

Abstract
Gaucher disease (GD), the commonest lysosomal storage disorder, results from the lack or functional deficiency of glucocerebrosidase (GCase) secondary to mutations in the GBA1 gene. There is an established association between GBA1 mutations and Parkinson's disease (PD), and indeed GBA1 mutations are now considered to be the greatest genetic risk factor for PD. Impaired lysosomal-autophagic degradation of cellular proteins, including α-synuclein (α-syn), is implicated in the pathogenesis of PD, and there is increasing evidence for this also in GD and GBA1-PD. Indeed we have recently shown in a Drosophila model lacking neuronal GCase, that there are clear lysosomal-autophagic defects in association with synaptic loss and neurodegeneration. In addition, we demonstrated alterations in mechanistic target of rapamycin complex 1 (mTORC1) signaling and functional rescue of the lifespan, locomotor defects and hypersensitivity to oxidative stress on treatment of GCase-deficient flies with the mTOR inhibitor rapamycin. Moreover, a number of other recent studies have shown autophagy-lysosomal system (ALS) dysfunction, with specific defects in both chaperone-mediated autophagy (CMA), as well as macroautophagy, in GD and GBA1-PD model systems. Lastly we discuss the possible therapeutic benefits of inhibiting mTOR using drugs such as rapamycin to reverse the autophagy defects in GD and PD.
Keywords: Gaucher disease, Parkinson's disease, Drosophila, autophagy, lysosome, glucocerebrosidase, GBA
The Link between Gaucher Disease and Parkinson's Disease
Homozygous loss-of-function mutations in the GBA1 gene cause Gaucher disease (GD), the commonest lysosomal storage disorder. This gene encodes glucocerebrosidase (GCase), a lysosomal enzyme responsible for the hydrolysis of the glycolipid substrate glucosylceramide (GlcCer) to ceramide and glucose. Reductions in enzymatic activity result in the lysosomal accumulation of GlcCer, as well as glucosylsphingosine, within reticulo-endoendothelial cells, leading to the systemic sequelae of GD, including organomegaly, bone disease, anaemia and thrombocytopenia. The clinical spectrum of GD has historically been sub-divided into Type 1 GD, the so-called non-neuronopathic milder form of the disease, and Types II and III GD, which are severe acute and chronic neuronopathic forms respectively (Cox, 2010). In recent years heterozygous GBA1 mutations have been identified as key genetic risk factors for Parkinson's disease (PD), and increase the risk of developing PD by approximately 20-fold (Sidransky et al., 2009; Migdalska-Richards and Schapira, 2016).
The Role of Autophagy in Cellular α-Synuclein Degradation
The neuropathological hallmark of PD is the presence of proteinacious intraneuronal inclusions, known as Lewy bodies, predominantly composed of aggregated α-synuclein (α-syn), in addition to other proteins such as ubiquitin and p62 (Zatloukal et al., 2002; Shults, 2006). α-syn is a presynaptic protein of unknown function, which is thought to play a central role in the pathogenesis of PD. Moreover, there is mounting evidence that soluble α-syn aggregation intermediates (so-called oligomeric or protofibrillar species) represent the most toxic form of the protein (Volles and Lansbury, 2003). α-syn is removed from the cell by both macroautophagy and chaperone-mediated autophagy (CMA) (Cuervo et al., 2004; Watanabe et al., 2012), and indeed α-syn aggregates accumulate in response to the pharmacological inhibition of autophagy in mice (Klucken et al., 2012). α-syn is a well-characterised substrate of CMA due to the presence of a CMA-specific pentapeptide sequence motif. It is selectively translocated across the lysosomal membrane in a complex with the heat shock cognate protein 70 (hsc70), a process dependent on lysosomal-associated membrane protein 2A (LAMP-2A). Furthermore pathogenic forms of α-syn inhibit CMA through the blockade of receptor-mediated uptake into the lysosome (Cuervo et al., 2004). Macroautophagy is a less selective degradative pathway, which is responsible for the bulk removal of defective organelles and mis-folded cytoplasmic proteins, including α-syn, from the cell. It involves the sequestration of the cytosolic contents into double-membrane autophagosomes, which are then delivered to the lysosome to form a single-membrane autophagolysosome. The cargo is then degraded by lysosomal hydrolases. Macroautophagy dysregulation is increasingly being recognized as a pathogenic factor in neurodegeneration, including in PD (Ravikumar et al., 2010). Consistent with this, the selective suppression of autophagy, through the neuronal loss of the autophagy genes atg7 or atg5, results in a number of phenotypes in mice, including locomotor defects, accumulation of polyubiquitinated proteins and neurodegeneration (Hara et al., 2006; Komatsu et al., 2006).
