

Abstract
Background
Novel fusion transcripts caused by chromosomal rearrangement are common factors in the development of cancers. We used massively parallel RNA-sequencing to identify new fusion transcripts in colon cancers.
Methods
RNA-seq and TopHat-Fusion were used to identify new fusion transcripts in colon cancers. We then investigated whether the novel fusion transcript NR5A2-KLHL29FT was transcribed from a genomic chromosomal rearrangement. Next, the expression of NR5A2-KLHL29FT was measured by quantitative RT-PCR in colon cancers and matched corresponding normal epithelia.
Results
We identified the fusion transcript NR5A2-KLHL29FT in normal and cancerous epithelia. While investigating this transcript, we unexpectedly found that it was due to an uncharacterized polymorphic germline insertion of NR5A2 sequence from chromosome 1 into the KLHL29 locus at chromosome 2, rather than a chromosomal rearrangement. This germline insertion, which occurred at a population frequency of 0.40, appeared to bear no relationship to cancer development. Moreover, NR5A2-KLHL29FT expression was validated in RNAs from samples with insertions of NR5A2 at the KLHL29 gene locus, but not from samples without this insertion. Notably, NR5A2-KLH29FT expression levels were significantly lower in colon cancers than in matched normal colonic epithelia (p = 0.029), suggesting potential participation of NR5A2-KLHL29FT in the origin or progression of this tumor type.
Conclusions
NR5A2-KLHL29FT was generated from a polymorphism insertion of NR5A2 sequence into the KLHL29 locus. NR5A2-KLHL29FT may influence the origin or progression of colon cancer. Moreover, researchers should be aware that similar fusion transcripts may occur due to trans-chromosomal insertions that are not correctly annotated in genome databases, especially with current assembly algorithms.
Keywords: TopHat-Fusion, KLHL29 insertion, fusion transcript, NR5A2-KLHL29FT, polymorphism
Introduction
Colorectal cancer (CRC) is the third most frequent cancer type worldwide1. Although the diagnosis and treatment of CRC have progressed rapidly, the overall 5-year survival rate of CRC remains low2. The underlying molecular mechanisms causing this high incidence and poor prognosis are not yet fully elucidated.
Cancer-related genetic alterations include amplification, deletion, duplication, and chromosomal rearrangement (viz., chromosome translocation, inversion and deletion). These aberrations can exert potent impacts on the development or progression of cancer3–4. Gene fusions are commonly caused by chromosomal rearrangement. Many gene fusions have been identified in leukemias, lymphomas and sarcomas, with the BCR-ABL fusion gene in chronic leukemia being a notable example5–6. In recent decades, gene fusions have also been discovered in epithelial cancers, including TMPRSS2-ERG in prostate cancer and EML4-ALK in non-small cell lung cancer7–9. For many years, gene fusions in epithelial cancers were not previously identified, possibly due to difficulties in detecting chromosomal aberrations in the chaotic karyotypic profiles typical of solid tumors10.
Next-generation whole genome sequencing can identify chromosomal rearrangements. However, this method is expensive, inefficient, and tedious for discovering oncogenic fusion gene events, since only a fraction of chromosomal rearrangements generate fusion mRNAs. In contrast, massively parallel RNA-sequencing (RNA-seq) can directly identify only those fusion genes that produce transcripts, providing an efficient approach to discover gene fusions10. Various algorithms have been used to detect gene fusions in RNA-seq datasets. TopHat-Fusion is a new algorithm developed to identify fusions11. This method is advantageous for finding novel splice variants and novel gene fusions because it detects individual and paired reads spanning a fusion point, and it does not rely on existing annotation.
Currently, several fusion transcripts have been shown to occur during colorectal carcinogenesis, including C2orf44-AKL, VTI1A-TCF7L2, NAV2-TCF7L1, EIF3E-RSPO2, PTPRK-RSPO3, and LACTB2-NCOA212–15. In the current study, we used TopHat-Fusion to identify novel fusion transcripts caused by chromosomal rearrangements in colon cancers. We found a previously undescribed fusion gene transcript resulting from an insertion at the Kelch-like family member 29 (KLHL29) gene locus. This fusion gene transcript was not tumor-specific and was expressed in all individuals with the genomic DNA KLHL29 insertion. Interestingly, downregulation of this fusion transcript was observed in colon cancers.
Methods
Tissue and plasma specimens
In this study, colon cancer samples and corresponding normal colon epithelia were obtained at colonoscopy, while gastric cancer samples were obtained at endoscopy. Plasma samples were collected from noncancer and cancer populations. Japanese gastric cancer samples were obtained at endoscopy in Japan. All patients and normal populations provided written informed consent under protocols approved by institutional review boards at The Johns Hopkins University School of Medicine, The University of Maryland, and Tohoku University in Japan. All specimens were stored in liquid nitrogen or at −80°C until DNA or RNA extraction.
