


Abstract
S4 is an automatically generated database of multiple structure-based sequence alignments of protein superfamilies in the SCOP database. All structural domains that do not share more than 40% sequence identity as defined by the ASTRAL compendium of protein structures are included. The alignments are constructed using pairwise structural alignments to generate residue equivalences that are then integrated into multiple alignments using sequence alignment tools. We describe the database and give examples showing how the automatically generated S4 alignments compare favourably to hand-crafted alignments. Available at: http://compbio.mds.qmw.ac.uk/S4.html.
INTRODUCTION
The comparison of sequences of related proteins, which are diverse at the sequence level, can reveal features that are important for both structure and function. When aligning distantly related proteins, the availability of structural information is particularly valuable.
The Structural Classification Of Proteins (SCOP) database (1,2) groups together protein structural domains in a hierarchical manner according to class, fold, superfamily and family. Importantly, relationships between proteins grouped at the superfamily level will often not be apparent from the consideration of sequence alone. The ASTRAL compendium (3) provides subsets of SCOP clustered at various levels of sequence identity and is useful for selecting domains that are diverse at the sequence level. S4 (structure-based sequence alignments of SCOP superfamilies) provides multiple structure-based alignments of SCOP (version 1.63) protein superfamilies where no two domains share more than 40% sequence identity as defined by ASTRAL. Although some superfamilies are highly populated many have only one domain (only the four main SCOP classes are considered in this paper, as they are the classes for which superfamily relationships can be considered meaningful). Despite this there are 456 superfamilies with more than one domain for which the database provides alignments.
Structural alignment and analysis of protein superfamilies have been carried out by Blundell and co-workers (4,5). CAMPASS (5) is a database of structurally aligned protein superfamilies, available via the Web, where no two proteins share a sequence identity more than 25%. The HOMSTRAD database (4) is a valuable resource containing aligned three-dimensional structures of homologous proteins. However, it is sometimes difficult to structurally align all the sequences in a family and HOMSTRAD may choose to split a group of proteins into two separate families, since the focus is to correctly align functionally and structurally important residues. A similar database to ours is PASS2 (6,7), which is generated using COMPARER (8) from initial equivalences generated by STAMP (9) or MALIGN (10).
Assessing the accuracy of alignments generated by automated procedures is very difficult due to a lack of hand-crafted alignments of sequence diverse proteins. Here, we show that our alignments are very similar to a handful of alignments produced manually by domain experts as evidence of the quality of the alignments. We also show that the alignments correctly equivalence known sequence signatures corresponding to a functional binding site.
PROTOCOL FOR CREATING ALIGNMENTS
The multiple structure-based alignments are constructed using the program SAP (11) for carrying out pairwise structural alignments and T-COFFEE (12) to perform a progressive hierarchical alignment using the information from the pairwise SAP scores. The T-COFFEE algorithm has the advantage that it allows information from all sequences to be considered at each alignment step in an hierarchical alignment procedure. The T-COFFEE method has been used (13) for constructing alignments for functional families as part of the DALI Domain Dictionary. The method has also been shown to be successfully used for a mixture of sequence and structural information (14,15). The protocol we have adopted is listed in detail:
- For each pair of domains a, b in a superfamily s:
- Use SAP to generate a structural superposition of the domains, which generates a list of the three-dimensional co-ordinates x of the Cα atoms for each domain in the superposition, xia and xjb, where i and j run over the lengths of the two domains; a list of pairs of residues indices {i,j}l, l = 1, …, N, where the algorithm has equivalenced N residues; and the overall RMSD of the alignment RMSDab.
-
Create a T-COFFEE library Lab for the superposition of a and b. The library has N entries, each entry specifies the equivalenced residues {i,j}l and the weight, wij that the T-COFFEE algorithm should give to the equivalencing of i and j. We calculate wij using the following formula:
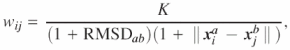 where K is constant.
where K is constant.
Now generate a multiple sequence alignment of superfamily s by running T-COFFEE with all libraries Lab with a ∈ s, b ∈ s and a ≠ b.
We have experimented with various formulas for the weighting function for calculating w and found this method to give high-quality results. The motivation behind the formula is that the certainty that the two residues are equivalent should be weighted by not only the global similarity of the two domains, but also how well the particular residues superpose. We use a value of K = 1000 to generate the alignments.
