

Abstract
Matrix metalloproteinase 9 (MMP9) is a member of a large family of proteases that are secreted as inactive zymogens. It is a key regulator of the extracellular matrix, involved in the degradation of various extracellular matrix proteins. MMP9 plays a pathological role in a variety of inflammatory and oncology disorders and has long been considered an attractive therapeutic target. GS-5745, a potent, highly selective humanized monoclonal antibody inhibitor of MMP9, has shown promise in treating ulcerative colitis and gastric cancer. Here we describe the crystal structure of GS-5745·MMP9 complex and biochemical studies to elucidate the mechanism of inhibition of MMP9 by GS-5745. GS-5745 binds MMP9 distal to the active site, near the junction between the prodomain and catalytic domain, and inhibits MMP9 by two mechanisms. Binding to pro-MMP9 prevents MMP9 activation, whereas binding to active MMP9 allosterically inhibits activity.
Keywords: allosteric regulation, antibody, enzyme inhibitor, matrix metalloproteinase (MMP), X-ray crystallography
Introduction
Human matrix metalloproteinase 9 (MMP9),3 also referred to as gelatinase B, is a member of the MMP family, which are secreted from cells as inactive zymogens. The MMP family consists of ∼25 members that share a common domain structure. The proenzyme (pro-MMP9) contains an N-terminal propeptide domain, a zinc-containing catalytic domain with an insertion of three fibronectin type II repeats, and a C-terminal hemopexin-like domain, which is connected to the catalytic domain by an O-glycosylated linker (1). The propeptide domain contains a conserved cysteine residue that coordinates the active site zinc, preventing substrate binding and catalysis. Stepwise proteolytic cleavage of the propeptide is required to generate catalytically competent MMP9 (2, 3). A related protease, MMP3, has been shown to activate MMP9 in vitro by cleaving between residues, Glu-59/Met-60 and Arg-106/Phe-107, releasing the prodomain (4, 5). Once active, MMP9 is capable of cleaving numerous substrates (6).
In vivo, MMP9 activity is critical in remodeling components of the extracellular matrix, including collagen IV and laminin in the basement membrane (7). In addition to its role in extracellular matrix remodeling, MMP9 regulates other cellular processes, and its expression and secretion are up-regulated in pathological states such as cancer and chronic inflammation (7–13). MMP9 has also been shown to activate latent cytokines and growth factors and alter both trafficking and cell surface protein expression of both myeloid and lymphoid cells (14–17).
Because of its association with disease and correlation with poor prognosis in ulcerative colitis and colorectal cancer patients, MMP9 has long been considered a potential target for therapeutic intervention. Efforts to discover and develop safe and effective drugs selectively targeting MMP9 have been difficult due to the high structural conservation of the protease active site among MMP family members. A clinical stage, broad-spectrum peptidomimetic MMP inhibitor, marimastat, failed to meet efficacy end points and led to musculoskeletal syndrome in some patients (12, 18).
Although zinc-chelating small-molecule peptidomimetic MMP inhibitors were neither specific nor well tolerated in clinical trials (8–10), a monoclonal antibody was identified that selectively inhibited MMP9 (19). This antibody and its humanized counterpart, GS-5745, were well tolerated and efficacious in preclinical models and human trials, respectively, of ulcerative colitis and cancer (19). Although GS-5745 was shown to be an allosteric inhibitor of human MMP9 activity (19), the mechanisms of specificity and inhibition remained elusive. In the current studies, we determined the high-resolution crystal structure of MMP9 in complex with GS-5745. GS-5745 binds to MMP9 at the junction between the propeptide and catalytic domains, distal to the active site. We further characterized the effect of GS-5745 on MMP9 activation in cell culture and with recombinant proteins. These results reveal that GS-5745 can inhibit MMP9 by two mechanisms: as an allosteric inhibitor of MMP9 activity and by preventing activation of the secreted zymogen. The latter mechanism is unique among inhibitory antibodies and represents a new approach for developing molecules that inhibit therapeutic targets.
Results
Characterization of activated MMP9
For biochemical and structural studies, human MMP9, spanning residues 20–707 followed by a C-terminal MYC and His6 tag (MMP9-MYC-His6) was expressed in Expi293 cells. The soluble, secreted protein was purified from the culture medium (supplemental Fig. 1). We also generated a truncated form of MMP9 (MMP9-pro-cat) containing a deletion at the N terminus of the prodomain, an internal deletion of the fibronectin type II-like domain, and a C-terminal deletion that removed the hemopexin domain. MMP9-pro-cat was refolded and purified from inclusion bodies (supplemental Fig. 1).
Several known activators of MMP9 have been reported, including MMP3, trypsin, and 4-aminophenylmercuric acetate (APMA) (4, 20, 21). APMA activation results in cleavage of MMP9 between Ala-93 and Met-94, whereas MMP3 and trypsin cleave MMP9 between Arg-106 and Phe-107. We were able to activate MMP9-MYC-His6 and MMP9-pro-cat with APMA to generate active enzyme (MMP9-MYC-His6 APMA and MMP9-pro-catAPMA). To study MMP9 that was activated by cleavage at a site identical to cleavage by MMP3, we activated MMP9-pro-cat with trypsin (MMP9-cat) and further purified MMP9-cat to remove residual trypsin and cleaved prodomain fragments. Treatment of MMP9-MYC-His6 with trypsin resulted in multiple cleavage events, and the resulting MMP9 digestion could not be purified to homogeneity (data not shown). N-terminal sequencing and mass spectrometry confirmed the cleavage sites of MMP9 by trypsin and MMP3 (data not shown).
We determined the binding affinity of GS-5745 to MMP9 using a ProteOn XPR36 biosensor to directly compare the interaction of GS-5745 with pro- and activated MMP9 proteins. The binding profiles of both MMP9-MYC-His6 and MMP9-pro-cat (supplemental Fig. 2, A, B, and F) showed no measureable decay over the course of a dissociation period of 1 h, placing a limit of ≤10−5 s−1 on the dissociation rate constant (koff). Together with the association rates (kon) for these constructs, the equilibrium dissociation constants (KD) were estimated to be ≤43 and 8 pm for MMP9-MYC-His6 and MMP9-pro-cat, respectively (Table 1). We were unable to observe significant dissociation between GS-5745 and pro-MMP9, and the affinity of GS-5745 toward pro-MMP9 is likely higher than what we currently report. Unlike the proform of MMP9, APMA- or trypsin-activated MMP9 showed weaker affinity to GS-5745 with measureable decay observed during the 1-h dissociation period (supplemental Fig. 2, C–F, and Table 1). GS-5745 showed nm affinity to MMP9-MYC-His6 APMA, MMP9-pro-catAPMA, and MMP9-cat (KD = 2.0, 6.6, and 3.3 nm, respectively).
Table 1.