DNA and RNA extraction
Genomic DNA was isolated from tissues and plasma samples using a DNeasy Tissue Kit (Qiagen) according to the manufacturer’s instructions. Total RNA was extracted from tissues using TRIzol reagent (Invitrogen). DNAs and RNAs were stored at −80°C before analysis.
Next-generation RNA sequencing
RNA-seq was performed according to existing protocols at the SKGCC core facility. TopHat Fusion was used to analyze RNA-seq data from colonic tissues to detect new fusion transcripts. TopHat-Fusion consists of two steps: (1) alignment and (2) identification of fusion transcripts. TopHat2 (version 2.0.10) was used to align RNA-seq reads to the human reference genome (GRCh37) [parameters: -p 12 –segment-mismatches 2 –fusion-min-dist 100000 –fusion-anchor-length 13 –max-intron-length 100000 -r 0 –mate-std-dev 80 –fusion-ignore-chromosomes MT] and tophat-fusion-post [parameters: -p 12 –num-fusion-reads 2 –num-fusion-pairs 0], which is included in the TopHat2 package, was used to identify fusion transcripts from the TopHat2 alignments. Further details about the TopHat-Fusion method are described previously11. The read length was 75bp. In addition, TopHat-Fusion was also used to detect the new fusion transcript in the ENCODE public RNA-seq data set from 135 experiments.
Genomic DNA polymerase chain reaction
DNAs were amplified using Platinum Taq DNA polymerase kit (Invitrogen). The 20-μl reaction volume contains 10ng DNA, 2μ 10xBuffer, 0.4μl 10mM d-NTP, 0.6 μl 50mM MgCl2, 0.2μl Taq Polymerase, 0.4μl Forward and Reverse primers (10μM) and water. The reaction conditions were 95°C for 30s, 35 cycles of 95°C for 30s, 60°C for 45s and 72°C for 1 min, followed by final elongation at 72°C for 10 min and stored at 4°C. The primers are shown in Supplementary Table S1. PCR products were loaded onto 1.5% agarose gels, stained with ethidium bromide, and visualized under UV Luminescent Image Analyzer (LAS-4000 Mini).
Quantitative real-time polymerase chain reaction (qRT-PCR)
During extraction of RNA from colon cancer and matched normal colon epithelia, DNAase (Invitrogen) was used to digest potential contaminating DNA. Then, random RT primers and SYBR Green Supermix (Bio-Rad Laboratory) were used to perform two-step qRT-PCR, as described previously16. The primers for the new fusion transcript and internal control (GAPDH) are described in Supplementary Table S1. Gene expression levels of the novel fusion transcript were normalized to GAPDH expression.
TOPO Cloning reaction and Sanger sequencing
PCR products were cloned into a TOPO vector and amplified using a TOPO Cloning TM Kit (Invitrogen), according to the manufacturer’s instructions. Specifically, PCR products were ligated to TOPO vector and transformed in E. coli DH5α by heat shock. Transformed cells were spread on LB agar plates with 50 μl/ml ampicillin and incubated overnight at 37°C. Transformants were selected and recombinant plasmids were extracted using a plasmid miniprep kit (Qiagen). Then, recombinant plasmids were sent to the GRCF core facility at The Johns Hopkins University School of Medicine for Sanger sequencing.
Statistical analyses
Relative expression levels of the fusion transcript between cancer and matched normal colonic epithelia groups were evaluated using the two-tailed Student’s t-test. The insertion rate in different groups was analyzed using Chi-squared testing. P < 0.05 was considered statistically significant. All statistical analyses were performed using IBM SPSS version 19.0.
Results
Next-generation RNA-seq and genomic analysis detect a novel fusion transcript (NR5A2-KLHL29FT) in normal colon epithelia
By using TopHat-Fusion alignment to analyze RNA-seq data from matched colon cancer and corresponding normal colon epithelia samples, only one normal colon epithelium (sample 1N) was found to contain fusion transcripts (FTs), while other samples did not detect any FTs due to either no new FTs or low read levels. In the normal colon epithelium (sample 1N) containing FTs, 11 candidate FTs were found, including 9 intra-chromosomal and 2 inter-chromosomal FTs (Supplementary Table S2). These candidate FTs were reviewed according to the UCSC genome database (hg38) and NCBI Blast. All intra-chromosomal FTs appeared to span short distances and were considered to represent unknown (long) introns or read-through transcription, thus not studied further. The two inter-chromosomal FTs occurred between chromosomes 5 and 12 and chromosomes 1 and 2, respectively. The chromosome 5–12 FT was deemed a false-positive because this FT involved both the SUDS3 (Suppressor of defective silencing 3 homolog) gene and a pseudogene of SUDS3, which have nearly identical sequences and could fool the aligner. Finally, the chromosome 1–2 FT was found to involve the sense strand of NR5A2 (Nuclear receptor subfamily 5, group A, member 2) gene on chromosome 1 and the antisense strand of KLHL29 gene on chromosome 2 (NR5A2-KLHL29FT) and was chosen for further study (Figure 1).
