% in the CuII(atsm)-treated mice at 3 dpi (Fig. 6b-c).
Copper Delivery by CuII(atsm) Reduces Additional Markers of Inflammation in the Ischemic Brain
To further assess the effect of copper delivery on the inflammatory milieu following ischemia, the immunoreactivity of p38 MAPK was analyzed by histochemical staining and the levels of six brain cytokines were analyzed by CBA. p38 MAPK is a key serine/threonine protein kinase that is an important contributor to increased microglial production of proinflammatory cytokines [36]. Staining for the active form of p38 MAPK in the brains of ischemic mice revealed that CuII(atsm) reduced its expression at 1 dpi (Fig. 7a-b).
Fig. 7.

Copper delivery by CuII(atsm) reduces markers of inflammation in the ischemic brain. a Typical example images of phosphorylated p38 MAPK immunoreactivity in the peri-ischemic brain area in vehicle-treated and CuII(atsm)-treated mouse brains at 1 day after pMCAO. Scale bar = 20 μm. b Quantitative analysis of phosphorylated p38 MAPK immunoreactivity in the peri-ischemic brain area at 1 dpi. Data are shown as means ± standard deviation (SD). Unpaired 2-tailed t-test, *p < 0.05. n = 8 in both groups. c The concentration of IL-12 in the peri-ischemic brain area was measured by CBA at 1 dpi. Data are shown as means ± SD. Unpaired 2-tailed t-test, *p < 0.05. n = 5 in vehicle and n = 6 in the CuII(atsm) group. d The concentration of IL-10 in the peri-ischemic brain area was measured by CBA at 3 dpi. Data are shown as means ± SD. Unpaired 2-tailed t-test, *p < 0.05. n = 5 in vehicle and n = 6 in the CuII(atsm) group
CBA analyses showed that CuII(atsm) treatment at 1 dpi did not affect the amount of IL-6, IL-10, MCP-1, IFN-γ or TNF in the ischemic brain. However, copper delivery by CuII(atsm) reduced the protein level of IL-12 by 54% in the peri-ischemic area of the brain at 1 dpi (Fig. 7c). In contrast, IL-10 levels were increased by CuII(atsm) treatment at 3 dpi (Fig. 7d).
Discussion
CuII(atsm) has been shown to have protective effects in multiple animal models of chronic neurodegenerative diseases, such as ALS and PD [18, 20]. The compound is about to be tested in clinical trials in ALS patients (ClinicalTrials.gov identifier: NCT02870634). Interestingly, CuII(atsm) has a tendency to deliver copper into cells that suffer from a dysfunctional electron transport chain, indicating optimal pharmacokinetics in the ischemic brain [37]. Here, we report for the first time that CuII(atsm) is protective in acute brain injury, in two in vivo models of ischemic stroke. The protective effect is accompanied by beneficial modulation of the inflammatory milieu during stroke.
We report that CuII(atsm) reduces lesion volume and improves functional outcome in both transient and permanent models of ischemic stroke in mice. Demonstration of the therapeutic effect in two different models shows that protection is not dependent on mouse strain, duration of ischemia, or location of lesion. In the transient model the doses sufficient to elicit therapeutic effects were about half of that required in the permanent model of cerebral ischemia. The difference in effective dose may be connected to access to the ischemic area, as the reperfusion period in the transient model opens a circulatory connection to the injured site, mimicking recanalization therapy received by a human patient. The fact that CuII(atsm) reduced lesion volume and promoted functional recovery in both transient and permanent ischemia models suggests that it is a potent approach given the heterogeneous stroke subtypes observed in humans. In addition, using both permanent and transient ischemia is a recommendation mentioned in STAIR criteria aiming to improve translation of preclinical stroke research [38].
As previously mentioned, studies with human patients have suggested an imbalance in copper homeostasis after cerebral ischemia [11, 12]. Elevated levels of copper were observed in the circulation of stroke patients in both studies, suggesting that copper is redistributed due to stroke-induced damage. Moreover, chronic intake of copper in the form of copper sulfate in the drinking water has been shown to exacerbate ischemic damage in mice, thereby indicating that increases in free copper exert toxic effects in the brain [13]. Here, we report that CuII(atsm) increased peri-ischemic concentrations of copper coincidentally with reduced lesion size and behavioural improvements. Formerly known as a hypoxia-imaging agent that releases copper mainly in the presence of a dysfunctional electron transport chain [37], we propose that short-term treatment with CuII(atsm) increased the copper content of the cells in the peri-ischemic resulting in observed protective and anti-inflammatory effects after stroke.
