


Abstract
The Human Anatomic Gene Expression Library (H-ANGEL) is a resource for information concerning the anatomical distribution and expression of human gene transcripts. The tool contains protein expression data from multiple platforms that has been associated with both manually annotated full-length cDNAs from H-InvDB and RefSeq sequences. Of the H-Inv predicted genes, 18 897 have associated expression data generated by at least one platform. H-ANGEL utilizes categorized mRNA expression data from both publicly available and proprietary sources. It incorporates data generated by three types of methods from seven different platforms. The data are provided to the user in the form of a web-based viewer with numerous query options. H-ANGEL is updated with each new release of cDNA and genome sequence build. In future editions, we will incorporate the capability for expression data updates from existing and new platforms. H-ANGEL is accessible at http://www.jbirc.aist.go.jp/hinv/h-angel/.
INTRODUCTION
Genome-scale analyses of gene expression have grown exponentially in the last few years, providing clues to the function of genes and genomes and helping our understanding of the molecular basis of health and disease. A growing number of technological platforms are available for conducting these studies, including solid-support approaches, such as oligonucleotide (1) and cDNA arrays (2,3), PCR-based high-throughput expression profiling methods such as introduced amplified fragment length polymorphism (iAFLP) (4) and random tag identification, such as the serial analysis of gene expression (SAGE) (5) or massively parallel signature sequencing (MPSS) (6). The possibility of integrating all the data already produced has the potential to provide unique insight into understanding the expression pattern of a whole genome. In order to assess the compatibility of data, several research groups have compared results from distinct types of high-throughput expression technologies (7,8).
Comparison has been done at the gene level and some groups have reported achieving good correlations between results produced by different techniques (9). Nonetheless, others have reported significant discrepancies between the output of certain techniques (10,11). However, no group has yet tried to integrate expression data at the resolution of transcript variation with the intention to resolve discrepancies in gene level comparison or across multiple platforms. The Human Anatomic Gene Expression Library (H-ANGEL) was developed for the first international annotation jamboree of the human transcriptome, entitled the Human Full-length cDNA Annotation Invitational (H-Invitational) (12). During the jamboree, the tool was used to present expression data from different methods and platforms in a manner that aided the manual annotation of predicted loci. We combined publicly available expressed sequence tag (EST), SAGE and microarray data with proprietary gene expression data that was generated and analyzed by members of the H-Invitational consortium.
H-ANGEL is the first step towards a global analysis (meta-analysis) of gene expression data, providing an overview of consistencies and discrepancies between expression data generated by different platforms. It is hoped that this display will help us to appreciate the fortes and caveats of the different technologies available, so that in future studies, the maximum amount of beneficial information can be derived from the appropriate use of each method.
DATABASE CONTENTS
Data resources
One of the distinctive features of H-ANGEL is that it contains a substantial amount of disparate and unique data brought together for the jamboree. A large proportion of publicly available data has been created to answer specific questions. For this reason, the experimental information associated with the data can vary in quality and is often brief in content or limited in utility because of intellectual property issues (13). Nevertheless, we found that the benefits of including such data outweighed the caveats inherent in performing an analysis involving this kind of proprietary data. Gene expression data were collected as follows: (i) iAFLP profiling data were generated as described previously (4) using 22 987 primers corresponding to 14 431 independent UniGene clusters for competitive RT–PCR using mRNA from 71 tissue samples. (ii) Long oligomer microarray (Oligoarray) data were generated by dual-color competitive hybridization. Under this process commercially available pools of human tissue RNAs were hybridized against custom-made oligomers of between 50 and 60 nt in length. (iii) MPSS data for human mRNAs were generated by the Lynx Corporation (6) for the National Institute of Genetics. The tag-to-gene mapping was also provided based on the position and direction of the tags by the manufacturer (14) but it may require further qualification. (iv) For cDNA array (custom-made cDNAarray), total and poly(A)+ RNAs were purchased from Clontech (Palo Alto, CA) and Stratagene (La Jolla, CA). Probes were prepared using a direct labeling protocol with a reference design experiment (e.g. each sample versus a universal reference design), and double color hybridizations on human cDNA glass slides Dye Swap was performed (15,16).