Figure 1. The deep sequence results that identify the new fusion transcript (NR5A2-KLHL29FT).
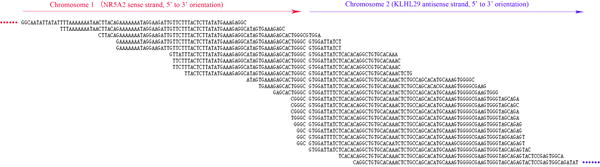
The read length was 75bp. NR5A2-KLHL29FT was a new antisense fusion transcript in the orientation of KLHL29 antisense strand.
Validation of a chromosomal alteration (a long insertion, rather than a chromosomal rearrangement) in clinical samples
Fusion transcripts are transcribed from genetic aberrations, specifically chromosomal rearrangements7. At first, we hypothesized that NR5A2-KLHL29FT was transcribed from chromosomal rearrangement between chromosome 1 and 2, with a breakpoint located in intron 4 of NR5A2 and intron 1 of KLHL29. The insertion site was only 175 bp away from KLHL29 exon 2, but much farther away from KLHL29 exon 1. A diagram of this hypothesized chromosomal rearrangement is displayed in Figure 2A. We designed four pairs of primers to amplify products spanning the breakpoint of this chromosomal rearrangement (i.e., the fusion point between NR5A2 and KLHL29; Figure 2A and Supplementary Table S1). However, only primer pairs 1 and 2 successfully amplified products covering the breakpoints, while primer pairs 3 and 4 failed to amplify any products using the same sample (1N) as DNA template. Identical results were found in several additional samples, including DNA from colon tumor samples and from noncancer patient white blood cells (Figures 2B/C). Thus, we hypothesized that DNA abnormalities could not have resulted from complete chromosome 1–2 rearrangement. The genomic DNA status of wild-type NR5A2 and wild-type KLHL29 was assessed using additional primers flanking the breakpoint in these two genes (Supplementary Table S1). As shown in Figure 2B, NR5A2 manifested only one wild-type genotype in all samples, while KLHL29 showed two alleles (one large band and one small band) and three possible genotypes (homozygous large band, heterozygous large-small bands, or homozygous small bands) in various patient samples. Every sample with products using primer pair 1/2 had at least one large allele of KLHL29. These results led to a new hypothesis as follows: KLHL29 had two types of alleles as a germline polymorphism: one, the wild-type KLHL29 allele, corresponding to genome references in the UCSC database (hg38); and two, a previously undescribed KLHL29 allele, resulting from an insertion of part of the NR5A2 gene (Figure 2D). Meanwhile, KLHL29 genotypes were predicted to be of three types: homozygous insertion, heterozygous insertion, or homozygous wild-type.
Figure 2. The identification of NR5A2 sequence insertion at KLHL29 gene locus.
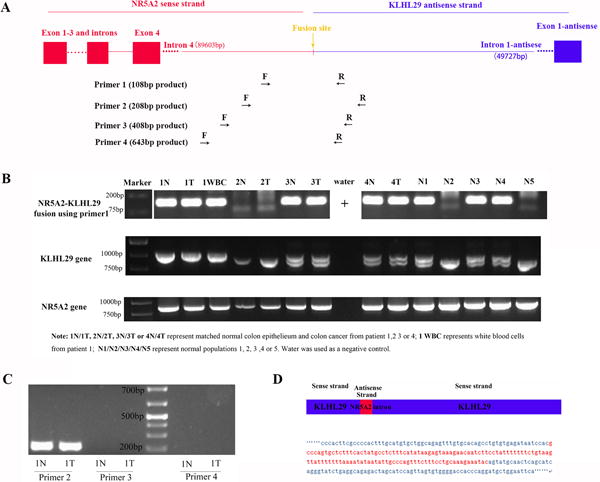
(A) We hypothesized that there was a chromosomal rearrangement between NR5A2 gene on chromosome 1 and KLHL29 gene on chromosome 2. Four primers were generated to validate the fusion gene. (B) The fusion gene using primer 1, KLHL29 gene and NR5A2 gene status were respectively studied in each sample. NR5A2 gene status was the same in all the samples. KLHL29 gene has three statuses: homozygous big band, heterozygous big band and homozygous small band. Both samples with homozygous or heterozygous big band showed the positive fusion bands. (C) Among primer 2, 3 and 4 amplifying the fusion gene, only primer 2 worked, while the other two primers did not amplify any bands, implying the fused NR5A2 sequence was not so long. (D) Based on B and C analysis, it is believed that the fusion gene is a polymorphism insertion instead of chromosomal rearrangement. By TOPO Cloning and Sanger sequence, we found that the insertion was the same in the different samples as shown here.