Inhibition of MMP9 by GS-5745
| Km | kcat-appa | IC50 | Inhibition | α | KD | |
|---|---|---|---|---|---|---|
| nm | nm | % | nm | |||
| MMP9-MYC-His6 | ≤0.043b | |||||
| MMP9-pro-cat | ≤0.008b | |||||
| MMP9-MYC-His6 APMA | 9.6 ± 0.1 | 55.3 ± 1.0 | 0.26 ± 0.02 | 56 ± 2 | 1.2 ± 0.0 | 2.0 ± 0.3 |
| MMP9-pro-catAPMA | 880 ± 160 | 3.1 ± 0.3 | 0.32 ± 0.01 | 88 ± 8 | NDc | 6.6 ± 1.3 |
| MMP9-cat | 710 ± 70 | 5.3 ± 0.1 | 1.3 ± 0.1 | 73 ± 0 | 1.1 ± 0.2 | 3.3 ± 0.6 |
akcat-app (relative fluorescence units × min−1 × nm−1) is defined as Vmax/[Etot].
bDissociation rates were below the limit of detection and could only be estimated at ≤10−5 s−1.
cα value for competitive inhibition could not be determined due to the low activity of MMP9-pro-catAPMA.
We tested the enzymatic activity of APMA- or trypsin-activated MMP9 using DQ gelatin as a substrate (22). DQ gelatin is a highly quenched, fluorescein-labeled gelatin that increases in fluorescence (excitation, 485 nm; emission, 520 nm) upon cleavage by MMP9. APMA- and trypsin-activated forms of MMP9 were both active against DQ gelatin, whereas untreated MMP9 showed no detectable activity (supplemental Fig. 3, A and B). The activity of MMP9-cat was not due to residual trypsin as aprotinin had no effect on the rate of cleavage of DQ gelatin (supplemental Fig. 4). MMP9-MYC-His6 APMA was ∼200-fold more active than MMP9-pro-catAPMA and ∼80-fold more active than MMP9-cat. To further characterize the different activated forms of MMP9, we measured the cleavage of varying concentrations of DQ gelatin by a fixed concentration of MMP9 (supplemental Fig. 3, C and D, and Table 1). The catalytic efficiency (kcat/Km) of MMP9-MYC-His6 APMA was ∼1600-fold higher than MMP9-pro-catAPMA and ∼800-fold higher than MMP9-cat.
GS-5745 is a non-competitive inhibitor of MMP9
Fig. 1A shows the activity of MMP9 in the presence of GS-5745. A fixed concentration of the activated forms of MMP9 was incubated with a fixed concentration of DQ gelatin and varying concentrations of GS-5745, and cleavage of DQ gelatin was monitored by fluorescence (Fig. 1A and Table 1). GS-5745 elicits a dose-dependent decrease in MMP9 activity with an IC50 of 0.26 and 0.32 nm against APMA-activated MMP9 (MMP9-MYC-His6 APMA and MMP9-pro-catAPMA, respectively). GS-5745 inhibition of trypsin-activated MMP9 (MMP9-cat) was similar with an IC50 of 1.3 nm. In comparison, marimastat inhibited all activated forms of MMP9 with an IC50 of 1–2 nm (supplemental Fig. 5).
Figure 1.

Inhibition of MMP9 by GS-5745. A, inhibition of activated MMP9 by GS-5745 was measured by incubating a fixed concentration of active MMP9 (0.5 nm MMP9-MYC-His6 APMA (black circles), 5 nm MMP9-pro-catAPMA (red squares), or 5 nm MMP9-cat (blue squares)) with various concentrations of GS-5745. DQ gelatin was added at 2 × Km (30 nm for MMP9-MYC-His6 APMA or 1500 nm for MMP9-pro-catAPMA and MMP9-cat), and activity was monitored by measuring the increase in fluorescence of DQ gelatin upon cleavage by MMP9. IC50 values were determined using a four-parameter curve fitting with GraphPad Prism 6. B and C, for competition studies, a fixed concentration of MMP9-MYC-His6 APMA (0.2 nm) (B) or MMP9-cat (5 nm) (C) was mixed with varying concentrations of GS-5745 (0–2 nm for MMP9-MYC-His6 APMA or 0–16 μm for MMP9-cat) and DQ gelatin (0–150 nm for MMP9-MYC-His6 APMA or 0–2.3 μm for MMP9-cat). The rate of DQ gelatin cleavage was monitored, and the data were fit as described under “Experimental procedures.” Each data point represents the average of duplicate experiments, and error bars represent S.D. RFU, relative fluorescence units.
Fig. 1, B and C, show the enzymatic activity of MMP9-MYC-His6 APMA and MMP9-cat in the presence of increasing concentrations of DQ gelatin and GS-5745. The data were fit globally to the competition equation described under “Experimental procedures.” The fit for α gave a value of 1.2 and 1.0 for MMP9-MYC-His6 APMA and MMP9-cat, respectively. The value of α ∼1 indicates that GS-5745 is a non-competitive inhibitor of both APMA- and trypsin-activated MMP9.
Structure of GS-5745 Fab′ fragment bound to MMP9
Crystallographic analysis was performed to structurally characterize the interactions between GS-5745 and human MMP9 and to aid in elucidating a plausible mechanism for allosteric inhibition of MMP9 by the therapeutic antibody. To facilitate structure determination, an antibody fragment (Fab′) of GS-5745 was generated and incubated with MMP9-pro-cat. MMP9-pro-cat had been previously shown to contain the entire binding epitope recognized by the antibody (19). Diffraction-quality crystals yielded a 3.0-Å-resolution structure containing two well ordered Fab′·MMP9-pro-cat complexes and a third disordered complex that could only be partially modeled (Fig. 2A). It is not clear why the density for the third complex is so weak, but crystal packing analysis suggests that it forms the fewest number of contacts between neighboring molecules compared with the other two complexes in the asymmetric unit. Although all three complexes have been included in the final model for accuracy and completeness, all structural analyses exclude the third complex.
Figure 2.

Structure of GS-5745·MMP9-pro-cat and MMP9-pro-cat. A, the GS-5745·MMP9-pro-cat complex is represented as a ribbon diagram. The prodomain (pro) of MMP9 is colored yellow, and the catalytic domain (cat) is colored green. The heavy chain (Hc) of GS-5745 is colored pink, and the light chain (Lc) is colored cyan. Calcium atoms are colored gray, and zinc atoms are colored magenta. B, superposition of apo MMP9-pro-cat (gray) with MMP9-pro-cat bound to GS-5745 (yellow and green).
The crystal structure of the apo form of MMP9-pro-cat was also determined to ascertain whether there were any significant conformational changes to the enzyme upon binding to GS-5745. The apo and antibody-bound MMP9-pro-cat structures show no significant differences and superimpose with a root-mean-square deviation of 0.25 Å (Fig. 2B). No significant displacements were observed for the structural metal ions or the active-site zinc.
An overlay of the GS-5745·MMP9-pro-cat complex with a structure of pro-MMP9 containing an intact fibronectin II-like domain cluster (Protein Data Bank code 1L6J) (2) reveals that there is no overlap between GS-5745 Fab′ residues and protein atoms in the fibronectin domains. This suggests that the presence of the fibronectin domains would not alter the complex as currently observed, and it is unlikely that the antibody would interact with the fibronectin II-like domains.