Data already in the public domain were processed as follows. In UniGene Release 157 (17), the number of EST clones from libraries representing normal adult tissues without normalization or subtraction steps amounted to 745 446 in total. They were combined with 91 509 tag sequences from BodyMap (18) which represented 53 normal adult tissues libraries. The counts of cognate clones were based on UniGene and BodyMap. SAGE tags were selected from GEO (http://www.ncbi.nlm.nih.gov/geo/) and processed in a manner similar to the ESTs. Tag-to-gene correspondence was achieved through the determination of virtual tags for all transcripts in our dataset, in a similar strategy as the one used for SAGE Genie (19). GeneChip data were obtained from the HuGEIndex site (http://zlab.bu.edu/HugeIndex/index.htm) and the Normal Tissue Database site (http://www2.genome.rcast.u-tokyo.ac.jp/tp/).
Data processing
In order to allow users to compare the results from different platforms in an intuitive way, hundreds of mRNA sources used in the original analysis were manually categorized into 40 practical tissue types, based almost entirely on existing tissue classes used by commercial manufacturers of mRNAs. A table cross-referencing the tissue category originally assigned for each dataset by its provider and the corresponding tissue category manually allocated by H-Invitational consortium members can be viewed at http://www.jbirc.aist.go.jp/hinv/h-angel/title/tissue_html_list.html.
Tags were counted according to the 40 tissue categories and counts were normalized by calculating the total tag counts from each of the 40 tissues. All those values representing relative expression levels across all tissues, and other relative expression values from arrays and iAFLP, were normalized to make the sum total of expression across 40 tissues equal to 1. In studies where expression was not measured in all 40 tissues, the sum of normalized values was given by the total number of tissues under test divided by 40. For example, when only 20 tissues were tested by one platform, the sum of normalized values was set to 0.5.
The Spartan distribution of expression data among some of the 40 tissue often makes direct comparison between tissues and across multiple platforms difficult. Owing to the inherent difficulties associated with direct comparison across the 40 tissue categories, we decided to create 10 supra-categories—groups representing related tissues (Figure S1). When amalgamating the expression data from 40 tissue categories to 10, the average normalized value was given for each category. For tag frequency data, tags were counted again for each category and normalized by sum for each category.
Linking to the full-length cDNA assembly in H-InvDB
Using accession number IDs, we were able to cross-refer the clones from the H-InvDB predicted loci with their counterparts from UniGene. If the corresponding UniGene clone had any SAGE or EST expression data linked to it, these data were then associated with the matching H-Inv clone. This association procedure was repeated for RefSeq sequences which were members of predicted loci. The number of loci that could be associated with SAGE and EST data in this way is shown in Table 1. A total of 18 897 H-Inv loci have associated expression data from at least a single platform. The number increases to 24 520 if we take into account those loci in which all the members are RefSeq sequences. Sequences of SAGE tags, Oligo arrays, ESTs and iAFLP primers were mapped onto individual full-length cDNAs collected for the jamboree and all possible relationships between expression patterns and full-length cDNA clones were described.
Table 1. Summary of expression data sources.