Neuronal excitotoxicity is one of the key components of stroke pathology leading to mitochondrial dysfunction [39]. It has been earlier described that copper has an essential function in protecting neurons against excitotoxicity through NMDAR receptor modulation [40]. In agreement with this, we demonstrate that the copper complex CuII(atsm) improved the viability of primary cortical neurons exposed to excitotoxic injury. However, it is likely that CuII(atsm) also affected secondary damage in the cells, rather than merely receptor activation, since copper delivery by CuII(atsm) is improved in the presence of mitochondrial dysfunction, and slight neuroprotection was also seen in OGD study using N2a cells [37]. For instance, studies with a transgenic mouse model of ALS have shown that CuII(atsm) treatment can increase both total amount of Cu/Zn superoxide dismutase (SOD1) and its copper content [41]. This might have a beneficial effect on the antioxidant defence of brain cells. In addition, lack of complete protection against excitotoxic insult and OGD might indicate a more complex scheme of protection in vivo, involving the interplay between neurons and microglia. Nevertheless, it is plausible that the neuroprotective effect seen in animal models of ischemia was at least partly caused by CuII(atsm)-mediated direct neuroprotective effect.
We have reported that the copper bis(thiosemicarbazonato) complexes mediate anti-inflammatory effects in neurodegenerative diseases (Choo et al., submitted). Thus, we characterized this anti-inflammatory effect in acute injury during stroke. Indeed, quantification of CD45 immunoreactivity revealed a significant reduction in this cell type in the ischemic core at 1 and 3 days after pMCAO, coinciding with reduced lesion volume and improved functional outcome. The cells in the ischemic core appeared CD45high-expressing, indicating that CuII(atsm) reduced the amount of infiltrating myeloid cells into the ischemic area [42]. Because reduced infiltration of myeloid cells indicated altered initiation of post-stroke inflammation, we used multiple markers to specify the effect of CuII(atsm) on different inflammatory cell types in the pMCAO model. Based on immunohistochemical analyses of the peri-ischemic brain area, we report a CuII(atsm)-mediated reduction in Iba1 immunoreactivity. While previous studies have shown that Iba1 can be expressed by both microglia and invading peripheral inflammatory cells, the early time point of 3 days after ischemia used in the present study suggests that the observed reduction may be caused by reduced activation of resident microglia [35, 43].
Since microglial morphology and function are usually linked to each other [44], we next used an unbiased automated reconstructive procedure as to digitally reconstruct microglial morphology of the Iba1 positive cells following ischemia. CuII(atsm) increased the size of the somas of Iba1 positive microglia without affecting their processes or branching. Increased soma sizes have been reported to correlate with increased microglial activation [30]. Histologically, there may be separate phenotypic populations of microglial cells responding to CuII(atsm): those at an undifferentiated state displaying low Iba1 expression, and those with high Iba1 expression. Morphological assessment of the Iba1high cells revealed that the Iba1high cells are actually in a more activated state in CuII(atsm)-treated animals. These kinds of spatiotemporal differences in microglial responses to experimental treatments have been described earlier in the context of ischemic stroke, and are thought to arise from the heterogeneous nature of microglia [44, 45].
To calculate the true proportions of each cell type in the ischemic hemisphere, we used flow cytometry for multiple cellular markers. We report that 3 days after permanent MCAO, there was a strong infiltration of CD45high CD11b+ Ly6G- myeloid cells, CD45+CD11b- lymphocytes and Ly6G+ neutrophils. Infiltration of monocytes and lymphocytes is known to take place at this time point, whereas some controversy remains about the latency of neutrophil infiltration [46, 47]. In line with results from immunohistochemistry, CuII(atsm) significantly increased the amount of cells characterized by CD45low CD11b+ Ly6G-, and reduced the proportion of CD45high CD11b+ Ly6G- cells .3 days after permanent MCAO. These results suggest that CuII(atsm) reduces the proportion of invading monocytes, and protects the endogenous microglia against ischemic insult. This may be related to the cellular protection observed in the quantification of the lesion size in MRI images.
