

Abstract
Interest in the human microbiome is at an all time high. The number of human microbiome studies is growing exponentially, as are reported associations between microbial communities and disease. However, we have not been able to translate the ever-growing amount of microbiome sequence data into better health. To do this, we need a practical means of transforming a disease-associated microbiome into a health-associated microbiome. This will require a framework that can be used to generate predictions about community dynamics within the microbiome under different conditions, predictions that can be tested and validated. In this review, using the gut microbiome to illustrate, we describe two classes of model that are currently being used to generate predictions about microbial community dynamics: ecological models and metabolic models. We outline the strengths and weaknesses of each approach and discuss the insights into the gut microbiome that have emerged from modeling thus far. We then argue that the two approaches can be combined to yield a community metabolic model, which will supply the framework needed to move from high-throughput omics data to testable predictions about how prebiotic, probiotic, and nutritional interventions affect the microbiome. We are confident that with a suitable model, researchers and clinicians will be able to harness the stream of sequence data and begin designing strategies to make targeted alterations to the microbiome and improve health.
Keywords: microbiome, ecological models, metabolic models, community metabolic models
INTRODUCTION
Enthusiasm for understanding the human microbiome’s effect on health is at an all-time high and appears to be growing. The microbiome has been featured in high-profile journals, such as Science and Nature; on the cover of the New York Times Magazine; and in bestsellers such as Gut [1] and The Microbiome Solution [2]. Meanwhile, the amount of microbiome data is exploding; a Google Scholar search reveals that 1,540 articles about the microbiome were published in 2005, whereas roughly 20,000 were published in 2015. Clinicians have begun to tap the potential of microbiome-based therapies: fecal transplant is now a well-established therapy for Clostridium difficile infection [3]. Unfortunately, despite this singular advance, the intense interest from both the research community and the public, and reams of data, we still lack an effective and palatable approach to microbiome manipulation. To transform the microbiome, and thus improve health, we require a theoretical framework that can be used to generate predictions about community dynamics within the microbiome under different conditions, predictions that can then be tested and validated. Until we possess such a framework, the promise of the microbiome and health interventions such as prebiotics, probiotics, and nutritional changes will go unrealized.
We already have a good idea about what the ideal framework for analyzing microbiome data and predicting community dynamics that promote health, looks like.. First, it should focus on microbe-microbe and microbe-host interactions. Complex behavior in other systems, such as the coordination of pacemaker cells in the heart or the synchronized flashing of fireflies, has been unraveled by focusing on such small-scale interactions [4]. Second, it should include enough detail on these interactions to yield accurate predictions, but not so much that researchers find it difficult to discern which variables are important drivers of community dynamics. Third, it should facilitate analysis of the high-throughput data emerging from microbiome studies.
Currently, two main classes of model are being used to study community dynamics within the microbiome: ecological models and metabolic models. In this review, we explore both approaches, including their strengths and weaknesses, focusing on the gut. We conclude by arguing that the two approaches can be combined to yield a community metabolic model that meets our criteria for the ideal analytic framework for yielding accurate and helpful predictions about microbial dynamics. Our goal is to demonstrate how models can be used to develop interventions that involve targeted alterations to the gut microbiome.