| Methods | Platforms | Technologies | Institutes | No. of H-Inv loci |
|---|---|---|---|---|
| PCR-based quantitative expression profiling | iAFLP | Introduced Amplified Fragment Length Polymorphism | JBIRC (Kousaku Okubo) | 11 827 |
| Osaka University (Kousaku Okubo) | 8360 | |||
| DNA arrays | Long oligomers | 80 nt length oligomer chip | JBIRC (Shinya Watanabe) | 12 852 |
| Short oligomers | Affymetrix GeneChip™ | Boston University (HugeIndex) | 3971 | |
| Tokyo University (LSBM) | 13 201 | |||
| cDNA array | cDNA nylon macroarrays and cDNA glass microarray | CNRS (Charles Auffray) | 7891 | |
| cDNA sequence tags | SAGE | Serial Analysis of Gene Expression | Ludwig Institute for Cancer Research | 17 827 |
| EST + BodyMap | Expressed Sequence Tags | NCBI | 19 515 | |
| 3′-directed cDNA library | BodyMap | |||
| MPSS | Massively Parallel Signature Sequencing | NIG (Kousaku Okubo) | 11 442 |
QUERYING THE DATABASE—THE FUNCTIONALITY OF H-ANGEL
We have developed a web interface to provide easy access to the data stored in the H-ANGEL database. The H-ANGEL home page provides access to two separate web interfaces. These are the ‘H-Inv Locus Search for Gene Expression’ and the ‘Expression Pattern search’. Using the ‘H-Inv Locus Search for Gene Expression’, the user can search all the expression data available in the database for a particular gene or a gene list using several identifiers, such as H-Inv Cluster ID (HIX), RefSeq/FLcDNA accession numbers from DDBJ/GenBank/EMBL International Nucleotide Sequence Database (INSD), UniGene IDs, LocusLink IDs, definition keywords or gene product name.
After the search has been performed, the resulting web page consists of the following three sections:
Display H-Inv Cluster ID Box. This section shows all the H-ANGEL entries (Figure S2A) corresponding to the submitted query. The users access expression data from a specific locus by selecting the corresponding HIX number and clicking the ‘Display’ button.
-
Expression Pattern View. This section is the main view of H-ANGEL that displays an overview of all the expression data stored in H-ANGEL according to classified tissue categories (Figure S2B). All the H-ANGEL expression data related to each HIX number is listed along with the type of platform used for the analysis and the cDNA clone which is most likely to correspond to a given piece of expression data. Additionally, for iAFLP, SAGE and MPSS data, users can see the position of all tags or probes in relation to the locus or cDNA along with the corresponding exon–intron structure.
For SAGE data, we display the location of any internal adenosine stretches to make the user aware of possible internal priming sites. For ESTs, the frequencies of exon coverage is shown. Gene expression patterns are displayed for both the 10 and 40 tissue category groups using a histogram. For each bar on the histogram, the user can see the tissue expression level as a percentile value by moving the mouse over the histogram bar.
Expression Information in Text. This section shows publicly available information related to the clones on the locus in text format. It also shows up only when a single H-Inv locus entry is selected to be displayed. In the ‘iAFLP information Box’, conditions of gene expression measured by the iAFLP experiment for each tissue for clones on the locus are reported. In the ‘UniGene information Box’, tissues in which clone(s) from the UniGene cluster corresponding to the locus are reported (Figure S2B). Via the ‘Expression Pattern Search View’ interface, the user can retrieve H-ANGEL entries using a similarity search based on expression patterns among distinct tissue categories (Figure S2C). Users can set an arbitrary expression pattern across 10 tissue categories as a query and choose platform(s) of interest to retrieve gene clusters with a similar expression pattern. The pairwise correlation between a query pattern and each entry in H-ANGEL is estimated using the cosine coefficient and Pearson's correlation coefficient (Supplementary Method). The retrieved expression patterns returned by the search will be more similar to the query pattern if a high correlation coefficient value is set.
EXAMPLES OF CONSISTENCY AND DISCREPANCY BETWEEN PLATFORMS
In some cases, alignment of multi-platform expression patterns, individually mapped onto distinct spliced forms, allows users to deduce the expression patterns for each spliced form. Figure 1A shows an H-ANGEL representation of the locus of a potassium channel, subfamily K, member 4 (KCNK4).
Figure 1.