The Role of GCase Deficiency in PD
Both loss-of-function and toxic gain-of-function hypothesizes have been put forward to explain the link between GBA1 mutations and PD (Kinghorn, 2011; Migdalska-Richards and Schapira, 2016). There is growing evidence supporting the role of GCase loss-of-function in PD. The majority of GBA1 mutations, including missense, nonsense and frame-shift mutations, insertions or deletions, are associated with reduced lysosomal GCase levels (Montfort et al., 2004; Sidransky et al., 2009). Moreover, milder mutations, associated with slightly diminished GCase levels, confer a much lower risk of PD than more severe mutations resulting in severe enzymatic dysfunction (Swan and Saunders-Pullman, 2013). Moreover, GCase protein levels and enzymatic activity are both decreased in the post-mortem brain tissue from patients with both idiopathic and GBA1-linked PD (Gegg et al., 2012). A number of mechanisms linking GCase loss-of-function with neurodegeneration have been demonstrated (Kinghorn, 2011; Migdalska-Richards and Schapira, 2016). Loss of GCase activity has been shown to lead to GlcCer accumulation, resulting in stabilisation of α-syn oligomers and α-syn accumulation. The subsequent increase in α-syn may inhibit GCase ER-Golgi trafficking and lysosomal GCase levels, thus creating a bidirectional pathogenic loop (Mazzulli et al., 2012).
The Emerging Role of Autophagy-lysosomal Dysfunction in GD and PD
We recently investigated the role of the autophagy-lysosomal (ALS) system in neuronopathic GD using Drosophila melanogaster (Kinghorn et al., 2016). Employing homologous recombination techniques we developed a fly model lacking GCase in the brain. GCase-deficient flies displayed reduced lifespan, age-dependent locomotor abnormalities, as well clear evidence of synaptic loss and neurodegeneration. In keeping with the hallmark lysosomal dysfunction seen in GD cells, staining with LysoTracker® revealed numerous abnormally enlarged lysosomes in the brains of flies lacking GCase. This abnormal lysosomal pathology was associated with the accumulation of GlcCer, similar to that seen in neuronopathic GD brains (Conradi et al., 1984) (Figure 1).
Figure 1.
