

Abstract
The phosphoinositide 3-kinase (PI3K) signaling pathway is the most commonly mutated pathway in head and neck squamous cell carcinoma (HNSCC). There are several drugs targeting members of the PI3K signaling pathway in development for HNSCC. In this article, we review the genetic alterations reported in the pathway pertinent to HNSCC, various agents in development targeting various mediators of the pathway, results from clinical trials, and remaining challenges in the development of PI3K pathway inhibitors.
Keywords: PI3K signaling, head and neck squamous cell carcinoma, PIK3CA mutation, PI3K inhibitor, personalized medicine
INTRODUCTION
The phosphoinositide 3-kinase (PI3K) signaling pathway is of particular importance in head and neck squamous cell carcinoma (HNSCC), as it is the most frequently mutated pathway [1, 2]. Under normal conditions, this signaling pathway serves to promote cell survival, growth, development, and differentiation [3, 4]. Recently, dysregulation of this pathway has been noted at the genomic and proteomic levels, with implications in both the pathogenesis of HNSCC and potential therapeutic targets. In addition, activation of the PI3K pathway is known to be involved in acquired resistance to anticancer therapy [5]. In this review, we discuss the dysregulation of the PI3K signaling pathway, target therapies, and implications for precision medicine in the context of HNSCC.
PI3K SIGNALING PATHWAY IN NORMAL PHYSIOLOGY
Key players of the PI3K pathway include receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR), G-protein coupled receptors (GPCRs), PI3Ks, phosphatidylinositol 4,5-bisphosphate (PIP2), phosphatidylinositol (3,4,5)-trisphosphate (PIP3), Akt, mTOR, and PTEN. RTKs and GPCRs bind growth factors and cytokines at the cell surface, and then transduce signals via a number of intracellular pathways, such as the PI3K pathway. PI3Ks are classified into three groups: classes I, II, and III. Class I is subdivided into class IA and class IB and they consist of a catalytic (p110) and regulatory (p85 for class IA and p101 and p87 for class IB) components. Class IA is activated by various types of cell surface receptors and has 3 isoforms, p110α, p110β, and p110δ. Class IB is activated primarily by G-protein coupled receptors and has one isoform, p110γ. Of these isoforms, class IA PI3Ks have been studied the most extensively. Binding of p85 to p110 stabilizes p110, inhibits their kinase activity, and recruits phosphotyrosine in activated receptors or adaptors. The engagement of p85 and phosphotyrosine activates the kinase activity of p110 by releasing the p85-mediated inhibition of p110 [6]. The activated catalytic subunit of PI3K (p110) phosphorylates PIP2, generating PIP3. PIP3 binds Akt (protein kinase B), a serine/threonine protein kinase, leading to the exposure of two amino acid residues requiring phosphorylation. PIP3 also binds phosphoinositide-dependent protein kinase-1 (PDK1), which phosphorylates those two residues, leading to the full activation of Akt. Activated Akt then phosphorylates other proteins and thus affects a number of cellular processes. mTOR, another serine threonine protein kinase, is activated by Akt and plays an important role in cell growth by monitoring cellular needs, such as nutrients, oxygen, and energy. mTOR forms two distinct complexes with other proteins and both complexes are involved in tumorigenesis [7, 8]. Akt activates mTORC1 indirectly by inactivating proline-rich Akt substrate 40 kDa (PRAS40) and tuberous sclerosis complex (TSC2), which inhibit mTORC1. mTORC1 then regulates protein translation, specifically by targeting ribosomal protein S6 (pS6) kinase and eukaryote initiation factor 4E-binding protein 1 (4E-BP1). mTOR complex 2 (mTORC2) is involved in activation of Akt by phosphorylation of Ser473 in Akt. The mechanism of mTORC2 activation is not clear. Lastly, PTEN (phosphatase and tensin homology deleted from chromosome 10) is an important negative regulator of this pathway, as it serves to dephosphorylate PIP3 to PIP2 [9, 10].
PI3K SIGNALING PATHWAY IN HNSCC
Recent studies have worked to identify mutations, amplifications, and overexpression of the different mediators/genes involved in the PI3K pathway (Table 1). Of these, PIK3CA, the gene that encodes for the catalytic component p110α, is the most commonly mutated component of this pathway.
Table 1. Dysregulations in the PI3K pathway in HNSCC.
| PI3K Pathway Component | Type of alteration | Tumor site |
|---|---|---|
| p85 | Mutation [2, 11, 12] | |
| PTEN | Mutation [2, 13–15] Loss of heterozygosity [16, 17] Reduced expression [16] |
Not specified [2, 13, 14] Oropharynx, hypopharynx, larynx [15] Tongue, larynx, oral cavity [17] Not specified [16] |
| AKT | Mutation [2] Activation [16, 18] Copy number alteration [19] Over-expression [20] |
|
| mTORC1 | Mutation [2] Activation [21] |
|
| EGFR | Mutation [22, 23] Activation [24] Overexpression [24] |
Laryngeal [23] Not specified [22] |
PIK3CA Mutations
In HNSCC, PIK3CA mutations tend to be heavily focused on the helicase (exon 9) and kinase (exon 20) domains [25–27], which also holds true for PIK3CA mutations in most sporadic cancers (Figure 1). In the past few years, genomic datasets of HNSCCs have expanded with efforts led by The Cancer Genome Atlas (TCGA) [28, 29]. In Figure 1, the PIK3CA point mutation frequencies of HNSCC, breast cancer, and colorectal cancers catalogued by the TCGA are compared. The frequencies of PIK3CA mutations were 17.5%, 36.4%, and 16.7% in HNSCC, breast, and colorectal cancer, respectively. Of note, the hot-spot mutations on exon 9 (corresponding to residues E542 and E545 in p110α) and exon 20 (residue H1047) are the most common alterations regardless of cancer type. E542 and E545 are frequently mutated to lysine and H1047 is frequently altered to arginine.
Figure 1. PIK3CA point mutation rates in TCGA cohorts.