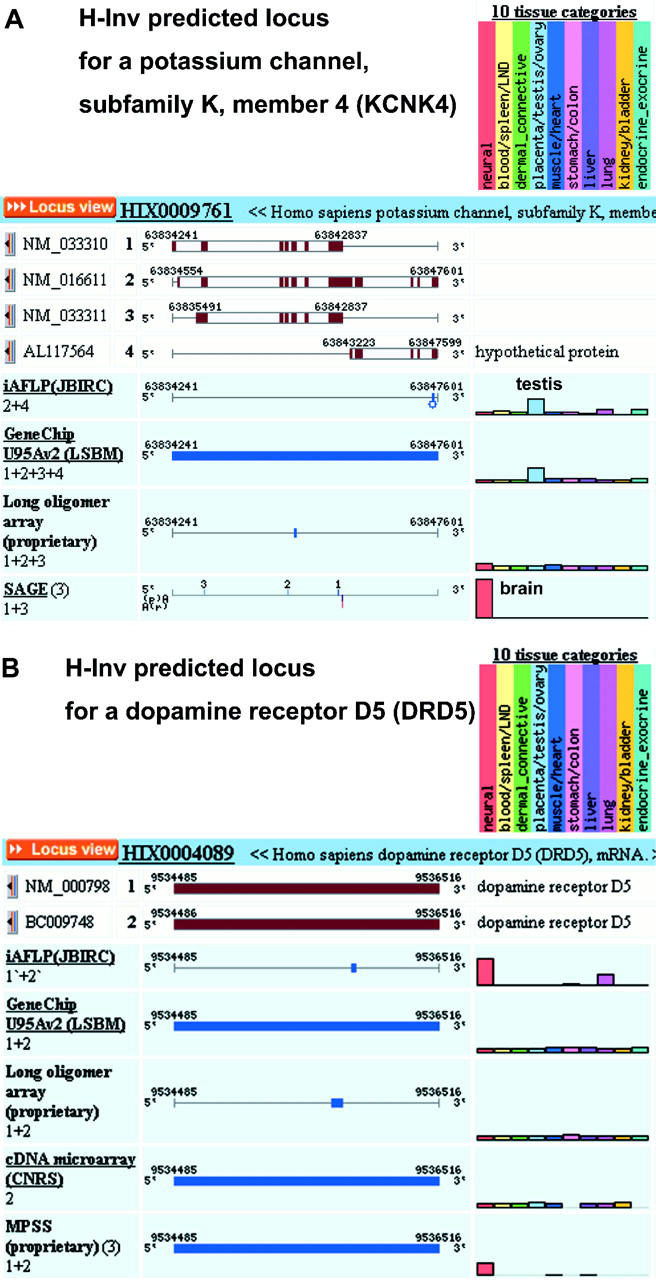
Examples of gene expression pattern results from H-ANGEL. (A) The upper panel shows the expression patterns of potassium channel, subfamily K, member 4 (KCNK4). In this locus, there are four clones, which are denoted by 1, 2, 3 and 4 for NM_033310, NM_016611, NM_033311 and AL117564, respectively. The sum of the expression levels of clones 1 and 3 was measured by SAGE and clones 2 and 4, which are mapped onto the 3′ end region of the locus, were done by iAFLP, respectively. The expression patterns of those transcripts are shown in a histogram in the right bottom sub-panel. (B) The lower panel shows the expression patterns of dopamine receptor D5 (DRD5). NM_000798 and BC009748 are denoted by 1 and 2, respectively. For more details, see those predicted H-Inv loci, HIX0009761 and HIX0004089, through the H-ANGEL website (http://www.jbirc.aist.go.jp/hinv/h-angel/).
In this predicted locus, two transcript variants are known among the three clustered RefSeq sequences (see NCBI LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=50801). Marked discrepancies among expression patterns for this locus suggest that the transcript 2 and 4 are mainly expressed in the testis and transcripts 1 and 3 are expressed mainly in the brain. This observation is generally consistent with the literature (20).
In Figure 1B, a disagreement in the expression patterns of the dopamine receptor D5 (DRD5) between platforms can be clearly observed. The three microarray-based methods report a low-uniform distribution with no high levels of expression in any one tissue. However, the two PCR-based techniques predict that DRD5 is more highly expressed in neural tissues than other tissue types. As DRD5 is a well-studied protein, we know from repeated northern blot analyses that the PCR-based results are in accord with those observed previously (21). This result can be due to the greater sensitivity in many cases of PCR techniques over microarray-based techniques (22).