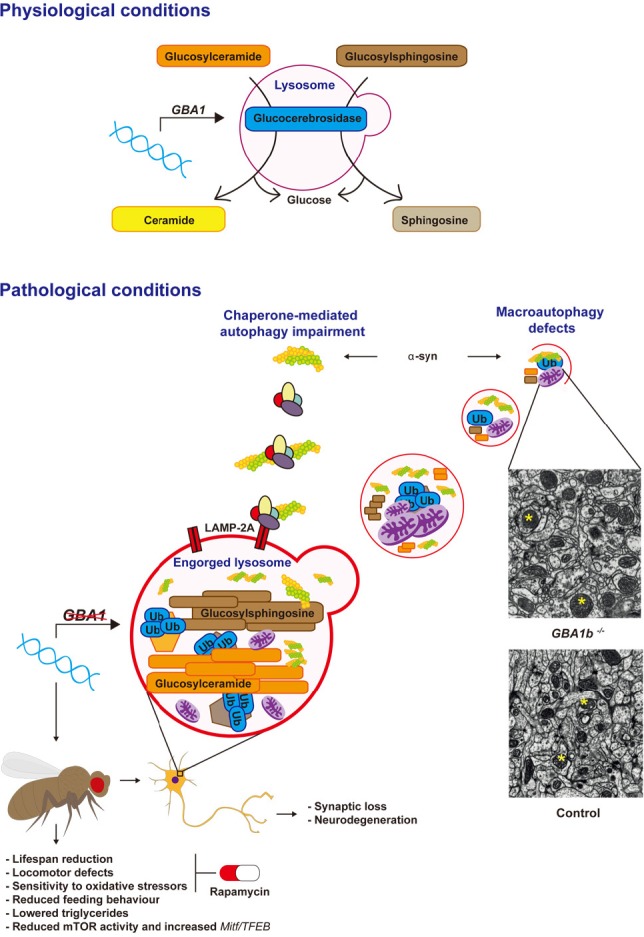
Glucocerebrosidase (GCase) deficiency results in autophagy-lysosomal system (ALS) dysfunction.
Under normal physiological conditions GCase hydrolyses glycolipid substrates (glucosylceramide and glycosylsphingosine) within the lysosome. Normal lysosomal function is required for the autophagic clearance of defective cellular organelles and mis-folded proteins. Mutations in the GBA1 gene result in GCase loss-of-function and accumulation of its substrates. This leads to lysosomal dysfunction and subsequent defects in both macroautophagy and chaperone-mediated autophagy (CMA), with the consequent accumulation of α-synuclein (α-syn). ALS dysfunction in a fly model of neuronal GCase deficiency was associated with the accumulation of abnormal giant mitochondria (representative giant mitochondria in GBA1 knockout (GBA1b−/−) and healthy mitochondria in wild type control fly brains are shown with yellow asterisks, shown under identical magnification), accumulation of autophagy substrates (p62 and polyubiquitinated proteins), in addition to neurodegeneration and synaptic loss. These neuropathological defects resulted in a number of neurotoxic phenotypes, including reduced lifespan, locomotor abnormalities and decreased resistance to oxidative stress. Likely as a compensatory response to the autophagy block, mammalian target of rapamycin (mTOR)-complex 1 (mTORC1) activity was decreased and Mitf/TFEB gene expression was up-regulated in the brains of GCase-deficient flies. The mTOR inhibitor rapamycin was able to functionally rescue the lifespan, locomotor and oxidative stress phenotypes of the GCase-deficient flies, highlighting the potential therapeutic benefits of rapamycin and other inhibitors of mTORC1 in Gaucher disease (GD) and Parkinson's disease (PD) (Kinghorn et al., 2016). LAMP-2A: Lysosomal-associated membrane protein 2.