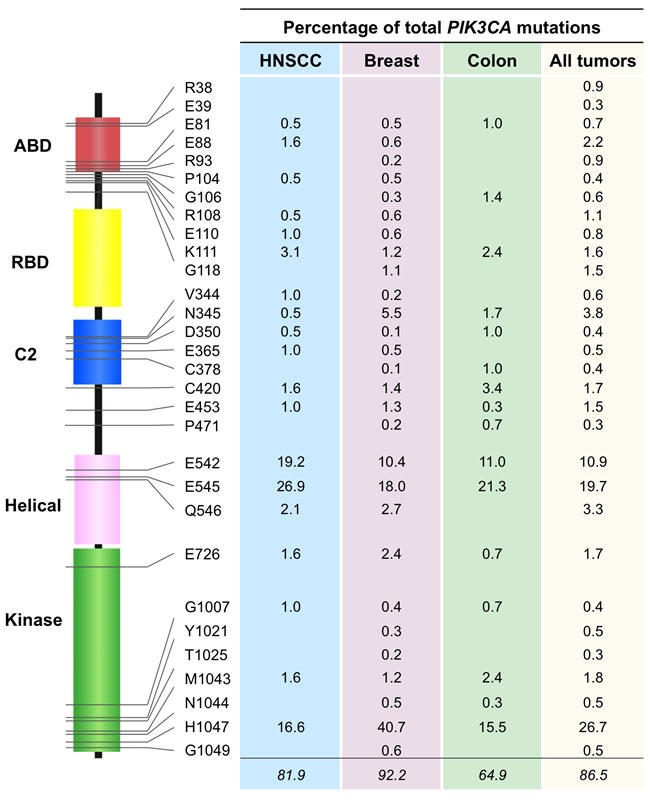
Analysis of TCGA cohorts of HNSCC, breast cancer, and colorectal cancer was performed to determine rates of point mutations in PIK3CA. The top 30 most frequent mutations when analyzing all TCGA tumor samples were selected. The residues affected by these mutations are depicted along with the corresponding rates of mutation. The mutation rates represent the percentage of total PIK3CA mutations affecting each residue for the various types of tumor. Abbreviations: ABD, adaptor-binding domain; RBD, RAS-binding domain; C2, protein-kinase-C homology-2 domain.
PIK3CA over-amplification [16, 20, 30] and copy number gain [20, 31] have also been reported in HNSCC. Moreover, PIK3CA overexpression and copy number gains in the PI3K pathway have been associated with HPV-positive HNSCC [32, 33]. Though copy number gains have been linked to increased transcription and expression of p110α [34], there are not necessarily correlations between PIK3CA gene amplification and p110α expression [16]. This suggests that there may be other pathways that interact with or influence p110α expression and play a role in tumorigenesis.
MUTATIONS IN OTHER COMPONENTS OF THE PI3K PATHWAY
p85, the regulatory subunit of Class IA PI3K, has three isoforms — p85α, p85β, and p55γ — that are encoded by the genes PIK3R1, PIK3R2, and PIK3R3, respectively. Mutants of p85 are oncogenic, as they have been shown to promote basal PI3K activity without stimulation by growth factors [35–37]. In HNSCC, PIK3R1 missense [12], in-frame insertion [12], nonsense [2], and non-synonymous [11] mutations have been noted, mostly in HPV-negative patients. Depending on whether p85 exists as a monomer or a dimer with p110, it plays different roles. As a monomer, it binds the cell surface receptor adaptor protein IRS-1 and limits downstream PI3K pathway signaling. As a dimer with p110, however, it serves to potentiate PI3K signaling. If a gene such as PIK3R1, which encodes for a component of p85, is dysfunctional, adequate p85 may not be produced, leading to hyperactivation of the PI3K pathway and resultant tumorigenesis [35]. In addition, PIK3R2 overexpression was noted in esophageal squamous cell carcinoma, and is thought to be related to under-expression of microRNA-126, which likely plays a tumor-suppressing role and targets PIK3R2 [38].
PTEN is a tumor suppressor that has been found to have a number of alterations in HNSCC, including nonsense [2, 13, 14], missense [2, 13, 15], loss of heterozygosity [16, 17], hemizygous deletion [39], intron [14], and splice site single nucleotide polymorphisms [2], as well as reduced expression [16]. Given the many different genomic and proteomic alterations seen with PTEN, its dysregulation in HNSCC might be attributed to multiple molecular mechanisms. With regard to genetic mutations, exon 5 of the PTEN gene is of particular interest, as it encodes for PTEN's lipid phosphatase catalytic domain, which plays a major role in tumor suppression [40]. Low PTEN protein expression (not necessarily linked to PTEN gene mutations) may be attributed to degradation of PTEN [41, 42] or gene silencing [43, 44].
Akt promotes cell survival and proliferation through the phosphorylation of various substrates and is encoded by 3 Akt genes (Akt1, Akt2, and Akt3) [45]. In HNSCC, Akt genes have been found to display missense mutations [2], copy number variations [19], increased activation [16, 18, 46], and overexpression [20]. Amplification and overexpression of Akt is thought to enable a cell to proliferate in conditions not normally conducive to proliferation [47]. mTOR is a downstream target of Akt that integrates signals from multiple pathways, including nutrients (e.g., amino acids and glucose), growth factors (e.g., insulin and insulin-like growth factor 1), hormones (e.g., leptin), and stresses (e.g., starvation, hypoxia, and DNA damage) to regulate a wide variety of eukaryotic cellular functions, such as translation, transcription, protein turnover, cell growth, differentiation, cell survival, metabolism, energy balance, and stress response. In HNSCC, mTOR was found to be activated [21, 48] and have missense mutations [2]. It has been demonstrated that these point mutations can lead to constitutive activation of mTOR, which enables cells to grow and proliferate in the absence of nutrients [49].
EGFR is an upstream activator of the PI3K signaling pathway that is frequently altered in cancer. It is a member of the RTK (receptor tyrosine kinases) that activates class IA PI3Ks, the most commonly mutated members of the PI3K pathway in cancer. It has been reported to display missense [23], in-frame deletion [22], and activating mutations [24], as well as overexpression [24] in HNSCC. An abnormally truncated EGFR mutant (deletion in exons 2-7) has been found to be constitutively active and potentiate the PI3K pathway signaling [50], and may play a role in tumorigenesis.
Clinical trials targeting the PI3K pathway in HNSCC
Treatment modalities for HNSCC are guided by site and stage of disease and include surgical resection, radiation, and chemotherapy. However, when tumors have developed resistance, relapse occurs and these treatments are no longer effective and new therapeutic options are needed. The PI3K/Akt/mTOR pathway is activated in many types of cancers and has been demonstrated to contribute to treatment resistance [5]. In addition, this pathway has been implicated in tumorigenic processes such as cell proliferation, invasion, angiogenesis, and metastasis [45, 51–53]. These factors make the PI3K/Akt/mTOR pathway an attractive target for cancer therapy. Significant efforts have been devoted to developing agents to target the pathway [54–56]. Many of these agents have shown promising results in preclinical in vitro and in vivo studies of various cancer types [57, 58], including HNSCC [59, 60]. Inhibition of this pathway can disrupt resistance acquired by cancer cells and sensitize cancers to antitumor agents of other modalities, reduce cell proliferation, and induce apoptosis [5, 61–63]. Furthermore, single inhibitors may not be enough to achieve sustained inhibition of the pathway. It has been shown that inhibition of PI3K pathway may trigger compensatory feedback [64]. Therefore, in the majority of the clinical trials, PI3K/Akt/mTOR inhibitors are used in combination with other agents or radiation with the goal of achieving a synergistic effect [65]. Here, we discuss the recent development of PI3K pathway inhibitors that have been or are being tested in clinical trials for HNSCC (Figure 2). The relevant clinical trials registered at ClinicalTrials.gov are listed in Table 2.
Figure 2. The PI3K signaling pathway's key players and inhibitors under investigation in HNSCC trials.