HOW METABOLISM SHAPES THE HUMAN GUT MICROBIOME: A PRIMER
The diverse microbes within the gastrointestinal tract encompass an equally diverse set of metabolic phenotypes [5], resulting in a tangled web of interdependence between different species within the community (Figure 1). Host epithelial cells excrete high levels of mucins: high-molecular-weight, heavily glycosylated proteins (glycans) that carry out a number of important physiologic functions, including lubrication, cell signaling, and mediation of pathogen binding [6,7]. Several well-studied microbial genera, including Akkermansia, Bacteroides, Bifidobacterium, Clostridium, Helicobacter, Prevotella, and Ruminococcus, produce glycosidases (e.g. fucosidase, sialidase, sulfatase) and proteases that degrade mucin into monosaccharides, oligosaccharides, sulfates, and amino acids. These metabolites, now in a form that more microbes can use, help determine the types of interactions that occur between the various species within the gut [7,8]. Microbes dependent on glycosidase-producing bacteria include groups as diverse as macromolecule degraders [9–11], nitrogen fixers [12], lactic acid bacteria [13], and various hydrogenotrophs [14], such as sulfate-reducing bacterium, methanogens, and acetogens. Thus, metabolism underlies many of the key interactions that connect the varied members of the gut microbiome. (Note: For a more detailed description of microbial metabolic relationships in the gut, we encourage the interested reader to peruse some of the many excellent reviews on this subject [15–18]).
Figure 1. The gut microbiome consists of a tangled web of interdependent microbial species.

Host epithelial cells produce mucin (top), which is metabolized by mucin-degrading microbes into molecules that other species can use, such as the short-chain fatty acid acetate. Other microbes produce molecules such as butyrate, formate, lactate, and methane, all of which are essential to the metabolism of certain species. Many species are both dependent on other species for their metabolic needs and depended on by other species; for instance, acetogens (bottom, middle) depend on butyric acid producers for lactate, and produce acetate used by methanogens.
Oxygen is an important determinant of microbial spatial structure and interactions within the gut—indeed, the steepest oxygen gradient in the body occurs in the intestine, with levels highest along the host cells lining the intestine and declining rapidly further into the gut [19,20]. Research on mice supports the hypothesis that the microbiome actually contributes to the oxygen gradient, with aerotolerant species colonizing the intestine after birth, creating a less oxygenated space in the lumen for less aerotolerant species[19]. In the mature gut, different species continue to position themselves along this radial oxygen gradient in the same manner, with bacteria capable of consuming oxygen concentrated near the oxygen-rich gut mucosa[21]. Importantly, research in mice shows that altering the host’s oxygen levels can modify the composition of the gut microbiota[21]; in humans, it has been proposed that diet may affect oxygenation levels in the gut by modulating the availability of antioxidants[22].
Many community traits cannot be explained as a sum of individual contributions; these so-called collective effects [23] are the result of interactions between community members. Because metabolism determines the nature of so many microbe-microbe and host-microbe interactions within the gut microbiome, any reasonable framework seeking to predict community traits must therefore take it into account. The importance of collective effects is evidenced by the metabolic behavior of microbes growing together. For example, work by Chiu et al. [24] suggests that emergent biosynthetic capacity is relatively common; that is, when grown in two-species culture, some bacteria are predicted to produce metabolites that neither species produces when grown individually. In another example, when exposed to one another, the microbes Eubacterium rectale and Bacteroides thetaiotaomicron alter their nutrient utilization in the gut [25]. Specifically, B. thetaiotaomicron signals the host to produce mucins that only it can use and upregulates expression of the genes needed to metabolize them. E. rectale, in turn, decreases production of mucin-degrading enzymes, instead focusing on other nutrients by increasing expression of select amino acid and sugar transporters. Given this evidence that collective effects are common within the gut, it is important to be able to model how metabolic interactions between multiple species determine the community traits of the microbiome, which may in turn affect health.
ECOLOGICAL MODELS OF THE GUT MICROBIOME
Theoretical ecologists have long studied the effects of species interactions on community dynamics, and basic ecological models can be applied to the study of the microbiome [26]. For example, the Lotka-Volterra model, originally applied to predator-prey dynamics [27], can easily be co-opted to investigate the dynamics of complex communities; variants of the basic model address spatial heterogeneity [28], lags in reproduction [29], and food-web dynamics [30]. Recently, Coyte et al. [31] published a generalized, unforced Lotka-Volterra model designed to analyze the microbiome. Essentially, this model uses between-species interaction terms to predict the rise and fall of different species within a community. This type of model is well suited for exploring general trends in the way that between-species interactions affect community dynamics. For example, Coyte et al. have already used their model to make the unintuitive prediction that cooperative networks of microbes can be unstable. One might expect that cooperative metabolism among microbes could promote stability, but their simulations indicate that dependence between microbes can reduce community stability because a decrease in the abundance of one species can pull other species down with it. This interesting and testable prediction highlights the contributions that an ecological model can make to the study of the microbiome.