Structural analysis of the well ordered complexes (Fig. 2) reveals that the antibody fragment binds to MMP9-pro-cat and buries ∼2130 Å2 of surface. The complex is stabilized by 11 hydrogen bonds, one salt bridge, and numerous, favorable hydrophobic and van der Waals interactions (Fig. 3, A and B). Asn-31 of CDR1 (VL chain) forms an H-bond with the carbonyl oxygen of Gln-126. Two tyrosine residues (Tyr-49 and Tyr-53) and Ser-50 on CDR2 (VL chain) form H-bonds via their side chain hydroxyl groups to Arg-162, Trp-124, and Gln-126, respectively. Interestingly, CDR3 of the VL chain does not contact the antigen directly. CDR1 and CDR2 of the VH chain form three hydrogen bonds with MMP9-pro-cat near the junction of the propeptide and catalytic domain (Fig. 3B). CDR3 (VH chain) contains another pair of tyrosine residues that interact with MMP9 as well as contribute to the lone salt bridge observed in the complex (MMP9-pro-cat, Arg-162; GS-5745, Asp-103). The distance between the nearest residue of GS-5745 (Thr-53 on CDR2 of the VH chain) to the active-site zinc of MMP9-pro-cat is ∼17 Å. GS-5745 does not occlude the active site (Fig. 3B), consistent with its proposed allosteric mode of inhibition (19).
Figure 3.

Interactions between GS-5745 and MMP9. A, Arg-162 of MMP9 forms a salt bridge, hydrogen bonds, and hydrophobic interactions with GS-5745. B, Gln-108 forms hydrogen bonds with the heavy chain of GS-5745. The position of the cleavage site of trypsin/MMP3 between Arg-106 and Phe-107 is indicated. C, superposition of MMP9 bound to GS-5745 and active MMP9 (Protein Data Bank code 1GKD). Lc, light chain; Hc, heavy chain; Ab, antibody.
Arg-162 of MMP9-pro-cat provides the most extensive set of interactions between the antibody and enzyme compared with any other residue (Fig. 3A). Arg-162 is located on a large surface loop connecting strands β2 and β3 of the central parallel β-sheet of MMP9. The long side chain extends into a deep crevice formed between CDR2 and CDR3 of the heavy and light chains, respectively. The backbone nitrogen and two of the side chain guanidinium nitrogens form three hydrogen bonds in addition to the salt bridge with the aspartic acid residue described earlier. The alkyl portion of the arginine side chain is sandwiched between two tyrosine rings (Tyr-99 VH and Tyr-49 VL) that provide favorable hydrophobic interactions. Interestingly, the residue corresponding to position 162 of human MMP9 is responsible for species-specific recognition by GS-5745. The antibody loses significant affinity for mouse MMP9, which contains a proline at this position (19).
GS-5745 inhibits pro-MMP9 activation
Based on the structure of the complex, GS-5745 binding does not result in significant conformational changes of MMP9 and does not appear to interfere directly with the catalytic domain. However, GS-5745 interacts directly with Gln-108 near the junction between the prodomain and catalytic domain at the site of MMP3 cleavage. We tested an additional possibility that GS-5745 could inhibit the activation of MMP9 by MMP3.
To determine whether GS-5745 could inhibit pro-MMP9 activation, we monitored the activation of pro-MMP9 in the presence of GS-5745 or a control IgG4. Fig. 4 shows activation of recombinant MMP9-pro-cat by MMP3 in the presence of GS-5745 or a control IgG4 as a function of time. In the presence of GS-5745, activation of MMP9-pro-cat by MMP3 was reduced, and there was only a slight increase in the amount of activated MMP9-pro-cat. In contrast, MMP9-pro-cat could be activated to a greater extent by MMP3 in the presence of a control IgG4. Extended incubation of MMP9 with MMP3 resulted in degradation of active MMP9 (data not shown), and we were unable to monitor the formation of active MMP9 past 4 h.
Figure 4.
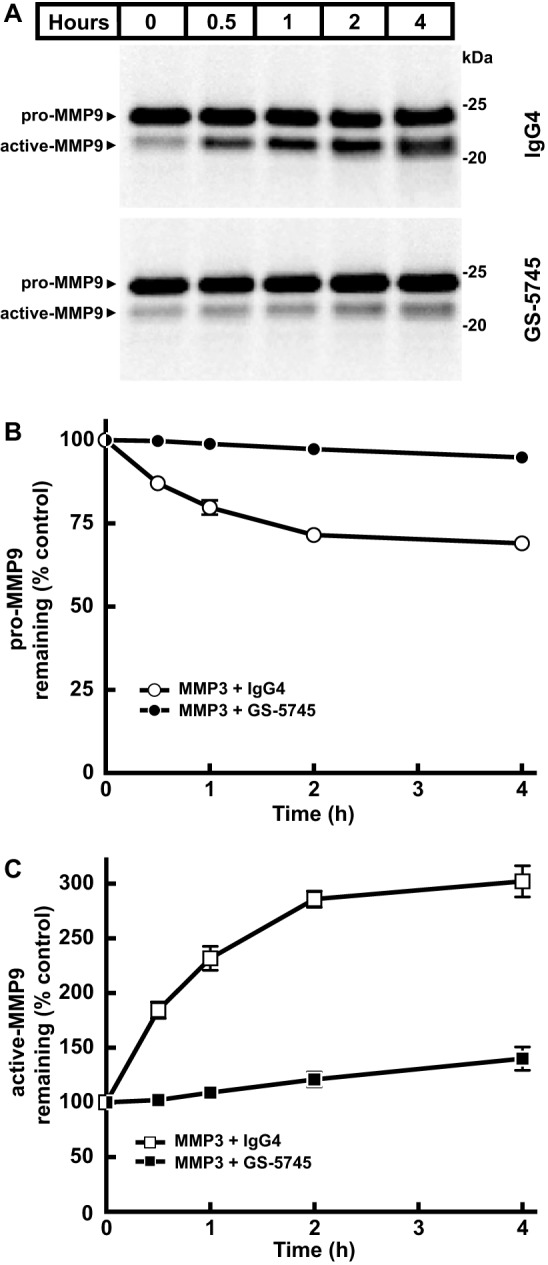
GS-5745 prevents activation of pro-MMP9 by MMP3. A, activation of pro-MMP9 by MMP3 was measured by incubating a fixed concentration pro-MMP9 (2.5 ng/μl MMP9-pro-cat) with MMP3 (0.25 ng/μl) in the presence of GS-5745 (30 ng/μl) or control IgG4 (30 ng/μl). Reactions were incubated at 37 °C for the indicated time, and pro- and active MMP9 were visualized by immunoblotting analysis using an anti-MMP9 antibody. B, the relative intensity of pro-MMP9 in the presence of control IgG4 (○) or GS-5745 (●) in A, normalized to the control lane (0 h), was quantified by densitometry. C, the relative intensity of active MMP9-G100L in the presence of control IgG4 (□) or GS-5745 (■) in A, normalized to the control lane (0 h), was quantified by densitometry. Each data point represents the average of duplicate experiments, and error bars represent S.D.
Fig. 5A shows the steady-state levels of pro-MMP9 and active MMP9 in the medium from a cell line stably expressing either wild-type or a mutant form of MMP9 (MMP9-G100L) (23). The weakened interaction between the prodomain and the catalytic domain of MMP9-G100L leads to increased activation of MMP9-G100L. Activation of wild-type pro-MMP9 in the same cellular background was undetectable. GS-5745 caused a decrease in the amount of active MMP9-G100L and an increase in the amount of pro-MMP9-G100L when compared with untreated cells. Marimastat had a negligible effect on MMP9-G100L processing, indicating that activation in our cell culture system was not the result of another MMP processing MMP9.
Figure 5.