To discover the exact sequence of the partial NR5A2 insertion into the KLHL29 gene, we separated several large and small KLHL29 bands amplified from different patients, using the KLHL29 gene primers flanking the fusion site, and then performed TOPO cloning and Sanger sequencing. All large bands contained the same sequence, showing that a portion of NR5A2 (121bp) had been inserted into KLHL29 at the breakpoint described above (Figure 2D). Meanwhile, NR5A2 products from all samples were identical to the sequence at the NR5A2 locus in the UCSC database (data not shown). Thus, this insertion represented a copy of part of NR5A2 intron 4 (from chromosome 1) inserted into KLHL29 intron 1 on chromosome 2. Because sample 1N was homozygous for the insertion at the KLHL29 locus, we reasoned that a new NR5A2-KLHL29 fusion transcript (NR5A2-KLHL29FT) had been detected by TopHat Fusion analysis, having been transcribed from the KLHL29 gene locus by an insertion of NR5A2, rather than from chromosomal rearrangement. Using NCBI-blast, we did not find this insertion sequence at any other loci besides NR5A2 on chromosome 1, demonstrating that it is a non-repetitive insertion from the NR5A2 gene to the KLHL29 locus.
The insertion is not related to cancer prevalence, and thus it represents a common polymorphism at the KLHL29 gene locus
According to the above results, the insertion was not a rare event at the KLHL29 gene locus in patients with cancers or in noncancer populations. Next, a larger cohort of samples, including 67 colon cancer patients, 45 gastric cancer patients, and 101 noncancer subjects, was evaluated to assess the prevalence of the insertion and its possible relevance to cancer risk. The polymorphism information content (PIC) of this polymorphism was 0.321, and heterozygosity at this locus was 0.352. As described in Table 1, neither the frequency nor carriage rate of the insertion showed any statistically significant difference between colon cancer, gastric cancer, and noncancer populations (P > 0.05). This result implied that KLHL29 insertion is a common polymorphism that is not associated with cancer occurrence. However, both the carrier rate (66.7%) and frequency rate (40.8%) of the insertion in Japanese gastric cancers were significantly higher than in American gastric cancers (43.3% and 23.1%) or American noncancer populations (41.6% and 22.8%) (p < 0.01). This phenomenon suggests that the insertion is more frequent in Asian populations, or possibly, that this insertion originally arose in ancient Asian populations and conferred an evolutionary advantage of some sort. However, our hypothesis needs to be verified by additional multi-institutional studies to assess this polymorphic insertion in future.
Table 1.
Polymorphism insertion frequency and carrier rate at the KLHL29 locus in control populations and in cancer patients.
| Samples and number | Homozygous KLHL29 insertions | Heterozygous KLHL29 insertions | Homozygous wild-type KLHL29 | Insertion carrier rate | P valuea | Insertion frequency | P valueb |
|---|---|---|---|---|---|---|---|
| USA noncancer controls (101) | 4 | 38 | 59 | 41.6% | / | 22.8% | / |
| USA colon cancers (67) | 2 | 27 | 38 | 43.3% | 0.827 | 23.1% | 0.938 |
| USA gastric cancers (45) | 2 | 13 | 30 | 33.3% | 0.345 | 18.9% | 0.456 |
| Japanese gastric cancers (60) | 9 | 31 | 20 | 66.7% | 0.002* 0.001*c |
40.8% | 0.001* 0.001*d |
p-value of difference in insertion carrier rate between control populations and cancer patients;
p-value of difference in insertion frequency between normal populations and cancer patients;
p-value of difference in insertion carrier rate between American gastric cancer and Japanese gastric cancer patients;
p-value of difference in insertion frequency between American gastric cancer and Japanese gastric cancer patients;
p-value less than 0.05.