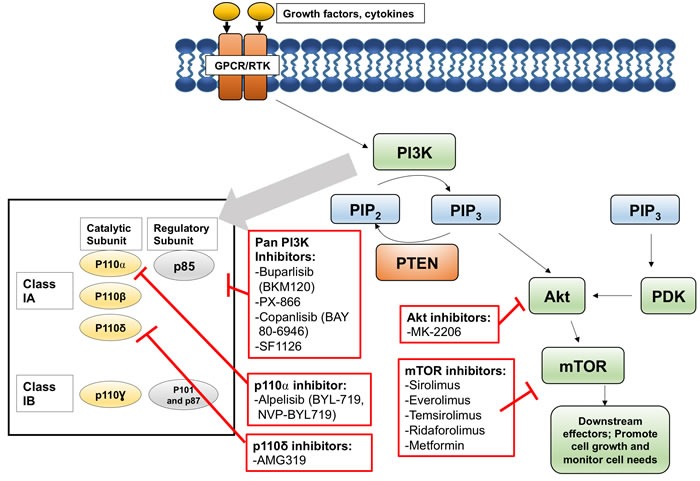
Table 2. Ongoing Clinical Trials of Agents Targeting the PI3K Pathway in Patients with HNSCC.
| Target | Agent | Other Interventions | Condition | Phase | Status | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| PI3K | Buparlisib (BKM120) | Advanced HNSCC | 2 | Unknown | NCT01527877 | |
| Cisplatin, IMRT | High risk LA HNSCC | 1b | Active | NCT02113878 | ||
| Paclitaxel | Pt pre-treated R/M HNSCC | 2 | Active | NCT01852292 | ||
| Cetuximab | R/M HNSCC | 1/2 | Active | NCT01816984 | ||
| R/M HNC | 2 | Active | NCT01737450 | |||
| PX-866 | Docetaxel | NSCLC, HNSCC | 1/2 | Completed | NCT01204099 | |
| Cetuximab | Metastatic CRC, R/M HNSCC | 1/2 | Completed | NCT01252628 | ||
| Copanlisib (BAY 80-6946) | Cetuximab | R/M HNSCC with PI3KCA mutation/amplification and/or PTEN loss | 1/2 | Active | NCT02822482 | |
| SF1126 | R/M SCCHN with mutation in PIK3CA and/or PI3K Pathway | 2 | Active | NCT02644122 | ||
| Alpelisib (BYL-719) | Cisplatin, IMRT | LA HNSCC | 1 | Active | NCT02537223 | |
| Paclitaxel | Breast cancer and HNC | 1 | Active | NCT02051751 | ||
| Cetuximab, IMRT | Stage III/IVb HNSCC | 1 | Active | NCT02282371 | ||
| Pt therapy failed, R/M HNSCC | 2 | Active | NCT02145312 | |||
| Cetuximab | R/M HNSCC | 1b/2 | Completed | NCT01602315 | ||
| AMG319 | HPV negative HNSCC | 2 | Active | NCT02540928 | ||
| AKT | MK2206 | R/M HNC | 2 | Completed | NCT01349933 | |
| mTOR | Sirolimus (rapamycin) | HNSCC | 1/2 | Completed | NCT01195922 | |
| Everolimus (RAD001) | Docetaxel | LA and R/M HNSCC | 1/2 | Terminated | NCT01313390 | |
| Carboplatin, cetuximab | Advanced HNC | 1/2 | Completed | NCT01283334 | ||
| Carboplatin, paclitaxel | LA HNC not removable by surgery | 1/2 | Completed | NCT01333085 | ||
| Erlotinib | Recurrent HNSCC | 2 | Completed | NCT00942734 | ||
| Erlotinib, radiation | R/M HNSCC treated with radiation | 1 | Withdrawn | NCT01332279 | ||
| Cetuximab | R/M colon cancer or HNC | 1 | Completed | NCT01637194 | ||
| Cetuximab, cisplatin, carboplatin | R/M HNSCC | 1/2 | Terminated | NCT01009346 | ||
| HNSCC | 2 | Active | NCT01133678 | |||
| HNSCC | 2 | Active | NCT01051791 | |||
| HNC | 2 | Active | NCT01111058 | |||
| Docetaxel, cisplatin | LA HNC | 1 | Completed | NCT00935961 | ||
| Cisplatin, radiation | LA, inoperable HNC | 1 | Terminated | NCT01057277 | ||
| Cisplatin, IMRT | LA HNC | 1 | Terminated | NCT01058408 | ||
| Cisplatin, IMRT | LA HNC | 1 | Completed | NCT00858663 | ||
| Ceritinib | HNC, NSCLC | 1/1b | Active | NCT02321501 | ||
| LA HNSCC | 2 | Active | NCT01133678 | |||
| Temsirolimus (CCI-779) | HNSCC | 2 | Completed | NCT01172769 | ||
| Cetuximab | R/M HNC not respond to therapy | 2 | Completed | NCT01256385 | ||
| Paclitaxel, carboplatin | R/M HNSCC | 1/2 | Active | NCT01016769 | ||
| Cisplatin, cetuximab | R/M HNSCC | 1/2 | Terminated | NCT01015664 | ||
| Advanced HNSCC | Completed | NCT00195299 | ||||
| Erlotinib | Pt-refractory or-ineligible, advanced SCC | 2 | Terminated | NCT01009203 | ||
| Cetuximab, cisplatin, radiation | Advanced HNC | pilot | Withdrawn | NCT01326468 | ||
| Ridaforolimus (AP23573, MK-8669, deforolimus) | Advanced HNC, NSCLC, colon cancer | 1 | Terminated | NCT01212627 | ||
| Metformin (glucophage) | Paclitaxel | R/M HNSCC | 2 | Terminated | NCT01333852 | |
| Cisplatin, radiation | LA HNSCC | 1 | Active | NCT02325401 | ||
| HNSCC | 0 | Active | NCT02402348 | |||
| HNSCC | 0 | Active | NCT02083692 | |||
| PI3K/AKT | Perifosine (KRX-0401) | R/M HNC | 2 | Terminated | NCT00062387 | |
| PI3K/PLK | Rigosertib (ON-01910) | Cisplatin, radiation | HNSCC | 1 | Completed | NCT02107235 |
Unknown: Status not verified over 2 years. Abbreviations: IMRT: Intensity-modulated radiotherapy. HNC: Head and Neck Cancer. SCC: Squamous cell carcinoma. HNSCC: Head and neck squamous cell carcinoma. NSCLC: Non small cell lung cancer. HPV: Human Papillomavirus. R/M: recurrent and/or metastatic
PAN-PI3K INHIBITORS
Pan-PI3K inhibitors refer to inhibitors of more than one of the p110 isoforms of PI3K.
Buparlisib (BKM120)
Buparlisib is an oral PI3K inhibitor and inhibits the activity of all four p110 isoforms of class I PI3K. Using cultured cell lines with wild-type or E542K, E545K, and H1047R hotspot mutations in PIK3CA, Kong et al. showed that buparlisib has comparable impact on the hotspot and wild-type PIK3CA [66]. It was also shown that a combination of buparlisib with the anti-EGFR monoclonal antibody cetuximab exerts a synergistic effect on tumor inhibition in wild-type or PIK3CA mutant HNSCC cell lines [67] as well as in a xenograft model of HNSCC [68]. However, the half maximal inhibitory concentration (IC50) of buparlisib is much higher than that of many of the PI3K inhibitors under investigation. The toxicity of doses required for PI3K inhibition in vivo is a concern in its clinical application. Five phase 1 and/or 2 clinical trials are ongoing to evaluate the efficacy and safety of buparlisib in combination with cisplatin and IMRT, paclitaxel, or cetuximab (Table 2). Results from these trials have not been posted.