GS-5745 stabilizes pro-MMP9. A, HEK293 cells stably expressing either MMP9 or MMP9-G100L were treated with 30 μg/ml GS-5745 or 200 nm marimastat for 16 h. The medium was harvested and subjected to SDS-PAGE. Pro- and active MMP9 were visualized by immunoblotting analysis using an anti-MMP9 antibody. B, the stability of pro- and active MMP9-G100L was monitored by measuring the half-life of the protein in the absence or presence of GS-5745. HEK293 cells stably expressing MMP9-G100L were incubated with 50 μg/ml cycloheximide (CHX) and either 30 μg/ml GS-5745 or control IgG4. The medium was collected at various times, and pro- and active MMP9-G100L were detected as described previously. C, the relative intensity of pro-MMP9-G100L in the presence of control IgG4 (○) or GS-5745 (●) in B, normalized to the control lane (0 h), was quantified by densitometry. D, the relative intensity of active MMP9-G100L in the presence of control IgG4 (□) or GS-5745 (■) in B, normalized to the control lane (0 h), was quantified by densitometry. Each data point represents the average of duplicate experiments, and error bars represent S.D.
To understand the kinetics of MMP9 processing in more detail, we determined the stability of pro- and active MMP9 in the presence of GS-5745. Cells were treated for up to 28 h with the protein synthesis inhibitor cycloheximide and either GS-5745 or a control human IgG4 (Fig. 5, B–D). In the presence of control IgG4, pro-MMP9-G100L levels decreased by ∼75% accompanied by a slight increase in active MMP9-G100L. In contrast, GS-5745 resulted in an ∼35% increase in pro-MMP9-G100L levels with no significant change in the levels of active MMP9-G100L. In the presence of either control IgG4 or GS-5745, wild-type MMP9 showed no detectable change in the levels of pro-MMP9, and active MMP9 was undetectable (data not shown).
Activation of pro-MMP9 in tissue is critical for subsequent proteolytic activity, but the full spectrum of activating proteases is unknown. The role of MMP9 in ulcerative colitis (UC) suggested that diseased colon tissue could contain high levels of active MMP9 and concomitantly an activator of MMP9.
Fig. 6A shows the levels of pro-MMP9 and active MMP9 from normal and UC tissue lysates as determined by Western blotting analysis. To measure the activity of MMP9 from tissue lysates, total MMP9 was captured with an antibody, and the activity of the captured enzyme was monitored using a fluorescent peptide substrate (Fig. 6B). Active MMP9 was identified in the diseased colon lysates but not normal colon tissue.
Figure 6.

GS-5745 prevents activation of pro-MMP9 by ulcerative colitis lysates. A, colon tissue was lysed, and MMP9 was visualized by immunoblotting. The upper band (molecular mass, ∼92 kDa) was detected in diseased and healthy colon lysate, whereas the lower band (∼82 kDa) was detected in only the ulcerative colitis lysate. B, endogenous MMP9 activity from each colon lysate was assessed. A standard curve of quantitatively activated MMP9 was used to determine the equivalents of active MMP9 in ng/ml. C, incubation of recombinant pro-MMP9 with ulcerative colitis lysates results in MMP9 activation. The activated MMP9 was detected by the neoepitope antibody (red), whereas both pro- and active MMP9 are detected by the total MMP9 antibody (green). MMP9 activation is inhibited by GS-5745 but not a human IgG4 isotype control antibody. D, the activity of MMP9 in C was measured with the Biotrak MMP9 assay kit. GS-5745, but not the isotype control IgG4, blocks this proteolysis. MMP9 was isolated from the lysate constituents prior to activity assessment. Each data point represents a single experiment, and error bars represent S.D.
To determine whether GS-5745 could block activation of pro-MMP9 by UC tissue lysates, MMP9-MYC-His6 was incubated with UC tissue lysates in the absence or presence of GS-5745 or control IgG4. GS-5745 did not interfere with the capture of MMP9 in this assay (data not shown). The UC tissue lysate was able to activate pro-MMP9, and this activation was inhibited by GS-5745 but not control IgG4 (Fig. 6, C and D). Western blotting analysis using an antibody that specifically recognizes the N terminus of MMP9 after cleavage between Arg-106 and Phe-107 (supplemental Fig. 6) demonstrated that UC tissue lysates can cleave MMP9 after Arg-106. This site is identical to the site cleaved by MMP3 (Fig. 6C). The appearance of the active MMP9 band correlated with MMP9 activity, and the addition of GS-5745 prevents both formation of the active MMP9 band and an increase in activity.
Discussion
Marshall et al. (19) recently showed that GS-5745 could inhibit APMA-activated MMP9 by a non-competitive mechanism. One caveat to APMA activation of MMP9 is that the resulting cleavage of MMP9 may not represent the physiologically relevant form of active MMP9. By using trypsin to activate truncated MMP9, we could mimic MMP3 activation of MMP9 (4, 20). Truncated MMP9 is nearly identical in all properties to full-length MMP9. GS-5745 binds full-length and truncated MMP9 with similar affinities (Table 1) and inhibits both APMA- and trypsin-activated full-length and truncated MMP9 with similar IC50 (Fig. 1A). The structure of GS-5745·MMP9 complex shows that the truncated MMP9 used in crystallization has a structure identical to the reported structure of pro-MMP9 (2).
GS-5745 binds to active MMP9 with ∼150–400-fold weaker affinity compared with pro-MMP9. The striking difference in the affinity of GS-5745 to pro-MMP9 compared with active MMP9 is consistent with the crystal structure of the GS-5745·MMP9 complex. The structure of the GS-5745·MMP9 complex reveals that MMP9 adopts the inactive conformation present in the pro-MMP9 structure (2), and GS-5745 binds residues exclusively within the catalytic domain. Our structure is consistent with previous site-directed mutagenesis studies that identified that residues critical for binding GS-5745 were located entirely within the catalytic domain (19). When compared with the structure of the MMP9 catalytic domain (24), MMP9 in the GS-5745·MMP9 complex is nearly identical. However, the region at the N terminus of the catalytic domain (residues 107–114) adopts an altered conformation (Fig. 3C). A key residue identified in the crystal structure of GS-5745·MMP9 complex is Gln-108. For Gln-108 in activated MMP9 to bind to GS-5745, this region would have to undergo a conformational change and translate ∼8.5 Å. The lower affinity observed for GS-5745 binding to active MMP9 may be the result of the loss of interaction of Gln-108 to GS-5745 in activated MMP9.
Analysis of the structure GS-5745·MMP9 complex and the biochemical data revealed that the interaction of GS-5745 with Gln-108 of MMP9, near the junction between the prodomain and catalytic domains (Fig. 3B), may be of particular relevance. Previous reports suggest one or multiple proteolytic cleavage sites on human MMP9 in this region that may be required for activation (4, 20, 21). MMP3 activates MMP9 by cleaving between Arg-106 and Phe-107. Based on the structure, it seems plausible that GS-5745 could bind to the inactive zymogen (pro-MMP9) and prevent or limit the ability of activating proteases to cleave at the necessary site or sites, thereby preventing activation of MMP9 altogether.