Validation of NR5A2-KLHL29FT on RNA levels in clinical samples
We then proceeded to validate the existence of NR5A2-KLHL29FT in sample 1N (normal colon epithelium). After eliminating potential residual DNA using DNase (Invitrogen) in RNA, we performed RT-PCR using primer 1 (Supplementary Table S1) and ran the products on gels. A clear band is shown in sample 1N. Meanwhile, we also detected NR5A2-KLHL29FT in colon cancer (1T) matched to sample 1N (Figure 3A). Based on the presence of NR5A2-KLHL29FT in both tumor and normal cDNA, this fusion transcript was germline rather than tumor-specific. Interestingly, when qRT-PCR was carried out to measure the expression of NR5A2-KLHL29FT, the expression level of NR5A2-KLHL29FT in colon cancer was lower than in matched normal colon epithelium (2−7.4186/1, or 0.058/1; Supplementary Table S3a). Subsequently, matched colon cancer-normal colon epithelial pairs from 21 additional colon cancer patients were enrolled to measure NR5A2-KLHL29FT expression by qRT-PCR. Only 14 matched cancer-normal colon epithelia were clearly detected with NR5A2-KLHL29FT by a single peak on a melting curve, and confirmed by clear bands on gels (Figure 3B, Supplementary Table S3a/b). Notably, all 14 of these matched pairs were KLHL29 insertion-positive (homozygous or heterozygous for the insertion allele), while the remaining 7 pairs were homozygous wild-type. Based on these results, only samples containing the insertion, either homozygous or heterozygous, could express NR5A2-KLHL29FT. In addition, in all 15 matched colon cancer-normal colon epithelia with expression of NR5A2-KLHL29FT, comprising sample 1N-1T pair and the additional 14 matched colon cancer-normal pairs, the expression level of NR5A2-KLHL29FT was decreased relative to normal colon epithelia in the majority (11/15, average fold-change 0.23, p-value = 0.029; Figure 3C and Supplementary Table S3a). Thus, we concluded that NR5A2-KLHL29FT is only expressed in subjects with the KLHL29 insertion. We also speculated that when NR5A2-KLHL29FT is downregulated, colon carcinogenesis may be supported. However, this hypothesis requires further experimental evidence.
Figure 3. The validation of new fusion transcript (NR5A2-KLHL29FT).
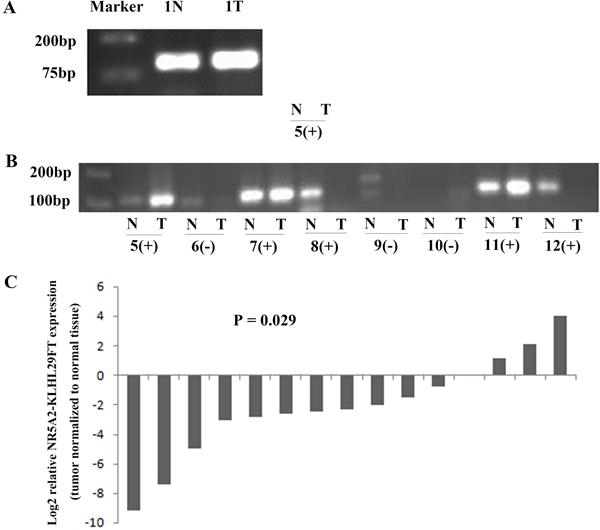
(A) NR5A2-KLHL29FT was validated in sample 1N (normal colon epithelium) and 1T (colon cancer). (B) NR5A2-KLHL29FT was also validated in other samples with KLHL29 insertion. + represents samples with insertion at KLHL29 locus, - represents samples without insertion at KLHL29 locus. (C) The expression level of NR5A2-KLHL29FT was significantly lower in colon cancers than matched normal colon epithelium.
Analysis of RNA-seq data from a public database using TopHat Fusion alignment to detect NR5A2-KLHL29FT
To confirm the prevalence of NR5A2-KLHL29FT in the general population, we re-analyzed ENCODE public RNA-seq data set from 135 experiments using TopHat-Fusion. Interestingly, the NR5A2-KLHL29 fusion was detected in only 2 of 135 cell lines, including ENCSR000AFK (described as thyroid gland/tissue/fetal) and ENCSR908ZAS (described as hepatocyte/in vitro differentiated cells/embryonic). The fusion point was exactly the same as what was detected in our first sample (sample 1N). Based on this analysis, the fusion transcript NR5A2-KLHL29FT does occur, but is not a common event in the ENCODE database. However, as described above, in our cohort of matched colon cancer-normal pairs, NR5A2-KLHL29FT was common and could be detected in all matched normal tissues containing the insertion. To explain this disparity, we re-reviewed expression levels of NR5A2-KLHL29FT by qRT-PCR in our cohort (Supplementary Table S3a). We hypothesized that only samples with relative high expression levels of NR5A2-KLHL29FT by qRT-PCR could be identified by TopHat Fusion, while samples with low expression levels could not.