P38 MAPK is a key mediator of microglial production of proinflammatory cytokines in the presence of multiple stressors [36]. In addition, inhibition of this kinase is protective in experimental models of stroke through multiple other pathways [48]. In the present study, we show that immunoreactivity of the active form of p38 was reduced in the ischemic CuII(atsm)-treated animals. Based on morphology, these cells were microglial cells, giving further evidence of a reduced inflammatory state of the cells in the ischemic brain. At the same time, we also saw a reduction in IL-12 levels and a later increase in IL-10 levels. These alleviations in inflammatory signaling might be an important feature in CuII(atsm)-mediated protection of higher brain functions, as IL-12 has been associated with cognitive decline in stroke patients [49]. In addition, IL-10 deficiency has been previously shown to exacerbate ischemic brain damage, whereas common protectants such as minocycline increase IL-10-signaling [50, 51]. Taken together, these results suggest that CuII(atsm) has several inflammation-modulating actions in the ischemic brain.
Available treatment options for stroke are inadequate and those available have limited efficacy. This study reports the therapeutic actions of CuII(atsm) in two preclinical in vivo models of stroke, and characterizes the effects of copper modulation on immune responses occurring during experimental stroke. These result suggest that similarly to models of AD and peripheral inflammation (Choo et al., submitted), the copper bis(thiosemicarbazonato) complexes confer their beneficial effects at least partly through the modulation of inflammation, and specifically microglia, in ischemic brain injury. Since a copper bis(thiosemicarbazonato) complex is going to be tested clinically for ALS (ClinicalTrials.gov identifier: NCT02870634), it may shed light for the complex to be tested against neuronal damage after ischemic stroke clinically. This study also provides further support for the neurotherapeutic potential of copper modulation in brain disorders.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 1224 kb)
(PDF 1197 kb)
(PDF 1225 kb)
Acknowledgements
We thank Ms Mirka Tikkanen for expert technical assistance. The study was supported by The Academy of Finland, The Sigrid Juselius Foundation, Orion Finland, the Australian Research Council, the National Health & Medical Research Council of Australia (NHMRC), the Cooperative Research Center for Mental Health, and National Natural Science Foundation of China (81271403, 81471304, 81571236).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Compliance with Ethical Standards
Competing Financial Interests
Dr. Bush is a shareholder in Prana Biotechnology Ltd, Cogstate Ltd, Mesoblast Ltd, NextVet Ltd, Brighton Biotech LLC, and Cogstate Ltd, and a consultant for Collaborative Medicinal Development Pty Ltd.
Footnotes
Mikko T. Huuskonen, Qing-zhang Tuo, Anthony R. White, Peng Lei and Katja M. Kanninen contributed equally to this work.
Contributor Information
Mikko T. Huuskonen, Email: mikko.huuskonen@uef.fi
Qing-zhang Tuo, Email: tqzh1220@163.com.
Sanna Loppi, Email: sanna.loppi@uef.fi.
Hiramani Dhungana, Email: hiramani.dhungana@uef.fi.
Paula Korhonen, Email: paula.korhonen@uef.fi.
Lachlan E. McInnes, Email: l.mcinnes2@student.unimelb.edu.au
Paul S. Donnelly, Email: pauld@unimelb.edu.au
Alexandra Grubman, Email: alexandra.grubman@monash.edu.
Sara Wojciechowski, Email: sara.wojciechowski@uef.fi.
Katarina Lejavova, Email: klejavova@yahoo.com.
Yuriy Pomeshchik, Email: yuriy.pomeshchik@uef.fi.
Laura Periviita, Email: laurap@student.uef.fi.
Lotta Kosonen, Email: lotkos@student.uef.fi.
Martina Giordano, Email: martigio@student.uef.fi.
Frederick R. Walker, Email: rohan.walker@newcastle.edu
Rong Liu, Email: rong.liu@hust.edu.cn.
Ashley I. Bush, Email: ashleyib@unimelb.edu.au
Jari Koistinaho, Email: jari.koistinaho@uef.fi.