In addition to blocking the activation of pro-MMP9, GS-5745 inhibits active MMP9 (Table 1). Although GS-5745 does not completely inhibit MMP9 activity and substrate binding, it may affect substrate orientation and subsequent cleavage. Compared with marimastat, which showed complete inhibition of active MMP9 (supplemental Fig. 5), GS-5745 only partially inhibited MMP9 (56%). In contrast, GS-5745 inhibited truncated forms of activated MMP9 to a greater extent (73–88%). An overlay of the GS-5745·MMP9-pro-cat structure and an MMP1 structure bound to a native triple-helical collagen peptide (25) suggests that GS-5745 binding distal to the MMP9 active site could sterically hinder but not completely block larger substrates, resulting in less efficient proteolysis (supplemental Fig. 7). The fibronectin type II and hemopexin domains have been shown to be important for binding gelatin (26, 27). Removal of these domains could explain the differences observed in the activity and extent of inhibition among the various forms of activated MMP9 used in this study. Further structural studies will be required to elucidate how MMP9 binds to its natural substrates and how GS-5745 could affect substrate binding.
GS-5745 is a promising therapeutic anti-MMP9 antibody for treatment of ulcerative colitis and gastric cancer, showing efficacy in phase I clinical trials (28). Our current data support a model whereby GS-5745 inhibits MMP9 by two mechanisms: 1) inhibiting the catalytic activity of MMP9 by sterically interfering with proteolysis of substrates and 2) inhibiting the processing of MMP9 to an active form by sterically preventing accessibility of activating proteases. This dual mode of inhibition and the high affinity of GS-5745 toward pro-MMP9 are unique among therapeutic antibodies and provide a significant advantage to GS-5745 inhibition of MMP9. By inhibiting the activation and activity of MMP9, GS-5745 both reduces the amount of circulating active MMP9 and inhibits any active MMP9 that is present. The extent to which these mechanisms contribute to GS-5745 efficacy in vivo is currently under investigation.
Experimental procedures
Materials and plasmids
pSecTag2-MMP9, encoding human MMP9 followed by a C-terminal MYC and His6 tag, was generated by amplifying the nucleotide sequence from residues 1–707 of MMP9 by PCR. The PCR fragment was digested with NheI and XhoI and ligated into pSecTag2 (Life Technologies). pSecTag2-MMP9-G100L was generated by site-directed mutagenesis (Stratagene). pet30a-MMP9-pro-cat (residues 39–215 and 391–444) was generated by deletion of residues 216–390 of MMP9 by sited-directed mutagenesis (Phusion site-directed mutagenesis, New England Biolabs). The resulting deletion mutant of MMP9 was used to amplify residues 39–444 by PCR. The PCR fragment was digested with NdeI and HindIII and ligated into pet30a (Novagen).
Stable cell lines expressing either full-length MMP9 or MMP9-G100L were generated by transfecting HEK293 cells with either pSecTag2-MMP9 or pSecTag2-MMP9-G100L using FuGENE HD (Roche Applied Science). Cells expressing MMP9 or MMP9-G100L were selected by culturing in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS (medium A) and 50 μg/μl hygromycin B (Life Technologies). Cells were plated on 100-mm dishes, and individual clones were expanded in medium A supplemented with 50 μg/μl hygromycin B.
DQ gelatin was obtained from Life Technologies and resuspended in water to 10 μm (assuming a molecular mass of 100,000 g/mol). Marimastat was obtained from Tocris Biosciences (Bristol, United Kingdom). APMA and aprotinin were obtained from Sigma-Aldrich. FabRicator (Bulldog-bio) was prepared according to the manufacturer's protocol. Sequencing-grade trypsin was obtained from Promega. Total MMP9 reagent antibody was obtained from Biolegend (San Diego, CA), and the human IgG4 isotype control was obtained from Eureka Therapeutics (Emeryville, CA).
Protein expression and purification
MMP9-MYC-His6 was expressed in Expi293 cells (Invitrogen). Cells were grown in Expi293 expression medium and harvested 5 days post-transfection. The conditioned medium was applied to a nickel-nitrilotriacetic acid-agarose column equilibrated in 25 mm Tris, pH 7.5, 150 mm NaCl (buffer A). MMP9-MYC-His6 was eluted using a linear gradient of 0–500 mm imidazole. Fractions containing MMP9-MYC-His6 were pooled, concentrated, and further purified by size-exclusion chromatography on a Superdex 200 column (GE Healthcare) equilibrated in buffer A.
MMP9-pro-cat was expressed in BL21(DE3) cells (Novagen). Cells were grown in 2× YT (1.6% (w/v) Tryptone, 1.0% (w/v) yeast extract, 0.5% (w/v) NaCl) medium at 37 °C, and expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside for 3 h. Cells were lysed in 50 mm Tris-HCl, pH 7.5, 0.5% Triton X-100 using a microfluidizer. Inclusion bodies containing MMP9-pro-cat were isolated by centrifugation at 27,000 × g for 30 min. The pelleted inclusion bodies were washed sequentially with 25 mm Tris-HCl, pH 8.0, 1 m urea and 25 mm Tris-HCl, pH 8, 1 m NaCl, 0.2% Triton X-100. The final washed inclusion bodies were resuspended in 25 mm Tris-HCl, pH 7.5, 10 mm DTT, 8 m urea and stirred for 12 h at room temperature. Following centrifugation at 27,000 × g for 1 h, the supernatant, containing denatured MMP9-pro-cat, was adjusted to 0.4 mg/ml and dialyzed against 12 liters of 20 mm Tris, pH7.5, 200 mm NaCl, 5 mm CaCl2, 1 mm ZnCl2, 2 m urea, 0.3 m l-arginine, 10 mm reduced glutathione, 1 mm oxidized glutathione at room temperature. After 12 h, MMP9-pro-cat was dialyzed against 40 liters of 25 mm Tris-HCl, pH7.5, 300 mm NaCl at 4 °C for 12 h. The refolded MMP9-pro-cat was purified by size-exclusion chromatography using a Sephacryl S200HR column (GE Healthcare) in 25 mm Tris-HCl, pH 7.5, 300 mm NaCl.
GS-5745 Fab′ was generated by digesting 1 mg of GS-5745 with ∼100 units of FabRicator for 3 h at 37 °C. The digest was passed through Protein A-agarose (GE Healthcare), and the F(ab′)2 was treated with 50 mm 2-mercaptoethanol amine for 2 h at 37 °C. Iodoacetamide was then added to a final concentration of 30 mm, and the resulting Fab′ was purified by size-exclusion chromatography using a Superdex 200 column in PBS.
GS-5745 Fab′ was incubated with a 1.5-fold excess of MMP9-pro-cat for 3 h at 4 °C. The complex was purified from excess MMP9-pro-cat by size-exclusion chromatography using a Superdex 200 column in 20 mm Tris-HCl, pH 8.0, 100 mm NaCl. Fractions containing purified complex were pooled and concentrated to 15 mg/ml.
Generation of antibody against activated MMP9
Rabbits were immunized with the peptide NH2-Phe-Gln-Thr-Phe-Glu-Gly-Asp-Cys conjugated to ovalbumin or keyhole limpet hemocyanin via thiol chemistry. Antibodies were enriched from crude serum by binding to the immunizing peptide conjugated to BSA and depleted with the peptide NH2-Val-Pro-Asp-Leu-Gly-Arg-Phe-Gln-Thr-Phe-Glu-Gly-Asp-Cys (also conjugated to BSA). The resulting polyclonal antibody was concentrated to 0.32 mg/ml at Abcam Burlingame.