Discussion
RNA-seq has been used widely to identify novel fusion transcripts in cancers17–18. We originally sought to detect novel fusion transcripts due to chromosomal rearrangements in colon cancers. To our surprise, a novel fusion transcript NR5A2-KLHL29FT was identified in a normal colon specimen (1N). In the process of validating this abnormality at the DNA level, NR5A2-KLHL29FT was unexpectedly shown to come from a polymorphic insertion at KLHL29 locus and to be expressed in both cancer and normal samples. These unexpected results imply that polymorphic insertion events at the DNA level represent a possible mechanism of novel fusion transcripts different from chromosomal rearrangement, being especially likely when novel fusion transcripts are identified in normal tissues by RNA-seq. We speculate that both heterogenous nuclear RNA (hnRNA) and mature fully processed mRNA could have been available for the construction of the RNA-seq library, and that the reads shown in Figure 1 were derived from both of these classes of molecules. In such cases, primers amplifying longer products that span both ends of an insertion junction should be generated to verify its origin.
We also asked how the non-repetitive sequence of NR5A2 gene could have become inserted into the KLHL29 gene. Retrotransposons, as transposable DNA elements, have the ability to duplicate themselves onto other regions of the genome. They mobilize in a ‘copy and paste’ manner involving reverse transcription of an RNA intermediate and insertion of its cDNA copy into a new locus19. Short interspersed elements (SINE, mainly Alu), long interspersed elements (LINE-1 or L1), and processed pseudogenes are three types of retrotransposons. However, both L1 and SINE are multiply-repeated mobile elements in the genome, while processed pseudogenes are characterized by a lack of introns. Thus, these three retrotransposons were unlikely mechanisms underlying the non-repetitive segmental duplication found in the current study. L1-mediated 3′ transduction can associate 3′ flanking DNA sequences as read-through transcripts and mobilize non-repetitive DNA sequences to a new genomic region20–21. These non-repetitive segmental duplications have some typical characteristics, including a poly-A tract, target site duplication flanking the insertion, and an L1 endonuclease site (5′-TTTT/AA-3′). Nevertheless, our insertion at KLHL29 gene locus did not exhibit these hallmarks of L1-mediated 3′ transduction, thus excluding the possibility of L1-mediated insertion. Recently, Onozawa experimentally confirmed that DNA double-strand breaks (DSBs) can be repaired via reverse transcription of an RNA template and insertion of cDNA, and he clinically validated the prevalence of polymorphic insertions related to DSBs using public databases22. Onozawa classified unknown insertions lacking common hallmarks of L1-mediated events as class 2 templated sequence insertion polymorphisms (TSIPs), which were considered microhomology-mediated annealing of mRNA, reverse transcription, and healing of DNA DSBs induced by physiologic or environment DNA damage. The insertion sequences at KLHL29 locus in our study have some microhomology, such as 5′ flanking sequence (AATCCAC at the KLHL29 sense strand and AATCCAA at the NR5A2 sense strand with opposite direction) and 3′ flanking sequence (A in both genes). Thus, we hypothesize that our insertion may be a class 2 TSIP, caused by DSB repair mechanism, including reverse transcription of a partial immature NR5A2 RNA and insertion of the corresponding cDNA into the KLHL29 locus, with KLHL29 intron 1 experiencing DNA damage due to unknown factors.
KLHL29 gene belongs to one member of the KLHL (Kelch-like) gene superfamily23. Four KLHL family members have been reported to be associated with cancers, including KLHL6 in chronic lymphocytic leukemia, KLHL19 in gallbladder and lung cancer, KLHL20 in prostate cancer, and KLHL37 in brain tumors24–28. There are no published papers about the KLHL29 gene, nor of the function of this polymorphic insertion at KLHL29 locus. Polymorphic insertions are associated with some diseases29. One example is the polymorphic insertion of varying numbers of an 86-bp tandem repeat in interleukin-1 receptor antagonist (IL-1RA) intron 2. By comparing differences in IL-1RA frequency in various groups, allele 2 of the IL-1RA gene (IL1RN*2) has been shown to be associated with ulcerative colitis and Crohn’s disease, systemic lupus erythematosus (SLE), and other illnesses30–32. Thus, we studied whether KLHL29 insertion polymorphism was associated with colon cancer. However, we did not find any difference in frequency or carrier rate of the insertion between noncancer and colon cancer cohorts. In addition, the insertion polymorphism was not correlated with gastric cancer occurrence. Based on these results, KLHL29 insertion polymorphism may represent a normal polymorphic event that occurred in the process of human evolution, unrelated to carcinogenesis. Nevertheless, this conclusion needs to be confirmed by analyzing a larger number of samples from other countries.