CONCLUSION AND FUTURE DEVELOPMENTS
Approximately 90% of H-Inv loci could be assigned expression data from at least one platform. In the majority of the predicted loci, some extent of discrepancy across platforms was noted. However, as shown in the examples, careful inspection using the viewer suggested that many of the discrepancies probably did not only represent simple errors in some measurements but that we may be seeing the effects of intrinsic factors associated with measuring expression using particular techniques coming into play. For example, there is growing concern among users of microarray technologies regarding disagreements between measurements due to alternative splicing (23–25), since several lines of evidence indicate that a large portion of our genes (40–60%) have alternatively spliced forms (26–28). Currently, even when adequate data are available to make an appropriate assessment an ‘informed decision’ made by a human is still required in order to confirm the most likely and logical expression pattern for an individual transcript.
The next step in the evolution of H-ANGEL will be to automate the process of these informed decisions so that H-ANGEL will be able to systematically deduce the most likely expression patterns for each transcript from conflicting expression data.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Toshiya Yamamoto for developing the H-ANGEL database and interface. This research is financially supported by the Ministry of Economy, Trade and Industry of Japan (METI), the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), and the Japan Biological Informatics Consortium (JBIC).
REFERENCES
- 1.Lockhart D.J., Dong,H., Byrne,M.C., Follettie,M.T., Gallo,M.V., Chee,M.S., Mittmann,M., Wang,C., Kobayashi,M., Horton,H. et al. (1996) Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol., 14, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 2.Lashkari D.A., DeRisi,J.L., McCusker,J.H., Namath,A.F., Gentile,C., Hwang,S.Y., Brown,P.O. and Davis,R.W. (1997) Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl Acad. Sci. USA, 94, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science, 270, 467–470. [DOI] [PubMed] [Google Scholar]
- 4.Kawamoto S., Ohnishi,T., Kita,H., Chisaka,O. and Okubo,K. (1999) Expression profiling by iAFLP: a PCR-based method for genome-wide gene expression profiling. Genome Res., 9, 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Serial analysis of gene expression. Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 6.Brenner S., Johnson,M., Bridgham,J., Golda,G., Lloyd,D.H., Johnson,D., Luo,S., McCurdy,S., Foy,M., Ewan,M. et al. (2000) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol., 18, 630–634. [DOI] [PubMed] [Google Scholar]
- 7.Kim H.L. (2003) Comparison of oligonucleotide-microarray and serial analysis of gene expression (SAGE) in transcript profiling analysis of megakaryocytes derived from CD34+ cells. Exp. Mol. Med., 35, 460–466. [DOI] [PubMed] [Google Scholar]
- 8.Ishii M., Hashimoto,S., Tsutsumi,S., Wada,Y., Matsushima,K., Kodama,T. and Aburatani,H. (2000) Direct comparison of GeneChip and SAGE on the quantitative accuracy in transcript profiling analysis. Genomics, 68, 136–143. [DOI] [PubMed] [Google Scholar]
- 9.Rhodes D.R., Barrette,T.R., Rubin,M.A., Ghosh,D. and Chinnaiyan,A.M. (2002) Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res., 62, 4427–4433. [PubMed] [Google Scholar]
- 10.Kothapalli R., Yoder,S.J., Mane,S. and Loughran,T.P.,Jr (2002) Microarray results: how accurate are they? BMC Bioinformatics, 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Pankratz,M. and Johnson,J.A. (2002) Differential gene expression patterns revealed by oligonucleotide versus long cDNA arrays. Toxicol. Sci., 69, 383–390. [DOI] [PubMed] [Google Scholar]