Normal lysosomal functioning is required for the formation of autolysosomes and thus the autophagic removal of misfolded proteins and defective organelles from the cell (Ravikumar et al., 2010). During macroautophagy LC3-I is converted into its lipidated form, LC3-II, which is recruited to autophagosomal membranes (Ravikumar et al., 2010). We therefore probed macroautophagy function by measuring the levels of the fly homologue of LC3, Atg8, by western blot analysis. This demonstrated that both Atg8(LC3)-I and Atg8(LC3)-II are increased in the GCase-deficient fly brain, in addition to the ratio of Atg-II/Atg-I, suggestive of increased autophagosome number and a block in autophagy flux. This mirrors the increased LC3-II levels seen in the substantia nigra of PD patients (Dehay et al., 2010). In addition, we demonstrated increased accumulation in GCase-deficient fly brains of autophagy substrates, namely polyubiquinated proteins and Ref(2)P, the Drosophila homologue of p62. Both ubiquitin and p62 are abundant constituents of protein inclusions associated with neurodegenerative diseases, including LBs in PD (Zatloukal et al., 2002; Bartlett et al., 2011). Further interrogation of autophagy at the regulatory level in fly brains revealed a decrease in the phosphorylation of p70S6 kinase, a substrate of the energy sensing mammalian target of rapamycin (mTOR)-complex 1 (mTORC1). mTORC1 plays a central role in macroautophagy by initiating the formation and elongation of the autophagosomal membrane (Ravikumar et al., 2010). Thus the decrease in mTORC1 activity seen in GCase-deficient fly brains likely represents a compensatory response to the autophagy block and is similar to that seen in fibroblasts derived from PD patients harbouring GBA1 mutations (Magalhaes et al., 2016). In keeping with the impaired autophagy, abnormal giant mitochondria were observed in the brains of GCase-deficient flies, a reflection of the inability of the cells to clear aged and defective mitochondria by autophagy (the process of mitophagy). Interestingly GCase-deficiency in flies appeared to phenocopy Mitf-knockdown. Mitf (microphthalmia transcription factor) is the fly homologue of mammalian TFEB (transcription factor EB), the master regulator of lysosomal biogenesis. It has many conserved functions in the fly including regulation of the lysosomal-autophagy pathway and lipid metabolism (Bouche et al., 2016). GCase deficiency in the fly was also associated with Mitf gene up-regulation, likely in response to the lysosomal-autophagy block. Lastly treatment of GCase-deficient flies with the mTOR inhibitor rapamycin resulted in a significant rescue of the lifespan and locomotor phenotypes, as well as resistance to oxidative stress (Figure 1).
ALS dysfunction is increasingly being implicated in the pathogenesis of GD and PD, and has been identified in several GD and GCase-deficient PD models. Autophagy defects, in association with inflammasone activation, were observed in GD macrophages (Aflaki et al., 2014). Pharmacological inhibition of GCase in mice, through the chronic systemic treatment with conduritol-β-epoxide, lead to the accumulation of insoluble α-syn aggregates and neurodegeneration, in addition to an increase in the CMA-associated protein LAMP-2A and the macroautophagy marker LC3-II (Rocha et al., 2015b). GCase siRNA knockdown in cell culture also inhibited macroautophagy flux with a rise in LC3-II and p62, and GBA1 knockout in mouse embryonic fibroblasts lead to a compromise in CMA. Furthermore the autophagy defects were associated with loss of autophagic lysosomal reformation and maturation of endosomes. They also observed a decrease in functional acidified lysosomes within GCase-deficient cells (Magalhaes et al., 2016). Enlargement of the lysosomal compartment, as observed in GCase-deficient fly brains, was also seen in GBA1 mutant PD induced pluripotent stem cell (iPSC)-derived dopamine neurons, in association with autophagy defects and accumulation of intracellular p62 (Fernandes et al., 2016).