PX-866
Wortmannin is a potent irreversible PI3K inhibitor that equally inhibits most PI3K isoforms and has been shown to have antitumor activity. However, it is not optimal for clinical applications due to its high liver and hematological toxicity as well as poor biological stability. PX-866 was derived from wortmannin to overcome these problems [69]. It can inhibit PI3K activity at 0.1 nM and was reported to enhance antitumor activity of other chemotherapeutic drugs and radiation in an ovarian cancer murine xenograft model [69]. However, phase 1 and 2 clinical trials of PX-866 in incurable recurrent or metastatic HNSCC patients have shown less promise. For example, a phase 2 randomized control trial using combination therapy with cetuximab showed no differences in disease control rate, median progression-free survival, or median overall survival when compared to the control group receiving cetuximab alone [70]. A phase 2 randomized control trial using combination therapy with docetaxel also showed no differences when compared to the control group receiving cetuximab [71]. Thus, the addition of PX-866 did not improve the efficacy in patients without molecular preselection. No active trial of PX-866 for HNSCC is currently registered.
Copanlisib (BAY 80-6946)
Copanlisib is a highly selective and potent intravenous pan-PI3K inhibitor with sub-nanomolar IC50 against isoforms p110α and p110δ [72]. The mean IC50s of copanlisib were 19 nM and 774 nM for PIK3CA mutant and wild type cell lines respectively, indicating superior antitumor activity. In non-small cell lung cancer xenograft models, combination of copanlisib and paclitaxel achieved a 100% sustained response. Copanlisib in combination with cetuximab is being evaluated in phase 1 and phase 2 trials in patients with recurrent and/or metastatic HNSCC harboring a PI3KCA mutation/amplification and/or a PTEN loss.
SF1126
LY294002 can inhibit both PI3K and mTOR but is generally considered a PI3K inhibitor. It has antitumor and antiangiogenesis activity in vivo, but is not a viable drug due to poor solubility and short half-life. SF1126 was designed as a prodrug of LY294002 with a small peptide tag on LY294002 to increase solubility and to target αvβ3 and α5β1 integrins [73]. SF1126 was able to significantly reduce tumor volumes in U87MG glioma and PC-3 prostate cancer xenograft models in nude mice. In addition, antiangiogenesis activity due to inhibition of the HIF-1/VEGF pathway activity in 6 of 11 xenograft models was observed. SF1126 is now being evaluated in a phase 2 trial in patients with recurrent or progressive HNSCC and mutations in PIK3CA and/or PI3K pathway genes.
ISOFORM-SPECIFIC PI3K INHIBITORS
Isoform-specific PI3K inhibitors are active against one of the p110 isoforms of class I PI3K. They are usually also active against other p110 isoforms to a lesser extent.
Alpelisib (BYL-719, NVP-BYL719)
Alpelisib was designed to selectively inhibit p110α, or PIK3CA [74]. In a PIK3CA-dependent murine xenograft model, alpelisib showed significant dose-dependent inhibition of tumor growth and a favorable safety profile [75]. These results suggest that alpelisib is a promising agent for treating tumors with PIK3CA mutations. Alpelisib is being evaluated in 5 clinical trials (Table 2). Preliminary results showed encouraging antitumor activity [76]. Results from a completed trial have not been posted.
Isoform p110δ of PI3K (PI3Kδ, AMG319)
p110δ is mostly confined to spleen, thymus, and peripheral blood leukocytes. Its dysregulation has been implicated in rheumatoid arthritis, systemic lupus erythematosus, and hematological malignancies. Inactivation of p110δ in regulatory T cells unleashes CD8+ cytotoxic T cells and induces tumor regression. Thus, p110δ inhibitors such as AMG319 can break tumor-induced immune tolerance [77]. Currently, AMG319 is in a double-blind, placebo-controlled phase 2a trial in patients with HPV-negative HNSCC.
AKT INHIBITORS
MK-2206
Akt activation and overexpression are often associated with resistance to chemotherapy or radiotherapy. Inhibition of Akt has great potential in cancer treatment. Many Akt inhibitors have been developed for cancer treatment [55]. Among them, MK-2206 is a highly potent and selective Akt inhibitor that has been shown to enhance the anti-tumor activity of several anticancer agents in vitro and in vivo [78]. MK-2206 was evaluated in a phase 2 trial in patients with recurrent or metastatic HNSCC. Nine out of 21 patients were alive and progression-free at the end of the trial (but final results have not been posted). Moreover, MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib was evaluated in a phase 1 trial in patients with advanced solid tumors [79]. Interestingly, two patients with HNSCC demonstrated a complete and partial response.
MTOR INHIBITORS
Rapamycin was isolated from Streptomyces hygroscopicus. It was initially developed as an anti-fungal agent and used as an immunosuppressant to prevent rejection in organ transplant. Later, it was found to inhibit mTOR and showed promising antitumor activity in many solid tumors. Rapamycin binds to the intracellular FK506-binding protein (FKBP12) to form a complex, which then binds to mTORC1 and interrupts its ability to signal to its downstream effectors [56, 80, 81]. Temsirolimus, everolimus, and deferolimus are analogs of rapamycin and they inhibit mTORC1 activity through the same mechanism.
Sirolimus (rapamycin)
Sirolimus has been tested for treatment of many types of tumors, including HNSCC, but has poor oral bioavailability and solubility (precluding intravenous administration). Analogs of sirolimus have been developed to overcome this problem. In addition, nanoparticle albumin-bound rapamycin has been developed and is currently being tested in a phase 2 trial that includes HNC.
Everolimus (RAD001)
Everolimus is a 2-hydroxyethyl derivative of sirolimus with a similar mechanism of action, but an improved oral bioavailability. Everolimus has been approved by the U.S. Food and Drug Administration (FDA) for treatment of several cancers including recent approval for the treatment of progressive, well differentiated, nonfunctional, neuroendocrine tumors of gastrointestinal or lung origin in unresectable, locally advanced, or metastatic disease. In a trial with recurrent or metastatic HNSCC patients, everolimus was not effective [82]. Everolimus, in combination with cetuximab/carboplatin, or cisplatin/IMRT, or cisplatin/docetaxel, has been evaluated in several early phase studies [83–85]. In a phase 2 study, everolimus in combination with erlotinib was evaluated in patients with platinum-resistant HNSCC with no significant benefit [86]. Currently, two other phase 2 trials are ongoing to evaluate the efficacy of everolimus in patients with refractory, recurrent, and locally advanced HNSCC and to study the correlation of everolimus treatment with tumor- and patient-associated markers of the EGFR-mTOR pathway.