Most genomic insertions of DNA fragments accumulate sequence substitutions and are not ultimately activated. Nevertheless, some insertions, especially repetitive transposable elements, develop new transcripts due to interaction with proximal genes, providing a transcription start site, polyadenylation site, or alternative splice site34. Novel transcripts are also derived from non-repetitive intronic genomic insertions by similar mechanisms to those shown for repeats33–35. In our study, since the insertion was presumably copied from intron 4 of NR5A2 gene to intron 1 of KLHL29, a new KLHL29 antisense transcript in the same orientation to the NR5A2 sense strand was identified by RNA-seq data and validated in various clinical samples. Perhaps a polyadenylation signal in the inserted NR5A2 sequence contributed to the generation of this novel alternative transcript (NR5A2-KLHL29FT). The new transcript was only transcribed from KLHL29 genes containing insertion sequence. Interestingly, since expression of NR5A2-KLHL29FT is lower in colon cancers than in matched normal tissues, we hypothesize that NR5A2-KLHL29FT may participate in the origin or progression of colon cancer. However, the precise mechanism and experimental validation require further study. Furthermore, because the orientation of the NR5A2 insertion is in the antisense direction relative to the sense direction of the KLHL29 gene, we reason that if the KLHL29 intron 1 is spliced out in the proper expected manner, this NR5A2 insertion will be removed. In this case, no novel fusion protein will be produced, since the alien sequence will have been removed during RNA splicing. However, since the insertion is located only 175 base pairs away from exon 2, it is conceivable that it may exert an effect on intron-exon splicing at this junction. Additional possible but unproven effects include the presence of cryptic DNA sequences containing an alternate promoter, a transcriptional enhancer element, or a motif affecting mRNA stability or molecular half-life. Thus, although there is no proven mechanism for this 121-base DNA insertion to alter the expression or function of the KLHL29 protein, this possibility still cannot be ruled out. Therefore, this insertion may still be functionally involved in the development or progression of colon cancer. Notably, many other details remain unanswered, including the full length of the new transcript, the exact genomic DNA splice site, and the exact mechanism leading to generation of the new transcript. We will continue to study these areas to clearly elucidate the function of the new transcript (NR5A2-KLHL29FT) in future studies. Meanwhile, trans-chromosomal insertions similar to the insertion at the KLHL29 locus are not fully recognized maybe because they always fool the aligners to be recognized as chromosome rearrangement in whole-genome sequencing. Thus, during clinical validation of novel fusion transcripts identified by RNA-seq, we should realize that similar fusion transcripts may be transcribed from trans-chromosomal insertions those are not correctly annotated in current genome databases as an alternative to chromosome rearrangement.
In conclusion, a novel transcript (NR5A2-KLHL29FT) was identified by RNA-seq and validated in colonic tissue samples. NR5A2-KLHL29FT is transcribed from a polymorphic insertion at the KLHL29 locus, rather than a chromosomal rearrangement between chromosomes 1 and 2. This polymorphic insertion does not appear to be related to colon cancer risk. However, primary data suggest that NR5A2-KLHL29FT may participate in the origin or progression of colon cancer. Further studies are needed to elucidate the precise function of NR5A2-KLHL29FT in colorectal and other cancers.
Supplementary Material
Two concise sentences.
A novel fusion transcript identified by RNA-seq, NR5A2-KLHL29FT, was generated from a polymorphism insertion at KLHL29 locus instead of chromosome rearrangement. The expression of NR5A2-KLHL29FT is lower in colon cancers than in matched normal tissues, suggesting potential participation of NR5A2-KLHL29FT in the origin or progression of colon cancer.
Acknowledgments
Funding sources: This work was supported by the National Institutes of Health [grant numbers CA133012, DK087454, CA190040 and CA200483].
Footnotes
Conflict of interests: None
Author contributions: S.J.M. and S.L.S. contributed to study concept and design; Z.S., X.K., Y.C., J.H.S. and B.H. performed clinical experiments; S.L.S., D.K., and V.A. contributed to the analysis of TopHat Fusion data; J.M.A. contributed to technical aspects of this study; S.I. collected clinical samples; Z.S., H.T., and S.J.M. performed statistical analysis. All authors contributed to the analysis and interpretation of data and drafting.