It has been suggested that both GD patients and GBA1 mutation carriers display a similar risk of developing PD (Schapira, 2015). Consistent with this, in vitro work has shown that the degree of autophagy inhibition and α-syn accumulation is comparable in iPSC-derived neurones from GD patients and GBA1 mutation carriers with PD (Schöndorf et al., 2014). Moreover, autophagic lysosomal reformation defects in GCase-deficient cellular models were also similar in heterozygote and homozygote mutant GBA1 models (Magalhaes et al., 2016). These observations are in keeping, at least in part, by the complex interplay between the loss and toxic gain-of-functions of GCase activity, the ageing cellular degradative machinery, including the ALS, and additional genetic risk factors predisposing to PD. Work in our GCase knockout fly model, as well as in other cellular and mouse models, has demonstrated that loss of GCase activity is sufficient to cause neurodegeneration, even in the absence of α-syn, when GCase levels are sufficiently low (Kinghorn et al 2016). As already mentioned, α-syn can further potentiate GCase deficiency through the generation of a bidirectional feedback loop, leading to lysosomal dysfunction and the generation of toxic oligomeric α-syn species (Mazzulli et al., 2012). In addition to the effects of GCase loss-of-function, the cellular sequelae of aging undoubtedly play a role. Indeed both lysosomal and autophagic function decline with age (Martinez-Vicente et al., 2005; Cuervo, 2008). During ageing GCase activity also declines in the substantia nigra and putamen of healthy controls, and is comparable to the enzymatic activity in PD patients with GBA1 mutations (Rocha et al., 2015a). This suggests that an age-dependent reduction in GCase activity may lower the threshold for developing PD, and contribute to the lysosomal dysfunction that occurs in ageing and PD. Therefore moderate reductions in GCase activity in GBA1 mutation carriers, in addition to the age-related decline in the ALS, and other cellular perturbations secondary to additional genetic and environmental risk factors, may be sufficient to promote α-syn accumulation and neurodegeneration. Further studies are required to better characterize the relationship between specific pathogenic GBA1 mutations, their corresponding GCase activity and ALS function, in addition to their dose effect and clinical PD phenotypes. Such studies will undoubtedly aid our understanding of the variable penetrance of GBA1 mutations, in both the heterozygous and homozygous state, in causing PD.
Targeting Autophagy in GD and PD using the mTOR Inhibitor Rapamycin
The beneficial effects of rapamycin, demonstrated in our GCase knockout fly model, suggest that the inhibition of mTORC1 may represent an additional therapeutic strategy in the treatment of GD and PD, as well as other neurodegenerative disorders, in which there is significant ALS dysfunction. In support of this, rapamycin increased the clearance of exogenously expressed human wild-type and mutant α-syn in an inducible cell model (Webb et al., 2003) as well as endogenous α-syn in GBA1-knockdown primary cortical neurons (Du et al., 2015). Rapamycin has also been shown to mediate neuroprotective effects in other neurodegenerative diseases, including in a TDP-43-expressing amyotrophic lateral sclerosis mouse model (Bové et al., 2011; Wang et al., 2012). However, inhibition of mTOR by rapamycin is not invariably protective in all GBA1 mutant models, and indeed rapamycin was found to trigger cell death in GD iPSC-derived neuronal cells harboring a GBA1 mutation (Awad et al., 2015). Furthermore chronic rapamycin treatment aggravated the pathology and muscle weakness in a mouse model of VCP (valosin containing protein)-associated myopathy, despite a defect in mTOR signaling (Ching and Weihl, 2013). Thus the effects of inhibiting mTORC1 activity appear to vary depending on disease pathology, the model organism, and also likely the timing of the therapy. Further studies in mammalian models are now required to verify the benefits of rapamycin, and other inhibitors of mTOR, in ameliorating the neuropathology in GD and PD. It is likely that the clinical success of pharmacological mTOR inhibitors will rely on the precise titration of mTOR activity, in order to stimulate autophagy appropriately in the face of ALS dysfunction, minimalizing unwanted side effects.
Footnotes
Conflicts of interest: None declared.