Temsirolimus (CCI-779)
Temsirolimus is hydrolyzed to form sirolimus quickly after intravenous administration but itself also has mTOR inhibitor activity. It has been approved by the FDA to treat renal cell carcinoma and evaluated in many trials of combination therapy for HNSCC. In a pharmacodynamic evaluation of temsirolimus in patients with newly diagnosed advanced HNSCC, Akt/mTOR pathway biomarkers were evaluated in tumor and peripheral blood mononuclear cells (PBMCs). Temsirolimus significantly decreased pS6 and p4E-BP1 in tumors, and pS6 and pAkt in PBMCs, indicating significant inhibition of the mTOR pathway in both tumors and PBMCs [87]. In addition, after only 2-3 doses, 8 of 14 patients’ tumors decreased in size on endoscopic evaluation [87]. In a different trial of patients with platinum- and cetuximab-refractory recurrent and/or metastatic HNSCC, treatment with temsirolimus resulted in disease stabilization in 58% and tumor shrinkage in 39% of 33 assessable patients [88]. However, combination therapy with temsirolimus may be limited by treatment toxicity. In trials studying combination therapy of temsirolimus, bevacizumab, and cetuximab or temsirolimus and erlotinib, numerous toxicities were reported [89, 90]. The combination of temsirolimus with weekly does of paclitaxel and carboplatin was evaluated in a phase 1 and 2 trial in patients with recurrent or metastatic HNSCC to establish recommended dosing for a phase 2 study and to determine the objective response rate.
Ridaforolimus (AP23573, MK-8669, deforolimus)
Ridaforolimus is available in oral and intravenous formulations. In combination with the Notch inhibitor MK-0752, ridaforolimus was evaluated in a phase 1 trial in patients with advanced tumors [91]. Fifteen of 30 enrolled patients had HNSCC. Among 10 HNSCC patients evaluated for tumor response, one had complete response and another had a confirmed partial response. However, there were a high number of adverse events at the maximum tolerated dose.
Metformin (glucophage)
Metformin is used in the treatment of type 2 diabetes. The use of metformin in diabetic patients has been associated with significantly lower risks of cancer incidence and mortality. In a retrospective study of 205 patients with laryngeal SCC [92], patients treated with metformin had more early stage tumors than untreated patients (48% vs 27%), but had fewer regional metastasis events (19% vs 50%) and a better survival rate (76% vs 41%). Yen et al. compared 66,600 diabetic patients either taking or not taking metformin and found the incidence of head and neck cancer (HNC) to be 34% lower in patients taking metformin [93]. Metformin indirectly inhibits mTOR by decreasing expression of Sp-regulated insulin-like growth factor-1 receptor and also inhibits Ras signaling by decreasing EGFR [94]. Skinner et al. investigated the role of TP53-disruptive mutations in radioresistance [95]. They found metformin potentiated the effects of radiation in the presence of a disruptive TP53 mutation in vitro and in vivo. Among patients treated with postoperative radiation therapy for HNSCC, patients taking metformin had a dramatically lower locoregional recurrence rate than did the control group. The five-year overall survival rate was 87% in patients taking metformin compared to 41% in the remaining patients [95]. The safety and efficacy of metformin, alone or in combination with radiation or other agents, are being evaluated in several early stage clinical trials in HNSCC patients (Table 2).
DUAL INHIBITORS
Perifosine (D-21266, KRX-0401, NSC 639966)
Perifosine inhibits both protein kinase B and Akt phosphorylation, but does not directly inhibit PI3K [96]. Though it inhibited tumor growth in various in vitro and in vivo studies [97], it showed little effect in a phase 2 trial of patients with recurrent or metastatic HNC that was terminated early [98]. Perifosine alone or in combination with other agents is currently being tested in several trials for cancers, but not in HNSCC.
Rigosertib (ON 01910.Na, estybon)
Polo-like kinases (Plks) are important regulators of the cell cycle and are new targets for cancer therapy [99, 100]. Rigosertib was developed to inhibit Plk [101] and was found to inhibit PI3K as well [102]. Rigosertib's antitumor activity was studied in 16 HPV-negative HNSCC cell lines and 8 direct patient tumor xenografts of HNSCC [103]. Rigosertib had potent antiproliferative effects on 11 of 16 HNSCC cell lines and inhibited growth reduction in 3 of 8 HNSCC xenografts. Biomarker analysis indicated that a combination of PI3K/TP53 events was necessary, but not sufficient, for rigosertib sensitivity. Safety and efficacy of rigosertib were evaluated in a recently completed phase 2 trial in patients with relapsed or metastatic, platinum-resistant, HPV-positive or HPV-negative SCC. Results from the trial have not been posted or published.
REMAINING CHALLENGES
Studies of PI3K pathway inhibitors on HNSCC cell lines and xenograft models have been very encouraging, but these agents have shown less promise in clinical trials to date. Several reasons may explain this discrepancy. As mentioned earlier, single agent therapy may have limited efficacy due to activation of compensatory feedback [64]. PI3K inhibition may also activate mitochondrial reprogramming that subsequently promotes tumor invasion and progression [104]. Yet, beyond unintended molecular signaling that can be induced by PI3K pathway inhibition, there are also challenges in maximizing clinical trial design. For one, non-optimized dosing schedules may lead to inadequate pharmacologic inhibition of the pathway [105].In addition, trial results may also be affected by inadequate patient preselection [57, 58]. Existing HNSCC trials of PI3K pathway inhibitors have stratified patients based on prior treatment failures [70, 71]. However, few published studies have examined trial results for a correlation between responsiveness and PI3K pathway mutational status. Furthermore, none - to our knowledge - have preselected HNSCC patients based on PI3K pathway mutation status, which likely has a significant effect on response to targeted therapies and trial results [58]. Below, we discuss the potential role of genomics in future HNSCC trials.
APPLYING PRECISION MEDICINE IN HNSCC TRIALS
In the era of genomics, next generation sequencing offers the ability to characterize the mutational profiles of patients on a genome-wide scale and the potential to enhance future trial design. Genomic sequencing can be readily performed on HNSCC tumor specimens. However, tumor specimens may not be available for all patients enrolled in HNSCC trials, would be costly to obtain, and could place patients at risk for procedural complications. Thus, it would be advantageous to utilize a non-invasive source of tumor samples for genetic profiling. “Liquid biopsies” using circulating tumor cells (CTCs) and tumor cell DNA extracted from the peripheral blood for biomarker analysis could provide a potential solution. Cancers are known to release CTCs and cell-free circulating tumor DNA (ctDNA), or cell-free nuclear acids (cfNA), into blood. Recently, their potential as diagnostic and monitoring tools for cancer has been extensively investigated [106, 107]. Combined with next generation sequencing technology, liquid biopsy can be used to personalize HNSCC patient treatments, evaluate tumor mutations throughout treatment, predict treatment response, and potentially elucidate novel biomarkers. Research on liquid biopsies in HNSCC has lagged behind that in more common cancer types, but is increasingly gaining more attention. In an ongoing trial (NCT02822482) of copanlisib with cetuximab in HNSCC patients with PI3K mutation/amplification and/or PTEN loss, mutational profiles of ctDNA at multiple time points will be analyzed to monitor disease progression.