- 12.Imanishi T., Itoh,T., Suzuki,Y., O'Donovan,C., Fukuchi,S., Koyanagi,K.O., Barrero,R.A., Tamura,T., Yamaguchi-Kabata,Y., Tanino,M. et al. (2004) Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol., 2, 856–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Killion P.J., Sherlock,G. and Iyer,V.R. (2003) The Longhorn Array Database (LAD): an open-source, MIAME compliant implementation of the Stanford Microarray Database (SMD). BMC Bioinformatics, 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Audic S. and Claverie,J.M. (1997) The significance of digital gene expression profiles. Genome Res., 7, 986–995. [DOI] [PubMed] [Google Scholar]
- 15.Pietu G., Eveno,E., Soury-Segurens,B., Fayein,N.A., Mariage-Samson,R., Matingou,C., Leroy,E., Dechesne,C., Krieger,S., Ansorge,W. et al. (1999) The genexpress IMAGE knowledge base of the human muscle transcriptome: a resource of structural, functional, and positional candidate genes for muscle physiology and pathologies. Genome Res., 9, 1313–1320. [PMC free article] [PubMed] [Google Scholar]
- 16.Pietu G., Mariage-Samson,R., Fayein,N.A., Matingou,C., Eveno,E., Houlgatte,R., Decraene,C., Vandenbrouck,Y., Tahi,F., Devignes,M.D. et al. (1999) The Genexpress IMAGE knowledge base of the human brain transcriptome: a prototype integrated resource for functional and computational genomics. Genome Res., 9, 195–209. [PMC free article] [PubMed] [Google Scholar]
- 17.Wheeler D.L., Church,D.M., Federhen,S., Lash,A.E., Madden,T.L., Pontius,J.U., Schuler,G.D., Schriml,L.M., Sequeira,E., Tatusova,T.A. et al. (2003) Database resources of the National Center for Biotechnology. Nucleic Acids Res., 31, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hishiki T., Kawamoto,S., Morishita,S. and Okubo,K. (2000) BodyMap: a human and mouse gene expression database. Nucleic Acids Res., 28, 136–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boon K., Osorio,E.C., Greenhut,S.F., Schaefer,C.F., Shoemaker,J., Polyak,K., Morin,P.J., Buetow,K.H., Strausberg,R.L., De Souza,S.J. et al. (2002) An anatomy of normal and malignant gene expression. Proc. Natl Acad. Sci. USA, 99, 11287–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesage F., Maingret,F. and Lazdunski,M. (2000) Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K+ channel. FEBS Lett., 471, 137–140. [DOI] [PubMed] [Google Scholar]
- 21.Tiberi M., Jarvie,K.R., Silvia,C., Falardeau,P., Gingrich,J.A., Godinot,N., Bertrand,L., Yang-Feng,T.L., Fremeau,R.T.,Jr and Caron,M.G. (1991) Cloning, molecular characterization, and chromosomal assignment of a gene encoding a second D1 dopamine receptor subtype: differential expression pattern in rat brain compared with the D1A receptor. Proc. Natl Acad. Sci. USA, 88, 7491–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taniguchi M., Miura,K., Iwao,H. and Yamanaka,S. (2001) Quantitative assessment of DNA microarrays—comparison with Northern blot analyses. Genomics, 71, 34–39. [DOI] [PubMed] [Google Scholar]
- 23.Kapranov P., Cawley,S.E., Drenkow,J., Bekiranov,S., Strausberg,R.L., Fodor,S.P. and Gingeras,T.R. (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science, 296, 916–919. [DOI] [PubMed] [Google Scholar]
- 24.Kampa D., Cheng,J., Kapranov,P., Yamanaka,M., Brubaker,S., Cawley,S., Drenkow,J., Piccolboni,A., Bekiranov,S., Helt,G. et al. (2004) Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res., 14, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson J.M., Castle,J., Garrett-Engele,P., Kan,Z., Loerch,P.M., Armour,C.D., Santos,R., Schadt,E.E., Stoughton,R. and Shoemaker,D.D. (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science, 302, 2141–2144. [DOI] [PubMed] [Google Scholar]
- 26.Modrek B., Resch,A., Grasso,C. and Lee,C. (2001) Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res., 29, 2850–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mironov A.A., Fickett,J.W. and Gelfand,M.S. (1999) Frequent alternative splicing of human genes. Genome Res., 9, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hide W.A., Babenko,V.N., van Heusden,P.A., Seoighe,C. and Kelso,J.F. (2001) The contribution of exon-skipping events on chromosome 22 to protein coding diversity. Genome Res., 11, 1848–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.