QUALITY OF ALIGNMENTS
In the absence of hand-crafted multiple alignments constructed by domain experts, it is difficult to access the accuracy of multiple structure-based alignments. We have examined the alignments created by Hill et al. (16). In their paper, three families are aligned: the long-chain four-helical cytokines, short-chain four-helical cytokines and the four-helical cytochromes. In aligning these families, Hill et al. examined not only RMSDs, but also hydrogen bonding, accessible surface areas and inter-residue contacts in producing pairwise alignments. A multiple structural alignment was then produced by merging the pairwise alignments. Sequence equivalences could then be generated from the structural alignment. The common core was defined as anything with an RMSD <3 Å. We use the alignments from this paper as reference alignments.
The four-helical cytokines have a unique up-up-down-down topology so far only observed in these cytokines. They represent a difficult family for structural alignment, since they are structurally diverse showing an average RMSD between members of the superfamily of 3.12 Å (from the SAP alignments). The SCOP superfamily has three families: long chain, short chain and interferon/interleukin 10 family. Hill et al. (16) provide alignments of the first two families and their common core. We selected the proteins aligned in both our alignments and the reference alignments, and marked on the common cores that should be aligned. The results of this can be seen in Figure 1. The figure shows that our protocol has mainly aligned the cores of these proteins. The major error is the alignment of 1scf, which fails to align to any core region. There are some more minor errors: part of the helix 3 of 1bgc has been misaligned, helix 2 of 1hul is also misaligned and most of the parts of the common core that are gaps are not correctly gapped. Despite this the alignment appears to be of high quality.
Figure 1.

Part of the multiple alignment of the four-helical cytokines showing those domains also aligned (16). The grey shading shows the cores of the families: light grey shows the long-chain core, medium grey the short-chain core and dark grey the common core. This dark grey shading should be aligned in four clear blocks. The boxes show parts of the core that are marked as gaps in the reference alignment, this makes visualization easier as all corresponding blocks are of the same length across each sequence. The figure was produced using ALSCRIPT (19).
A similar analysis was performed for the four-helical cytochromes. We found that the four-helical cytochromes were aligned in a way that the core agreed completely with the reference alignment. These data are summarized in Table 1, which shows alignment accuracies compared between PASS2 (database version August 20, 2003) and S4. What is evident is that S4 alignments are closer to the reference alignments than PASS2 in the three cases studied.
Table 1. Comparison of accuracies for PASS2 and S4 on the reference alignments.
| PASS2 | S4 | |||
|---|---|---|---|---|
| Alignment | ACa | ACw | ACa | ACw |
| Long-chain cytokines | 0.23 | 0 | 0.95 | 0.86 |
| Short-chain cytokines | 0.02 | 0 | 0.38 | 0 |
| All cytokines | 0.15 | 0 | 0.79 | 0 |
| Four-helical cytochromes | 0.95 | 0.9 | 1 | 1 |
Accuracy measures are as described previously (18): ACw is the accuracy of the whole alignment, i.e. number of correct positions divided by the length of the alignment; ACa is the average alignment accuracy over all possible pairs of sequences in the alignment. The score is only calculated over regions marked as core in the reference alignments. Note, ACw is quite ‘brittle’ decaying quickly since, for a position to be correct, all sequences must be aligned correctly in that position.
It is also important that an alignment correctly aligns the functionally important residues in a superfamily. For example, the cytochrome c superfamily covalently binds haem. The active residues for this binding form a CxxCH signature. Inspection of the S4 alignment shows that this signature is aligned across every member of the superfamily, in contrast to the PASS2 alignment that fails to align this signature.
We are aware, however, that different protocols may perform better with certain types of superfamilies and our study only covers three reference alignments. We would envisage these different resources to be complementary.
THE S4 DATABASE
All the alignments in the S4 database can be downloaded as both clustal and fasta files, or the database can be browsed through our website. The website provides additional markup to the downloadable alignments: the alignments can be displayed with either sequence or structure annotation. The sequence annotation uses Mview (17) to colour residues according to their physiochemical properties, allowing sequence conservation patterns to be seen. The structural annotation shows the alignments with helix and strand positions colour-coded. The consensus group and median distance between equivalenced residues are also shown at each position (for all residues equivalenced in a column of the multiple alignment, the distance between residues in all the pairwise alignments are calculated, and the median of the distances calculated; the median is taken to lessen the effect of outliers from any poor pairwise structural alignments). Links to the current SCOP location are provided from each alignment. There is also a search facility into which a user can specify a given SCOP superfamily, SCOP domain name or PDB name.