CONCLUSIONS
Recent studies on HNSCC continue to support the PI3K pathway as a promising target for future HNSCC therapies. Currently, there are multiple targeted therapies against this pathway under investigation. However, trial results to date have yet to show the same degree of efficacy as have been demonstrated in in vitro or in vivo studies. Several challenges may be limiting trial success. The results from ongoing trials, such as those with patient recruitment based on mutational profiles of the PI3K pathway or comparisons between single and multiple agent therapies, will be eagerly anticipated and may provide additional guidance on designing future trials .
Footnotes
CONFLICTS OF INTEREST
There is no conflict of interest.
REFERENCES
- 1.Iglesias-Bartolome R, Martin D Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov. 2013;7:722–725. doi: 10.1158/2159-8290.CD-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;7:761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman JA, Luo J Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;8:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 4.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;5573:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 5.Burris HA., 3rd Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;4:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;5:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 7.Laplante M Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;Pt20:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laplante M Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;2:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Broek R, Mohan S, Eytan DF, Chen Z Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;7:815–825. doi: 10.1111/odi.12206. [DOI] [PubMed] [Google Scholar]
- 10.Liu P, Cheng H, Roberts TM Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, Lawrence MS, Getz G, Bragelmann J, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;3:632–641. doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;6046:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chau NG, Li YY, Jo VY, Rabinowits G, Lorch JH, Tishler RB, Margalit DN, Schoenfeld JD, Annino DJ, Goguen LA, Thomas T, Becker H, Haddad T, et al. Incorporation of Next-Generation Sequencing into Routine Clinical Care to Direct Treatment of Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2016;12:2939–2949. doi: 10.1158/1078-0432.CCR-15-2314. [DOI] [PubMed] [Google Scholar]
- 14.Okami K, Wu L, Riggins G, Cairns P, Goggins M, Evron E, Halachmi N, Ahrendt SA, Reed AL, Hilgers W, Kern SE, Koch WM, Sidransky D, et al. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res. 1998;3:509–511. [PubMed] [Google Scholar]
- 15.Poetsch M, Lorenz G Kleist B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet Cytogenet. 2002;1:20–24. doi: 10.1016/s0165-4608(01)00509-x. [DOI] [PubMed] [Google Scholar]
- 16.Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, Gonzalez MV. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;2:242–248. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 17.Shao X, Tandon R, Samara G, Kanki H, Yano H, Close LG, Parsons R Sato T. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int J Cancer. 1998;5:684–688. doi: 10.1002/(sici)1097-0215(19980831)77:5<684::aid-ijc4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 18.Segrelles C, Moral M, Lara MF, Ruiz S, Santos M, Leis H, Garcia-Escudero R, Martinez-Cruz AB, Martinez-Palacio J, Hernandez P, Ballestin C, Paramio JM. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene. 2006;8:1174–1185. doi: 10.1038/sj.onc.1209155. [DOI] [PubMed] [Google Scholar]
- 19.Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J, Cortez E, Xie TX, Zhang D, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;7:770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenic I, Steger K, Gruber C, Arens C Woenckhaus J. Analysis of PIK3CA and Akt/protein kinase B in head and neck squamous cell carcinoma. Oncol Rep. 2007;1:253–259. [PubMed] [Google Scholar]
- 21.Garcia-Carracedo D, Angeles Villaronga M, Alvarez-Teijeiro S, Hermida-Prado F, Santamaria I, Allonca E, Suarez-Fernandez L, Victoria Gonzalez M, Balbin M, Astudillo A, Martinez-Camblor P, Su GH, Pablo Rodrigo J, et al. Impact of PI3K/AKT/mTOR pathway activation on the prognosis of patients with head and neck squamous cell carcinomas. Oncotarget. 2016;7:29780–93. doi: 10.18632/oncotarget.8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Soung YH, Kim SY, Nam HK, Park WS, Nam SW, Kim MS, Sun DI, Lee YS, Jang JJ, Lee JY, Yoo NJ, Lee SH. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;8:2879–2882. doi: 10.1158/1078-0432.CCR-04-2029. [DOI] [PubMed] [Google Scholar]
- 23.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, Hilbe W, Schwentner I, Sprinzl GM, Utermann G, Zwierzina H. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. Eur J Cancer. 2006;1:109–111. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 24.Willmore-Payne C, Holden JA Layfield LJ. Detection of EGFR- and HER2-activating mutations in squamous cell carcinoma involving the head and neck. Mod Pathol. 2006;5:634–640. doi: 10.1038/modpathol.3800552. [DOI] [PubMed] [Google Scholar]
- 25.Qiu W, Schonleben F, Li X, Ho DJ, Close LG, Manolidis S, Bennett BP, Su GH. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;5:1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruckman KC, Schonleben F, Qiu W, Woo VL Su GH. Mutational analyses of the BRAF, KRAS, and PIK3CA genes in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;5:632–637. doi: 10.1016/j.tripleo.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu W, Tong GX, Manolidis S, Close LG, Assaad AM, Su GH. Novel mutant-enriched sequencing identified high frequency of PIK3CA mutations in pharyngeal cancer. Int J Cancer. 2008;5:1189–1194. doi: 10.1002/ijc.23217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;269:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;5:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yen CC, Chen YJ, Pan CC, Lu KH, Chen PC, Hsia JY, Chen JT, Wu YC, Hsu WH, Wang LS, Huang MH, Huang BS, Hu CP, et al. Copy number changes of target genes in chromosome 3q25.3-qter of esophageal squamous cell carcinoma: TP63 is amplified in early carcinogenesis but down-regulated as disease progressed. World J Gastroenterol. 2005;9:1267–1272. doi: 10.3748/wjg.v11.i9.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin SC, Liu CJ, Ko SY, Chang HC, Liu TY, Chang KW. Copy number amplification of 3q26-27 oncogenes in microdissected oral squamous cell carcinoma and oral brushed samples from areca chewers. J Pathol. 2005;4:417–422. doi: 10.1002/path.1790. [DOI] [PubMed] [Google Scholar]
- 32.Lechner M, Frampton GM, Fenton T, Feber A, Palmer G, Jay A, Pillay N, Forster M, Cronin MT, Lipson D, Miller VA, Brennan TA, Henderson S, et al. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV- tumors. Genome Med. 2013;5:49. doi: 10.1186/gm453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yarbrough WG, Whigham A, Brown B, Roach M Slebos R. Phosphoinositide kinase-3 status associated with presence or absence of human papillomavirus in head and neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys. 2007;2(Suppl):S98–101. doi: 10.1016/j.ijrobp.2007.04.082. [DOI] [PubMed] [Google Scholar]