Indication of whether the paper was invited: No
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Huynh R, Abraham I, Kim E, Kumar RR. Number of lymph nodes examined and its impact on colorectal cancer staging. Am Surg. 2006;72:902–905. [PubMed] [Google Scholar]
- 3.Luo JH, Liu S, Zuo ZH, Chen R, Tseng GC, Yu YP. Discovery and Classification of Fusion Transcripts in Prostate Cancer and Normal Prostate Tissue. Am J Pathol. 2015;185:1834–1845. doi: 10.1016/j.ajpath.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Klein A, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 6.Maher CA, Palanisamy N, Brenner JC, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci USA. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 9.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 10.Edgren H, Murumagi A, Kangaspeska S, et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011;12:R6. doi: 10.1186/gb-2011-12-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biology. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bass AJ, Lawrence MS, Brace LE, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W, Bhagat TD, Yang X, et al. Hypomethylation of noncoding DNA regions and overexpression of the long noncoding RNA, AFAP1-AS1, in Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 2013;144:956–966. doi: 10.1053/j.gastro.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger MF, Levin JZ, Vijayendran K, et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20:413–427. doi: 10.1101/gr.103697.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salzman J, Marinelli RJ, Wang PL, et al. ESRRA-C11orf20 is a recurrent gene fusion in serous ovarian carcinoma. PLoS Biol. 2011;9:e1001156. doi: 10.1371/journal.pbio.1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 20.Ejima Y, Yang L. Trans mobilization of genomic DNA as a mechanism for retrotransposon-mediated exon shuffling. Hum Mol Genet. 2003;12:1321–1328. doi: 10.1093/hmg/ddg138. [DOI] [PubMed] [Google Scholar]
- 21.Solyom S, Ewing AD, Hancks DC, et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Hum Mutat. 2012;33:369–371. doi: 10.1002/humu.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onozawa M, Zhang Z, Kim YJ, et al. Repair of DNA double-strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc Natl Acad Sci USA. 2014;111:7729–7734. doi: 10.1073/pnas.1321889111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS. Update on the Kelch-like (KLHL) gene family. Hum Genomics. 2013;7:13. doi: 10.1186/1479-7364-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroll J, Shi X, Caprioli A, et al. The BTB-kelch protein KLHL6 is involved in B-lymphocyte antigen receptor signaling and germinal center formation. Mol Cell Biol. 2005;25:8531–8540. doi: 10.1128/MCB.25.19.8531-8540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 26.Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 28.Liang XQ, Avraham HK, Jiang S, Avraham S. Genetic alterations of the NRP/B gene are associated with human brain tumors. Oncogene. 2004;23:5890–5900. doi: 10.1038/sj.onc.1207776. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Huang S, Chen W, Song Z, Cai S. NFKB1 -94 insertion/deletion polymorphism and cancer risk: a meta-analysis. Tumour Biol. 2014;35:5181–5187. doi: 10.1007/s13277-014-1672-z. [DOI] [PubMed] [Google Scholar]
- 30.Mansfield JC, Holden H, Tarlow JK, et al. Novel genetic association between ulcerative colitis and the anti-inflammatory cytokine interleukin-1 receptor antagonist. Gastroenterology. 1994;106:637–642. doi: 10.1016/0016-5085(94)90696-3. [DOI] [PubMed] [Google Scholar]
- 31.Tountas NA, Casini-Raggi V, Yang H, et al. Functional and ethnic association of allele 2 of the interleukin-1 receptor antagonist gene in ulcerative colitis. Gastroenterology. 1999;117:806–813. doi: 10.1016/s0016-5085(99)70338-0. [DOI] [PubMed] [Google Scholar]
- 32.Blakemore AI, Tarlow JK, Cork MJ, Gordon C, Emery P, Duff GW. Interleukin-1 receptor antagonist gene polymorphism as a disease severity factor in systemic lupus erythematosus. Arthritis Rheum. 1994;37:1380–1385. doi: 10.1002/art.1780370917. [DOI] [PubMed] [Google Scholar]
- 33.Kim DS, Hahn Y. Identification of human-specific transcript variants induced by DNA insertions in the human genome. Bioinformatics. 2011;27:14–21. doi: 10.1093/bioinformatics/btq612. [DOI] [PubMed] [Google Scholar]
- 34.Alekseyenko AV, Kim N, Lee CJ. Global analysis of exon creation versus loss and the role of alternative splicing in 17 vertebrate genomes. RNA. 2007;13:661–670. doi: 10.1261/rna.325107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–1067. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.