Activation of recombinant pro-MMP9
MMP9-cat was generated by digesting 1 mg of MMP9-pro-cat with 10 μg of trypsin for 20 h at room temperature. Trypsin was removed by passing the digest through soybean trypsin inhibitor-agarose (Sigma-Aldrich), and MMP9-cat was further purified by size-exclusion chromatography using a Superdex 75 column (GE Healthcare) in 20 mm Tris-HCl, pH 7.5, 300 mm NaCl. MMP9-MYC-His6 APMA and MMP9-pro-catAPMA were generated by incubating MMP9-MYC-His6 or MMP9-pro-cat with 1 mm APMA for 15 h at 37 °C.
Activation of pro-MMP9 by ulcerative colitis lysates
Ulcerative colitis tissue was obtained after surgical colectomy (Folio Biosciences). Specimens were selected based on disease severity scored by a pathologist on matched formalin-fixed paraffin-embedded tissues. Frozen fragments of ∼50 mg were lysed in 10 mm HEPES, pH 7.5, 150 mm NaCl, 10 mm CaCl2, 0.05% Brij-35 on a TissueLyser (Qiagen). 2300 ng/ml recombinant pro-MMP9 was incubated with 2 μl of the resulting lysate in a total volume of 30 μl. Activity was assessed via immunoblotting on a 4–12% Tris-glycine gel with anti-MMP9 for total MMP9 (Biolegend) or the neoepitope antibody (described above; Abcam Burlingame). Activity was assessed with the MMP9 Biotrak kit (GE Healthcare) in the absence of APMA.
Surface plasmon resonance binding
Label-free binding studies were performed on a ProteOn XPR36 using GLM chips (Bio-Rad) at 25 °C. Each chip was preconditioned using sequential 1-min pulses of 0.5% SDS, 50 mm NaOH, and 100 mm HCl in both the horizontal and vertical directions. The six horizontal channels were activated with an injection of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysulfosuccinimide (EDC/sulfo-NHS) (Bio-Rad) followed by immobilization of goat anti-human Fc antibody (GAHFc, Invitrogen) in 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% Tween 20. The surfaces were then deactivated by injection of ethylenediamine (Sigma-Aldrich). All subsequent capture and analyte injections steps were performed in 10 mm HEPES, pH 7.4, 150 mm NaCl, 10 mm CaCl2, 0.8 mg/ml bovine serum albumin, 0.03% Tween 20. GS-5745 was captured at different concentrations on all six channels in the vertical direction to achieve ligand densities between 20 and 160 response units, accommodating an optimal maximal binding signal (Rmax) of 20–40 response units. To stabilize the system, buffer was injected three times horizontally prior to injection of analyte. MMP9 analyte was then injected in the horizontal direction for 8 min. This was followed by a dissociation phase of 1 h. MMP9 was injected at concentrations of 0, 1.24, 3.70, 11.1, 33.3, and 100 nm for all forms of MMP9 used except MMP9-pro-cat, which was injected at 0, 0.39, 1.56, 6.25, and 25 nm. Injections and dissociations were performed at a flow rate of 50 μl/min. At the end of each cycle, five 18-s pulses of 0.85% phosphoric acid (Sigma-Aldrich) were used to regenerate the GAHFc surfaces. Data were analyzed using the ProteOn Manager Software v.3.1.0.6 (Bio-Rad). GAHFc horizontal interspots were used to subtract background binding, and when necessary drift was corrected using a 0 nm protein injection. Data were fit to a simple 1:1 interaction model with single association and dissociation rate constants.
Enzymatic activity of MMP9
All assays were carried out in 96-well solid, black microplates (Corning) at 24 °C. DQ gelatin cleavage by MMP9 was monitored by measuring the increase in fluorescence (excitation, 485 nm; emission, 520 nm) on an Infinite M1000 Pro Reader (Tecan, Switzerland) over the course of 2 h.
A fixed concentration of active MMP9 (0.5 nm MMP9-MYC-His6 APMA, 5 nm MMP9-pro-catAPMA, or 5 nm MMP9-cat) was mixed with increasing concentrations of DQ gelatin (0–5 μm) in a final volume of 100 μl of 50 mm Tris-HCl, pH 7.5, 10 mm CaCl2, 150 mm NaCl, 0.05% (v/v) Brij-35 (buffer B). Cleavage of DQ gelatin was monitored by an increase in fluorescence, and initial rates were determined. Fluorescence increase was measured continuously over 2 h. Km and Vmax were determined by fitting data to the Michaelis-Menten equation (29) with GraphPad Prism 6. To test for residual trypsin activity, various concentrations of active MMP9-cat (0–5 nm) were mixed with a fixed concentration of DQ gelatin (100 nm) and aprotinin (0.05 mg/ml) in a final volume of 100 μl of buffer B.
IC50 determination for GS-574
For IC50 determinations, a fixed concentration of active MMP9 (0.5 nm MMP9-MYC-His6 APMA, 5 nm MMP9-pro-catAPMA, or 5 nm MMP9-cat) was incubated with varying concentrations of GS-5745, control IgG4, or marimastat. Reactions with marimastat contained 1% DMSO. DQ gelatin was added at 2 × Km (30 nm for MMP9-MYC-His6 APMA or 1500 nm for MMP9-pro-catAPMA and MMP9-cat), and cleavage of DQ gelatin was monitored as described above. Reaction rates were determined based upon progress curves after reaching steady state. IC50 values were determined using a four-parameter curve fitting with GraphPad Prism 6.
Competition of GS-5745 with DQ gelatin
Active MMP9 (0.2 nm MMP9-MYC-His6 APMA or 5 nm MMP9-cat) was mixed with varying concentrations of GS-5745 (0–2 nm for MMP9-MYC-His6 APMA or 0–16 μm for MMP9-cat). Varying concentrations of DQ gelatin (0–150 nm for MMP9-MYC-His6 APMA or 0–7 μm for MMP9-cat) were added, and cleavage of DQ gelatin was monitored as described above. The rate data were fit globally. Data analysis was performed with GraphPad Prism software with the following equation,
| (Eq. 1) |
where ν is the observed reaction rate, Vmax is the maximum reaction rate, [S] is the concentration of DQ gelatin, Km is the Michaelis constant for DQ gelatin, b is the value related to partial inhibition, [I] is the concentration of GS-5745, Ki is the inhibition constant for GS-5745, and α is a multiplier for Ki that describes the competition behavior. Values of α ≫1 are indicative of competitive inhibition, values of α ∼1 imply non-competitive inhibition, and values of α ≪1 are characteristic of uncompetitive inhibition.
Activation of MMP9 by MMP3 in the presence of GS-5745
Pro-MMP9 (2.5 ng/μl MMP9-pro-cat) was incubated with MMP3 (0.25 ng/μl) in the presence of GS-5745 (30 ng/μl) or control IgG4 (30 ng/μl). Reactions were incubated at 37 °C for various times, and pro- and active MMP9 were visualized by immunoblotting analysis using a rabbit monoclonal antibody raised against MMP9 (Abcam).