- 34.Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999;1:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 35.Luo J Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it's implication in cancer. Cell Cycle. 2005;10:1309–1312. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 36.Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennstrom S, von Kobbe C, Toran JL, L RB, Calvo V, Copin SG, Albar JP, Gaspar ML, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998;3:743–753. doi: 10.1093/emboj/17.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ Phillips WA. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;20:7426–7429. [PubMed] [Google Scholar]
- 38.Nie ZC, Weng WH, Shang YS, Long Y, Li J, Xu YT Li Z. MicroRNA-126 is down-regulated in human esophageal squamous cell carcinoma and inhibits the proliferation and migration in EC109 cell via PI3K/AKT signaling pathway. Int J Clin Exp Pathol. 2015;5:4745–4754. [PMC free article] [PubMed] [Google Scholar]
- 39.Xu B, Wang L, Borsu L, Ghossein R, Katabi N, Ganly I, Dogan S. A proportion of primary squamous cell carcinomas of the parotid gland harbour high-risk human papillomavirus. Histopathology. 2016;69:921–929. doi: 10.1111/his.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;23:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma A, Guha S, Wang H, Fok JY, Koul D, Abbruzzese J Mehta K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 2008;7:1997–2005. doi: 10.1158/1078-0432.CCR-07-1533. [DOI] [PubMed] [Google Scholar]
- 42.Torres J Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;2:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 43.Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am J Pathol. 2000;4:1123–1128. doi: 10.1016/S0002-9440(10)64627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;2:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osaki M, Oshimura M Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;6:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 46.Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, Sausville EA, Molinolo AA, Gutkind JS. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004;12(Pt 1):4029–4037. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 47.Altomare DA Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;50:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Carracedo D, Villaronga MA, Alvarez-Teijeiro S, Hermida-Prado F, Santamaria I, Allonca E, Suarez-Fernandez L, Gonzalez MV, Balbin M, Astudillo A, Martinez-Camblor P, Su GH, Rodrigo JP, et al. Impact of PI3K/AKT/mTOR pathway activation on the prognosis of patients with head and neck squamous cell carcinomas. Oncotarget. 2016;20:29780–29793. doi: 10.18632/oncotarget.8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato T, Nakashima A, Guo L, Coffman K Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;18:2746–2752. doi: 10.1038/onc.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moscatello DK, Holgado-Madruga M, Emlet DR, Montgomery RB, Wong AJ. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem. 1998;1:200–206. doi: 10.1074/jbc.273.1.200. [DOI] [PubMed] [Google Scholar]
- 51.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J Zhang LJ. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;2:177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM, Chen H, Hortobagyi GN, Mills GB, Meric-Bernstam F. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011;6:1093–1101. doi: 10.1158/1535-7163.MCT-10-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, Miyazaki J, Suda T, Itoh H, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 2005;17:2054–2065. doi: 10.1101/gad.1308805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;4:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Pal SK, Reckamp K, Yu H Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;11:1355–1366. doi: 10.1517/13543784.2010.520701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng Y Jiang Y. mTOR Inhibitors at a Glance. Mol Cell Pharmacol. 2015;2:15–20. [PMC free article] [PubMed] [Google Scholar]
- 57.Massacesi C, di Tomaso E, Fretault N, Hirawat S. Challenges in the clinical development of PI3K inhibitors. Ann N Y Acad Sci. 2013:19–23. doi: 10.1111/nyas.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016:203–210. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Isaacsson Velho PH, Castro G, Jr, Chung CH. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am Soc Clin Oncol Educ Book. 2015:123–128. doi: 10.14694/EdBook_AM.2015.35.123. [DOI] [PubMed] [Google Scholar]
- 60.Simpson DR, Mell LK Cohen EE. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015;4:291–298. doi: 10.1016/j.oraloncology.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 61.Niessner H, Schmitz J, Tabatabai G, Schmid AM, Calaminus C, Sinnberg T, Weide B, Eigentler TK, Garbe C, Schittek B, Quintanilla-Fend L, Bender B, Mai M, et al. PI3K Pathway Inhibition Achieves Potent Antitumor Activity in Melanoma Brain Metastases In Vitro and In Vivo. Clin Cancer Res. 2016;23:5818–5828. doi: 10.1158/1078-0432.CCR-16-0064. [DOI] [PubMed] [Google Scholar]
- 62.Peddi PF Hurvitz SA. PI3K pathway inhibitors for the treatment of brain metastases with a focus on HER2+ breast cancer. J Neurooncol. 2014;1:7–13. doi: 10.1007/s11060-014-1369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;19:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 64.Garrett JT, Chakrabarty A, Arteaga CL. Will PI3K pathway inhibitors be effective as single agents in patients with cancer? Oncotarget. 2011;12:1314–1321. doi: 10.18632/oncotarget.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.LoPiccolo J, Blumenthal GM, Bernstein WB Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;1-2:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong D, Yamori T, Yamazaki K Dan S. In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA. Invest New Drugs. 2014;6:1134–1143. doi: 10.1007/s10637-014-0152-z. [DOI] [PubMed] [Google Scholar]
- 67.Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G, Merlano M Lo Nigro C. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Invest New Drugs. 2015;2:310–320. doi: 10.1007/s10637-015-0210-1. [DOI] [PubMed] [Google Scholar]
- 68.Bozec A, Ebran N, Radosevic-Robin N, Chamorey E, Yahia HB, Marcie S, Gautier M, Penault-Llorca F Milano G. Combination of phosphotidylinositol-3-kinase targeting with cetuximab and irradiation: A preclinical study on an orthotopic xenograft model of head and neck cancer. Head Neck. 2017;39:151–159. doi: 10.1002/hed.24560. [DOI] [PubMed] [Google Scholar]
- 69.Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L Powis G. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;7:763–772. [PubMed] [Google Scholar]
- 70.Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, Cline-Burkhardt V, Winquist E, Hausman D, Walker L Cohen RB. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol. 2015;3:556–561. doi: 10.1093/annonc/mdu574. [DOI] [PubMed] [Google Scholar]
- 71.Jimeno A, Bauman JE, Weissman C, Adkins D, Schnadig I, Beauregard P, Bowles DW, Spira A, Levy B, Seetharamu N, Hausman D, Walker L, Rudin CM, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;4:383–388. doi: 10.1016/j.oraloncology.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, Fracasso PR, Wilkie DP, Hentemann M, Wilhelm SM, Scott WJ, Mumberg D, Ziegelbauer K. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;11:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 73.Garlich JR, De P Dey N, Su JD, Peng X, Miller A, Murali R, Lu Y, Mills GB, Kundra V, Shu HK, Peng Q, Durden DL. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;1:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 74.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D Caravatti G. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;13:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 75.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;5:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 76.Razak ARA AM, Yen C, et al. Phase Ib/II study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN) J Clin Oncol. 2014;5(supp) [Google Scholar]