References
- Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, Marugan J, Patnaik S, Dutra A, Southall N, Zheng W, Tayebi N, Sidransky E. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci Transl Med. 2014;6:240ra73. doi: 10.1126/scitranslmed.3008659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad O, Sarkar C, Panicker LM, Miller D, Zeng X, Sgambato JA, Lipinski MM, Feldman RA. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum Mol Genet. 2015;24:5775–5788. doi: 10.1093/hmg/ddv297. [DOI] [PubMed] [Google Scholar]
- Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A, Finley KD. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy. 2011;7:572–583. doi: 10.4161/auto.7.6.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 2016;12:484–498. doi: 10.1080/15548627.2015.1134081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- Ching JK, Weihl CC. Rapamycin-induced autophagy aggravates pathology and weakness in a mouse model of VCP-associated myopathy. Autophagy. 2013;9:799–800. doi: 10.4161/auto.23958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradi NG, Sourander P, Nilsson O, Svennerholm L, Erikson A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol. 1984;65:99–109. doi: 10.1007/BF00690463. [DOI] [PubMed] [Google Scholar]
- Cox TM. Gaucher disease: clinical profile and therapeutic developments. Biologics. 2010;4:299–313. doi: 10.2147/BTT.S7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du TT, Wang L, Duan CL, Lu LL, Zhang JL, Gao G, Qiu XB, Wang XM, Yang H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy. 2015;11:1803–1820. doi: 10.1080/15548627.2015.1086055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, Bogetofte H, Lang C, Ryan BJ, Sardi SP, Badger J, Vowles J, Evetts S, Tofaris GK, Vekrellis K, Talbot K, Hu MT, James W, Cowley SA, Wade-Martins R. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson's iPSC-derived dopamine neurons. Stem Cell Rep. 2016;6:342–356. doi: 10.1016/j.stemcr.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol. 2012;72:455–463. doi: 10.1002/ana.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Kinghorn KJ. Pathological looping in the synucleinopathies: investigating the link between Parkinson's disease and Gaucher disease. Dis Model Mech. 2011;4:713–715. doi: 10.1242/dmm.008615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn KJ, Gronke S, Castillo-Quan JI, Woodling NS, Li L, Sirka E, Gegg M, Mills K, Hardy J, Bjedov I, Partridge L. A drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered mtor signalling and is functionally rescued by rapamycin. J Neurosci. 2016;36:11654–11670. doi: 10.1523/JNEUROSCI.4527-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, Schlötzer-Schrehardt U, Hyman BT, McLean PJ, Masliah E, Winkler J. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy. 2012;8:754–766. doi: 10.4161/auto.19371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 2016;25:3432–3445. doi: 10.1093/hmg/ddw185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40:622–633. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher's disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2012;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migdalska-Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem Suppl. 2016;1:77–90. doi: 10.1111/jnc.13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills K, Eaton S, Ledger V, Young E, Winchester B. The synthesis of internal standards for the quantitative determination of sphingolipids by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:1739–1748. doi: 10.1002/rcm.1977. [DOI] [PubMed] [Google Scholar]
- Montfort M, Chabás A, Vilageliu L, Grinberg D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum Mutat. 2004;23:567–575. doi: 10.1002/humu.20043. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-arencibia M, Green-thompson ZW, Jimenez-sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DCO, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, Isacson O. Progressive decline of glucocerebrosidase in aging and Parkinson's disease. Ann Clin Transl Neurol. 2015a;2:433–438. doi: 10.1002/acn3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Graham A-R, Brown E, McLean JR, Hayes MA, Beagan J, Izen SC, Perez-Torres E, Hallett PJ, Isacson O. Sustained systemic glucocerebrosidase inhibition induces brain α-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid Redox Signal. 2015b;23:550–564. doi: 10.1089/ars.2015.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. Glucocerebrosidase and Parkinson disease: Recent advances. Mol Cell Neurosci. 2015;66:37–42. doi: 10.1016/j.mcn.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, Hedrich U, Berg D, Shihabuddin LS, Hu J, Pruszak J, Gygi SP, Sonnino S, Gasser T, Deleidi M. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014;5:4028. doi: 10.1038/ncomms5028. [DOI] [PubMed] [Google Scholar]
- Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, et al. Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan M, Saunders-Pullman R. The association between beta-glucocerebrosidase mutations and parkinsonism. Curr Neurol Neurosci Rep. 2013:13. doi: 10.1007/s11910-013-0368-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CKJ. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109:15024–15029. doi: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, Nakagawa M, Tanaka M. p62/SQSTM1-dependent autophagy of Lewy body-like α-synuclein inclusions. PLoS One. 2012;7:e52868. doi: 10.1371/journal.pone.0052868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]