Processing of secreted MMP9 in the presence of GS-5745
On day 0, HEK293 cells stably expressing MMP9 or MMP9-G100L were plated at a density of 0.4 × 106 cells/well in a 6-well plate in DMEM supplemented with 10% FBS. On day 1, the cells were changed to serum-free medium containing 30 μg/ml GS-5745 or 200 nm marimastat. After 16 h, the medium was collected and clarified by centrifugation at 17,000 × g for 30 min at 4 °C. Protein concentration was measured by BCA assay (Pierce), and equal amounts of protein were subjected to 6% SDS-PAGE and transferred to nitrocellulose membranes (Life Technologies). The membranes were incubated in Odyssey blocking buffer (LI-COR Biosciences) followed by incubation with a rabbit monoclonal antibody raised against MMP9 (Abcam). To detect MMP9, the membranes were incubated with IRDye680-conjugated anti-rabbit antibody and visualized with a LI-COR Biosciences scanner. The Western blots were quantified using Image Studio software (LI-COR Biosciences).
To determine the half-life of MMP9, HEK293 cells stably expressing MMP9 or MMP9-G100L plated on day 0 at a density of 0.5 × 106 cells/well in a 6-well plate in DMEM supplemented with 10% FBS. On day 1, cells were changed to serum-free medium containing 50 μg/ml cycloheximide and either 30 μg/ml GS-5745 or control IgG4. Medium was collected at various times, and MMP9 was detected as described above.
Crystallization and structure determination
Apo crystals of purified MMP9-pro-cat were grown from a 1:1 mixture of protein solution (12.8 mg/ml in 0.02 m Tris, pH 8.0, 0.3 m NaCl) to precipitant solution (4% (v/v) PEG 8000, 0.3 m lithium sulfate, 0.1 m sodium acetate, pH 5.0) that was equilibrated against 100 μl of reservoir solution (12% (v/v) PEG 8000, 0.3 m lithium sulfate, 0.1 m sodium acetate, pH 5.0). Crystals grown for approximately 2 weeks were harvested and flash frozen in cryogenic artificial mother liquor (12% (v/v) PEG 8000, 30% (v/v) ethylene glycol, 0.2 m lithium sulfate, 0.1 m sodium acetate, pH 5.0).
X-ray diffraction data were collected on a single frozen (100 K) MMP9-pro-cat crystal on beamline 5.0.1 at the Advanced Light Source (Berkeley, CA) to a resolution of 1.7 Å. Data were processed with HKL2000 and scaled with Scalepack (30) in the primitive monoclinic space group P21. A search model constructed using Protein Data Bank code 1L6J (deleting coordinates for residues 21–39 at the N terminus and residues 216–390 to remove the three fibronectin type II repeats from the catalytic domain) was used in the molecular replacement program Phaser (31), which found four independent molecules in the asymmetric unit (arbitrarily assigned molecules A, B, C, and D). Subsequent rounds of refinement using the PHENIX (32) suite of programs with manual model fitting and the addition of ordered solvent molecules brought the R-factor and Rfree down to 20.8 and 23.5%, respectively. The final protein model includes four MMP9-pro-cat molecules.
Crystals of the purified GS-5745 Fab′·MMP9-pro-cat complex were grown from a 6:1 mixture of protein solution (22.4 mg/ml in 0.02 m Tris, pH 8.0, 0.1 m NaCl) to precipitant solution (8% (v/v) PEG 8000, 0.1 m HEPES, pH 7.5, 50% (v/v) Silver Bullet 21 (Hampton Research)) that was equilibrated against 100 μl of reservoir solution (22.5% (v/v) PEG 8000, 0.1 m HEPES, pH 7.5). Crystals grown for approximately 2 weeks were harvested and flash frozen in cryogenic artificial mother liquor (20% (v/v) PEG 8000, 14% (v/v) glycerol, 0.05 m NaCl, 0.025 m HEPES, pH 7.5, 0.006 m hexamine cobalt(III) chloride).
X-ray diffraction data were collected on a single frozen (100 K) GS-5745 Fab′·MMP9-pro-cat complex crystal on beamline 5.0.1 at the Advanced Light Source to a resolution of 3.0 Å. Data, processed with HKL2000 and scaled with Scalepack (30), suggested a primitive tetragonal lattice. Molecule A from the apo-MMP9-pro-cat structure described above and a polyalanine antibody Fab′ fragment generated from Protein Data Bank code 1F8T were used as search models in the molecular replacement program Phaser (31) where the MMP9-pro-cat and polyalanine Fab′ models were treated as independent rigid bodies and searched for sequentially. All possible space groups in the primitive tetragonal point group P422 were searched with only the space group P43212 yielding a solution with two Fab′·MMP9-pro-cat complexes (complex A and complex B) in the asymmetric unit.
The initial density maps after least-square refinement using the PHENIX (32) suite of programs were of high quality and allowed the side chains to be added to the GS-5745 Fab′ model. Subsequent rounds of refinement with manual model fitting brought the R-factor and Rfree down to 24.0 and 28.4%, respectively. At this point in the refinement process, it was clear that there was weak density suggesting the presence of a third molecule of MMP9-pro-cat in the crystal. An additional MMP9-pro-cat was added to the current model and fit as a rigid-body component during additional rounds of refinement, bringing the R-factor and Rfree down to 22.7 and 26.5%. Even weaker density was now evident for an additional Fab fragment bound to the third MMP9-pro-cat, and therefore a third complete complex (complex C) was added to the model but refined as a three-component rigid body throughout the remainder of the refinement process. The model is currently refined to an R-factor of 21.8% and an Rfree of 25.6%. Data collection and refinement statistics for the apo- and antibody-bound MMP9-pro-cat structures are summarized in supplemental Table 1.
Author contributions
H. J. K., V. S., and R. S. conceived and coordinated the study. X. L. and H. J. K. designed the MMP9 construct for crystallization. R. W., M. H., and H. J. K. expressed and purified MMP9 and GS-5745. T. C. A. and A. G. V. crystallized the proteins and determined the structures. A. L. and B. E. S. designed and performed enzymatic assays. M. H. W. and G. A. P. designed and performed binding assays. M. V. and A. E. G. performed the cell culture and tissue experiments. T. C. A., A. E. G., and H. J. K wrote the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Erik Huntzicker and Jacky Woo for assistance identifying and analyzing ulcerative colitis tissues; Pierre Lee at Abcam Burlingame for assistance with the anti-neoepitope antibody; and Kathy Brendza, Wanchi Fung, Debi Jin, Andrew Post, Leanna Lagpacan, Michael Lee, Anita Majka, Mark Nagel, and Loredana Serafini for assistance with protein purification and characterization.
This work was funded by Gilead Sciences, Inc. All authors are employees of Gilead Sciences, Inc.
This article contains supplemental Figs. 1–7 and Table 1.
The atomic coordinates and structure factors (codes 5TH6 and 5TH9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MMP
- matrix metalloproteinase
- APMA
- 4-aminophenylmercuric acetate
- UC
- ulcerative colitis
- DQ
- dye-quenched
- GAHFc
- goat anti-human Fc antibody.