- 77.Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;7505:407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;7:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 79.Molife LR, Yan L, Vitfell-Rasmussen J, Zernhelt AM, Sullivan DM, Cassier PA, Chen E, Biondo A, Tetteh E, Siu LL, Patnaik A, Papadopoulos KP, de Bono JS, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014:1. doi: 10.1186/1756-8722-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hartford CM Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther. 2007;4:381–388. doi: 10.1038/sj.clpt.6100317. [DOI] [PubMed] [Google Scholar]
- 81.Nguyen SA, Walker D, Gillespie MB, Gutkind JS, Day TA. mTOR inhibitors and its role in the treatment of head and neck squamous cell carcinoma. Curr Treat Options Oncol. 2012;1:71–81. doi: 10.1007/s11864-011-0180-2. [DOI] [PubMed] [Google Scholar]
- 82.Geiger JL, Bauman JE, Gibson MK, Gooding WE, Varadarajan P, Kotsakis A, Martin D, Gutkind JS, Hedberg ML, Grandis JR, Argiris A. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck. 2016 doi: 10.1002/hed.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fury MG, Lee NY, Sherman E, Ho AL, Rao S, Heguy A, Shen R, Korte S, Lisa D, Ganly I, Patel S, Wong RJ, Shaha A, et al. A phase 1 study of everolimus + weekly cisplatin + intensity modulated radiation therapy in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2013;3:479–486. doi: 10.1016/j.ijrobp.2013.06.2043. [DOI] [PubMed] [Google Scholar]
- 84.Fury MG, Sherman E, Ho AL, Xiao H, Tsai F, Nwankwo O, Sima C, Heguy A, Katabi N, Haque S Pfister DG. A phase 1 study of everolimus plus docetaxel plus cisplatin as induction chemotherapy for patients with locally and/or regionally advanced head and neck cancer. Cancer. 2013;10:1823–1831. doi: 10.1002/cncr.27986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saba NF, Hurwitz SJ, Magliocca K, Kim S, Owonikoko TK, Harvey D, Ramalingam SS, Chen Z, Rogerio J, Mendel J, Kono SA, Lewis C, Chen AY, et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer. 2014;24:3940–3951. doi: 10.1002/cncr.28965. [DOI] [PubMed] [Google Scholar]
- 86.Massarelli E, Lin H, Ginsberg LE, Tran HT, Lee JJ, Canales JR, Williams MD, Blumenschein GR, Jr, Lu C, Heymach JV, Kies MS Papadimitrakopoulou V. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2015;7:1476–1480. doi: 10.1093/annonc/mdv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ekshyyan O, Mills GM, Lian T, Amirghahari N, Rong X, Lowery-Nordberg M, Abreo F, Veillon DM, Caldito G, Speicher L, Glass J Nathan CO. Pharmacodynamic evaluation of temsirolimus in patients with newly diagnosed advanced-stage head and neck squamous cell carcinoma. Head Neck. 2010;12:1619–1628. doi: 10.1002/hed.21374. [DOI] [PubMed] [Google Scholar]
- 88.Grunwald V, Keilholz U, Boehm A, Guntinas-Lichius O, Hennemann B, Schmoll HJ, Ivanyi P, Abbas M, Lehmann U, Koch A, Karch A, Zorner A, Gauler TC. TEMHEAD: a single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO) Ann Oncol. 2015;3:561–567. doi: 10.1093/annonc/mdu571. [DOI] [PubMed] [Google Scholar]
- 89.Liu X, Kambrick S, Fu S, Naing A, Subbiah V, Blumenschein GR, Glisson BS, Kies MS, Tsimberidou AM, Wheler JJ, Zinner RG, Hong DS, Kurzrock R, et al. Advanced malignancies treated with a combination of the VEGF inhibitor bevacizumab, anti-EGFR antibody cetuximab, and the mTOR inhibitor temsirolimus. Oncotarget. 2016;17:23227–23238. doi: 10.18632/oncotarget.7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bauman JE, Arias-Pulido H, Lee SJ, Fekrazad MH, Ozawa H, Fertig E, Howard J, Bishop J, Wang H, Olson GT, Spafford MJ, Jones DV, Chung CH. A phase II study of temsirolimus and erlotinib in patients with recurrent and/or metastatic, platinum-refractory head and neck squamous cell carcinoma. Oral Oncol. 2013;5:461–467. doi: 10.1016/j.oraloncology.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Piha-Paul SA, Munster PN, Hollebecque A, Argiles G, Dajani O, Cheng JD, Wang R, Swift A, Tosolini A, Gupta S. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer. 2015;14:1865–1873. doi: 10.1016/j.ejca.2015.06.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sandulache VC, Hamblin JS, Skinner HD, Kubik MW, Myers JN Zevallos JP. Association between metformin use and improved survival in patients with laryngeal squamous cell carcinoma. Head Neck. 2014;7:1039–1043. doi: 10.1002/hed.23409. [DOI] [PubMed] [Google Scholar]
- 93.Yen YC, Lin C, Lin SW, Lin YS, Weng SF. Effect of metformin on the incidence of head and neck cancer in diabetics. Head Neck. 2015;9:1268–1273. doi: 10.1002/hed.23743. [DOI] [PubMed] [Google Scholar]
- 94.Nair V, Sreevalsan S, Basha R, Abdelrahim M, Abudayyeh A, Rodrigues Hoffman A Safe S. Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer: role of specificity protein (Sp) transcription factors. J Biol Chem. 2014;40:27692–27701. doi: 10.1074/jbc.M114.592576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;1:290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;11:1093–1103. [PubMed] [Google Scholar]
- 97.Hilgard P, Klenner T, Stekar J, Nossner G, Kutscher B Engel J. D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur J Cancer. 1997;3:442–446. doi: 10.1016/s0959-8049(97)89020-x. [DOI] [PubMed] [Google Scholar]
- 98.Argiris A, Cohen E, Karrison T, Esparaz B, Mauer A, Ansari R, Wong S, Lu Y, Pins M, Dancey J Vokes E. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol Ther. 2006;7:766–770. doi: 10.4161/cbt.5.7.2874. [DOI] [PubMed] [Google Scholar]
- 99.Liu X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl Oncol. 2015;3:185–195. doi: 10.1016/j.tranon.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Strebhardt K, Becker S Matthess Y. Thoughts on the current assessment of Polo-like kinase inhibitor drug discovery. Expert Opin Drug Discov. 2015;1:1–8. doi: 10.1517/17460441.2015.962510. [DOI] [PubMed] [Google Scholar]
- 101.Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;3:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 102.Prasad A, Park IW, Allen H, Zhang X, Reddy MV, Boominathan R, Reddy EP, Groopman JE. Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells. Oncogene. 2009;12:1518–1528. doi: 10.1038/onc.2008.502. [DOI] [PubMed] [Google Scholar]
- 103.Anderson RT, Keysar SB, Bowles DW, Glogowska MJ, Astling DP, Morton JJ, Le P Umpierrez A, Eagles-Soukup J, Gan GN, Vogler BW, Sehrt D, Takimoto SM, et al. The dual pathway inhibitor rigosertib is effective in direct patient tumor xenografts of head and neck squamous cell carcinomas. Mol Cancer Ther. 2013;10:1994–2005. doi: 10.1158/1535-7163.MCT-13-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caino MC Altieri DC. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin Cancer Res. 2016;3:540–545. doi: 10.1158/1078-0432.CCR-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dienstmann R, Rodon J, Serra V Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;5:1021–1031. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 106.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;8:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 107.Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;1:112–123. doi: 10.1373/clinchem.2014.222679. [DOI] [PubMed] [Google Scholar]