Tarja Malm, Email: tarja.malm@uef.fi.
Anthony R. White, Phone: +61-7-33620360, Email: tony.white@qimrberghofer.edu.au, Email: arwhite@unimelb.edu.au
Peng Lei, Phone: +86-28-87636824, Email: peng.lei@scu.edu.cn, Email: peng.lei@florey.edu.au, Email: plei1984@163.com.
Katja M. Kanninen, Phone: +358-40-1354377, Email: katja.kanninen@uef.fi
References
- 1.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 2009;7:97-5876-7-97. [DOI] [PMC free article] [PubMed]
- 2.Solano Fonseca R, Mahesula S, Apple DM, Raghunathan R, Dugan A, Cardona A, et al. Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev. 2016;25(7):542–555. doi: 10.1089/scd.2015.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 4.Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32(30):10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015;129(2):279–295. doi: 10.1007/s00401-014-1372-1. [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol 2016. [DOI] [PubMed]
- 7.Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57(8):835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- 8.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zucconi GG, Cipriani S, Scattoni R, Balgkouranidou I, Hawkins DP, Ragnarsdottir KV. Copper deficiency elicits glial and neuronal response typical of neurodegenerative disorders. Neuropathol Appl Neurobiol. 2007;33(2):212–225. doi: 10.1111/j.1365-2990.2006.00793.x. [DOI] [PubMed] [Google Scholar]
- 10.Schrag M, Crofton A, Zabel M, Jiffry A, Kirsch D, Dickson A, et al. Effect of cerebral amyloid angiopathy on brain iron, copper, and zinc in Alzheimer's disease. J Alzheimers Dis. 2011;24(1):137–149. doi: 10.3233/JAD-2010-101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodali P, Chitta KR, Landero Figueroa JA, Caruso JA, Adeoye O. Detection of metals and metalloproteins in the plasma of stroke patients by mass spectrometry methods. Metallomics. 2012;4(10):1077–1087. doi: 10.1039/c2mt20092a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai M, Wang D, Lin Z, Zhang Y. Small molecule copper and its relative metabolites in serum of cerebral ischemic stroke patients. J Stroke Cerebrovasc Dis. 2016;25(1):214–219. doi: 10.1016/j.jstrokecerebrovasdis.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, Wang LP, Dong XH, Cai J, Jiang GJ, Zhang C, et al. Trace amounts of copper in drinking water aggravate cerebral ischemic injury via impairing endothelial progenitor cells in mice. CNS Neurosci Ther. 2015;21(8):677–680. doi: 10.1111/cns.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grubman A, White AR, Liddell JR. Mitochondrial metals as a potential therapeutic target in neurodegeneration. Br J Pharmacol. 2014;171(8):2159–2173. doi: 10.1111/bph.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, et al. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J Biol Chem. 2008;283(8):4568–4577. doi: 10.1074/jbc.M705957200. [DOI] [PubMed] [Google Scholar]
- 16.Torres JB, Andreozzi EM, Dunn JT, Siddique M, Szanda I, Howlett DR, et al. PET imaging of copper trafficking in a mouse model of Alzheimer disease. J Nucl Med. 2016;57(1):109–114. doi: 10.2967/jnumed.115.162370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci USA. 2009;106(2):381–386. doi: 10.1073/pnas.0809057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soon CP, Donnelly PS, Turner BJ, Hung LW, Crouch PJ, Sherratt NA, et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286(51):44035–44044. doi: 10.1074/jbc.M111.274407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllum EJ, Lim NK, Hickey JL, Paterson BM, Donnelly PS, Li QX, et al. Therapeutic effects of CuII(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7-8):586–590. doi: 10.3109/21678421.2013.824000. [DOI] [PubMed] [Google Scholar]
- 20.Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR, et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson's disease. J Exp Med. 2012;209(4):837–854. doi: 10.1084/jem.20112285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89(2):271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 22.Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 24.Dhungana H, Malm T, Denes A, Valonen P, Wojciechowski S, Magga J, et al. Aging aggravates ischemic stroke-induced brain damage in mice with chronic peripheral infection. Aging Cell. 2013;12(5):842–850. doi: 10.1111/acel.12106. [DOI] [PubMed] [Google Scholar]
- 25.Gingras BA, Suprunchuk T, Bayley CH. The preparation of some thiosemicarbazones and their copper complexes: Part III. 40(6) 1962;v62-161(Canadian Journal of Chemistry):1053-1059.