References
- 1. Nagase H., and Woessner J. F. Jr. (1999) Matrix metalloproteinases. J. Biol. Chem. 274, 21491–21494 [DOI] [PubMed] [Google Scholar]
- 2. Elkins P. A., Ho Y. S., Smith W. W., Janson C. A., D'Alessio K. J., McQueney M. S., Cummings M. D., and Romanic A. M. (2002) Structure of the C-terminally truncated human proMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. D Biol. Crystallogr. 58, 1182–1192 [DOI] [PubMed] [Google Scholar]
- 3. Vandooren J., Van den Steen P. E., and Opdenakker G. (2013) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit. Rev. Biochem. Mol. Biol. 48, 222–272 [DOI] [PubMed] [Google Scholar]
- 4. Ogata Y., Enghild J. J., and Nagase H. (1992) Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J. Biol. Chem. 267, 3581–3584 [PubMed] [Google Scholar]
- 5. Shapiro S. D., Fliszar C. J., Broekelmann T. J., Mecham R. P., Senior R. M., and Welgus H. G. (1995) Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP). J. Biol. Chem. 270, 6351–6356 [DOI] [PubMed] [Google Scholar]
- 6. Van den Steen P. E., Dubois B., Nelissen I., Rudd P. M., Dwek R. A., and Opdenakker G. (2002) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 37, 375–536 [DOI] [PubMed] [Google Scholar]
- 7. Ram M., Sherer Y., and Shoenfeld Y. (2006) Matrix metalloproteinase-9 and autoimmune diseases. J. Clin. Immunol. 26, 299–307 [DOI] [PubMed] [Google Scholar]
- 8. Overall C. M., and López-Otín C. (2002) Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer 2, 657–672 [DOI] [PubMed] [Google Scholar]
- 9. Coussens L. M., Fingleton B., and Matrisian L. M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 [DOI] [PubMed] [Google Scholar]
- 10. Hidalgo M., and Eckhardt S. G. (2001) Development of matrix metalloproteinase inhibitors in cancer therapy. J. Natl. Cancer Inst. 93, 178–193 [DOI] [PubMed] [Google Scholar]
- 11. Lakatos G., Sipos F., Miheller P., Hritz I., Varga M. Z., Juhász M., Molnár B., Tulassay Z., and Herszényi L. (2012) The behavior of matrix metalloproteinase-9 in lymphocytic colitis, collagenous colitis and ulcerative colitis. Pathol. Oncol. Res. 18, 85–91 [DOI] [PubMed] [Google Scholar]
- 12. Peterson J. T. (2006) The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc. Res. 69, 677–687 [DOI] [PubMed] [Google Scholar]
- 13. Gao Q., Meijer M. J., Kubben F. J., Sier C. F., Kruidenier L., van Duijn W., van den Berg M., van Hogezand R. A., Lamers C. B., and Verspaget H. W. (2005) Expression of matrix metalloproteinases-2 and -9 in intestinal tissue of patients with inflammatory bowel diseases. Dig. Liver Dis. 37, 584–592 [DOI] [PubMed] [Google Scholar]
- 14. Gallea-Robache S., Morand V., Millet S., Bruneau J. M., Bhatnagar N., Chouaib S., and Roman-Roman S. (1997) A metalloproteinase inhibitor blocks the shedding of soluble cytokine receptors and processing of transmembrane cytokine precursors in human monocytic cells. Cytokine 9, 340–346 [DOI] [PubMed] [Google Scholar]
- 15. McGeehan G. M., Becherer J. D., Bast R. C. Jr., Boyer C. M., Champion B., Connolly K. M., Conway J. G., Furdon P., Karp S., Kidao S., McElroy A. B., Nichols J., Pryzwansky K. M., Schoenen F., Sekut L., et al. (1994) Regulation of tumour necrosis factor-α processing by a metalloproteinase inhibitor. Nature 370, 558–561 [DOI] [PubMed] [Google Scholar]
- 16. Müllberg J., Durie F. H., Otten-Evans C., Alderson M. R., Rose-John S., Cosman D., Black R. A., and Mohler K. M. (1995) A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 155, 5198–5205 [PubMed] [Google Scholar]
- 17. Sheu B. C., Hsu S. M., Ho H. N., Lien H. C., Huang S. C., and Lin R. H. (2001) A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 61, 237–242 [PubMed] [Google Scholar]
- 18. Sparano J. A., Bernardo P., Stephenson P., Gradishar W. J., Ingle J. N., Zucker S., and Davidson N. E. (2004) Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J. Clin. Oncol. 22, 4683–4690 [DOI] [PubMed] [Google Scholar]
- 19. Marshall D. C., Lyman S. K., McCauley S., Kovalenko M., Spangler R., Liu C., Lee M., O'Sullivan C., Barry-Hamilton V., Ghermazien H., Mikels-Vigdal A., Garcia C. A., Jorgensen B., Velayo A. C., Wang R., Adamkewicz J. I., and Smith V. (2015) Selective allosteric inhibition of MMP9 is efficacious in preclinical models of ulcerative colitis and colorectal cancer. PLoS One 10, e0127063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duncan M. E., Richardson J. P., Murray G. I., Melvin W. T., and Fothergill J. E. (1998) Human matrix metalloproteinase-9: activation by limited trypsin treatment and generation of monoclonal antibodies specific for the activated form. Eur. J. Biochem. 258, 37–43 [DOI] [PubMed] [Google Scholar]
- 21. Okada Y., Gonoji Y., Naka K., Tomita K., Nakanishi I., Iwata K., Yamashita K., and Hayakawa T. (1992) Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J. Biol. Chem. 267, 21712–21719 [PubMed] [Google Scholar]
- 22. Vandooren J., Geurts N., Martens E., Van den Steen P. E., Jonghe S. D., Herdewijn P., and Opdenakker G. (2011) Gelatin degradation assay reveals MMP-9 inhibitors and function of O-glycosylated domain. World J. Biol. Chem. 2, 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher K. E., Fei Q., Laird E. R., Stock J. L., Allen M. R., Sahagan B. G., and Strick C. A. (2002) Engineering autoactivating forms of matrix metalloproteinase-9 and expression of the active enzyme in cultured cells and transgenic mouse brain. Biochemistry 41, 8289–8297 [DOI] [PubMed] [Google Scholar]
- 24. Rowsell S., Hawtin P., Minshull C. A., Jepson H., Brockbank S. M., Barratt D. G., Slater A. M., McPheat W. L., Waterson D., Henney A. M., and Pauptit R. A. (2002) Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J. Mol. Biol. 319, 173–181 [DOI] [PubMed] [Google Scholar]
- 25. Manka S. W., Carafoli F., Visse R., Bihan D., Raynal N., Farndale R. W., Murphy G., Enghild J. J., Hohenester E., and Nagase H. (2012) Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. U.S.A. 109, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collier I. E., Krasnov P. A., Strongin A. Y., Birkedal-Hansen H., and Goldberg G. I. (1992) Alanine scanning mutagenesis and functional analysis of the fibronectin-like collagen-binding domain from human 92-kDa type IV collagenase. J. Biol. Chem. 267, 6776–6781 [PubMed] [Google Scholar]
- 27. Roeb E., Schleinkofer K., Kernebeck T., Pötsch S., Jansen B., Behrmann I., Matern S., and Grötzinger J. (2002) The matrix metalloproteinase 9 (MMP-9) hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J. Biol. Chem. 277, 50326–50332 [DOI] [PubMed] [Google Scholar]
- 28. Bendell J. C., Starodub A., Shah M. A., Sharma S., Wainberg Z. A., and Thai D. L. (2015) Phase I study of GS-5745 alone and in combination with chemotherapy in patients with advanced solid tumors. ASCO Meet. Abstr. 33, 4030 [Google Scholar]
- 29. Michaelis L., Menten M. L., Johnson K. A., and Goody R. S. (2011) The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50, 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otwinowski Z., and W M. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.