SUMMARY
Do we really need another structural alignment database? Our survey of existing resources revealed that they either only covered a subset of the available proteins in a superfamily that were structurally conserved or showed significant differences to reference alignments. The use of a handful of reference alignments should not be considered a comprehensive assessment of the accuracy of the protocol we have used. In particular, our reference alignments are from the all alpha class—testing against alignments from other classes would be informative. Nevertheless, we are encouraged by the cases we have looked at.
We envisage the alignments contained in S4 will complement similar resources (4,5,7). The analysis of such alignments provides insight into protein sequence structure relationships. We have experimented with using profiles built from our structure-based alignments in profile–profile searching methods for detecting remote homologues and this approach shows some promise. The alignments may be useful for protein modelling as an aid in aligning the sequence to be modelled with a collection of diverse templates.
The database generation procedure is automated and we therefore anticipate updating the database on a periodic basis to reflect newer releases of the SCOP database.
REFERENCES
- 1.Lo Conte L., Ailey,B., Hubbard,T.J., Brenner,S.E., Murzin,A.G. and Chothia,C. (2000) SCOP: a structural classification of proteins database. Nucleic Acids Res., 28, 257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 3.Brenner S.E., Koehl,P. and Levitt,M. (2000) The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res., 28, 254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizuguchi K., Deane,C.M., Blundell,T.L. and Overington,J.P. (1998) HOMSTRAD: a database of protein structure alignments for homologous families. Protein Sci., 7, 2469–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sowdhamini R., Burke,D.F., Huang,J.F., Mizuguchi,K., Nagarajaram,H.A., Srinivasan,N., Steward,R.E. and Blundell,T.L. (1998) CAMPASS: a database of structurally aligned protein superfamilies. Structure, 6, 1087–1094. [DOI] [PubMed] [Google Scholar]
- 6.Bhaduri A., Pugalenthi,G. and Sowdhamini,R. (2004) PASS2: an automated database of protein alignments organised as structural superfamilies. BMC Bioinformatics, 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallika V., Bhaduri,A. and Sowdhamini,R. (2002) PASS2: a semi-automated database of protein alignments organised as structural superfamilies. Nucleic Acids Res., 30, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sali A. and Blundell,T.L. (1990) Definition of general topological equivalence in protein structures. A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming. J. Mol. Biol., 212, 403–428. [DOI] [PubMed] [Google Scholar]
- 9.Russell R.B. and Barton,G.J. (1992) Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins, 14, 309–323. [DOI] [PubMed] [Google Scholar]
- 10.Johnson M.S., Overington,J.P. and Blundell,T.L. (1993) Alignment and searching for common protein folds using a data bank of structural templates. J. Mol. Biol., 231, 735–752. [DOI] [PubMed] [Google Scholar]
- 11.Taylor W.R. (2000) Protein structure comparison using SAP. Methods Mol. Biol., 143, 19–32. [DOI] [PubMed] [Google Scholar]
- 12.Notredame C., Higgins,D.G. and Heringa,J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217. [DOI] [PubMed] [Google Scholar]
- 13.Dietmann S., Park,J., Notredame,C., Heger,A., Lappe,M. and Holm,L. (2001) A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3. Nucleic Acids Res., 29, 55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Sullivan O., Suhre,K., Abergel,C., Higgins,D.G. and Notredame,C. (2004) 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol., 340, 385–395. [DOI] [PubMed] [Google Scholar]
- 15.Poirot O., Suhre,K., Abergel,C., O'Toole,E. and Notredame,C. (2004) 3DCoffee@igs: a web server for combining sequences and structures into a multiple sequence alignment. Nucleic Acids Res., 32, W37–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill E.E., Morea,V. and Chothia,C. (2002) Sequence conservation in families whose members have little or no sequence similarity: the four-helical cytokines and cytochromes. J. Mol. Biol., 322, 205–233. [DOI] [PubMed] [Google Scholar]
- 17.Brown N.P., Leroy,C. and Sander,C. (1998) MView: a web-compatible database search or multiple alignment viewer. Bioinformatics, 14, 380–381. [DOI] [PubMed] [Google Scholar]
- 18.Raghava G.P., Searle,S.M., Audley,P.C., Barber,J.D. and Barton,G.J. (2003) OXBench: a benchmark for evaluation of protein multiple sequence alignment accuracy. BMC Bioinformatics, 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton G.J. (1993) ALSCRIPT—a tool to format multiple sequence alignments. Prot. Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]