- 26.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 27.Bargiotas P, Krenz A, Monyer H, Schwaninger M. Functional outcome of pannexin-deficient mice after cerebral ischemia. Channels (Austin) 2012;6(6):453–456. doi: 10.4161/chan.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24(1):117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 29.Shuaib A, Xu Wang C, Yang T, Noor R. Effects of nonpeptide V(1) vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke. 2002;33(12):3033–3037. doi: 10.1161/01.str.0000039405.31526.06. [DOI] [PubMed] [Google Scholar]
- 30.Kongsui R, Johnson SJ, Graham BA, Nilsson M, Walker FR. A combined cumulative threshold spectra and digital reconstruction analysis reveal structural alterations of microglia within the prefrontal cortex following low-dose LPS administration. Neuroscience. 2015;310:629–640. doi: 10.1016/j.neuroscience.2015.09.061. [DOI] [PubMed] [Google Scholar]
- 31.Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, et al. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun. 2016;54:95–109. doi: 10.1016/j.bbi.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lubjuhn J, Gastens A, von Wilpert G, Bargiotas P, Herrmann O, Murikinati S, et al. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J Neurosci Methods. 2009;184(1):95–103. doi: 10.1016/j.jneumeth.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 33.Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci. 2007;27(14):3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 2015;12:106-015-0329-1. [DOI] [PMC free article] [PubMed]
- 35.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32(5):1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 36.Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J Neuroinflammation 2011;8:79-2094-8-79. [DOI] [PMC free article] [PubMed]
- 37.Donnelly PS, Liddell JR, Lim S, Paterson BM, Cater MA, Savva MS, et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII. Proc Natl Acad Sci U S A. 2012;109(1):47–52. doi: 10.1073/pnas.1116227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci USA. 2009;106(24):9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015;22(9):772–784. doi: 10.1089/ars.2014.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts BR, Lim NK, McAllum EJ, Donnelly PS, Hare DJ, Doble PA, et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2014;34(23):8021–8031. doi: 10.1523/JNEUROSCI.4196-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greter M, Lelios I, Croxford AL. Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol. 2015;6:249. doi: 10.3389/fimmu.2015.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miro-Mur F, Perez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun. 2016;53:18–33. doi: 10.1016/j.bbi.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Z, Chopp M, Powers C. Temporal profile of microglial response following transient (2 h) middle cerebral artery occlusion. Brain Res. 1997;744(2):189–198. doi: 10.1016/S0006-8993(96)01085-2. [DOI] [PubMed] [Google Scholar]
- 45.Schroeter M, Jander S, Witte OW, Stoll G. Heterogeneity of the microglial response in photochemically induced focal ischemia of the rat cerebral cortex. Neuroscience 1999;89(4):1367-1377. [DOI] [PubMed]
- 46.Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23(1):34–44. doi: 10.1111/j.1750-3639.2012.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu HX, Kim HA, Lee S, Moore JP, Chan CT, Vinh A, et al. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab. 2014;34(3):450–459. doi: 10.1038/jcbfm.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang XM, Zhang L, Wang G, Niu W, He Z, Ding L, et al. Suppression of mitochondrial fission in experimental cerebral ischemia: The potential neuroprotective target of p38 MAPK inhibition. Neurochem Int. 2015;90:1–8. doi: 10.1016/j.neuint.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 49.Narasimhalu K, Lee J, Leong YL, Ma L, De Silva DA, Wong MC, et al. Inflammatory markers and their association with post stroke cognitive decline. Int J Stroke. 2015;10(4):513–518. doi: 10.1111/ijs.12001. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y, Salayandia VM, Thompson JF, Yang LY, Estrada EY, Yang Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflammation 2015;12:26-015-0245-4. [DOI] [PMC free article] [PubMed]
- 51.Perez-de Puig I, Miro F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Marquez-Kisinousky L, et al. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab. 2013;33(12):1955–1966. doi: 10.1038/jcbfm.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)
(PDF 1197 kb)
(PDF 1225 kb)