Whereas the Coyte et al. model is very general and can be applied to any microbiome, Lotka-Volterra–type models have also been designed to investigate more specific scenarios. For example, Stein et al. [32] recently used data on community membership at different time points in C. difficile-challenged mice to derive the interaction terms in a modified Lotka-Volterra model. By fitting the model with empirical data, they were able to simulate the specific conditions present in their system of interest: the gut microbiomes of mice that had been pre-treated with antibiotics vs. those that had not, post-pathogen exposure. Their results indicated that the gut microbiome is intrinsically stable, but antibiotic perturbation dramatically increases susceptibility to C. difficile infection. Furthermore, this analysis implicated a subnetwork of bacterial groups in protection against the pathogen. This example demonstrates the ability of basic ecological models to provide insight into both broad community dynamics and, when paired with empirical data, the roles of specific species in creating those dynamics.
Although ecological models are very helpful for elucidating broad trends in community dynamics, they have several important drawbacks. One drawback is that the interaction terms are purely descriptive and thus provide little practical insight into the mechanisms underlying the trends observed. In the models discussed above, a single interaction term exists for each two-species pairing; the values for these terms can be inferred by observing changes in empirical populations over time and working backward, as described for the Stein et al. [32] study. However, the interaction term is essentially a black box; we know little about the molecular mechanisms—such as the metabolic dependence of one microbe on another—that drive the interaction between two-species. Moreover, because these interaction terms are often estimated based on correlations between population numbers, they are prone to error, as correlations do not necessarily imply that two species are interacting [33]. Without detailed knowledge of the interactions taking place in a community, it is difficult to translate a model’s findings into designer probiotics, prebiotics, or nutritional interventions. Another drawback is that a model’s parameters must be estimated anew for each community studied, which also limits this approach’s practical utility. Suppose, for example, that we want to determine the effects of a nutritional intervention—adding starch to a diet—on the gut microbiome. To investigate this problem using an ecological model, we would have to give subjects in the study starch, document changes in their microbiome over time, and use correlation coefficients between species to infer interaction parameters specific to this dietary intervention (inferred from correlation coefficients between species). Only then could we begin our simulations. Moreover, should we want to study a different nutritional component, such as fat, we would need to repeat this process over again. Thus, a great deal of time, effort, and money is required to generate predictions using this type of model.
METABOLIC MODELS OF THE GUT MICROBIOME
Unlike ecological models, metabolic models contain a great deal of mechanistic detail. Based largely on biochemistry, they include information on the metabolic reactions that represent the basis for species-species interactions [34]. Whereas ecological models infer the strength and type of species-species interactions from time series data, metabolic modeling starts with sequence data. Sequence data can be converted into metabolic networks that are then used to predict the interactions between species present in the gut, and thus community dynamics. Here, we give an overview of how metabolic models are constructed and describe the two most popular approaches for analyzing the gut microbiome: topological and constraint-based models.
RECONSTRUCTING METABOLIC NETWORKS
Genome-scale models (GEMs) are the basis of every type of metabolic model. Any organism’s metabolism can be represented by a GEM, which is the mathematical format of an organism’s genome-scale metabolic network reconstruction [35]. GEMs provide a mechanistic framework for integrating data from multi-omics studies, providing the structure onto which proteomic and transcriptomic data can be mapped—the so-called “context for content” [36]. Essentially, they can be used to unify genomic, transcriptomic, proteomic, metabolomic, and biochemical kinetic information relevant to metabolic models [37–40]. (See, for example, the recent study by Noecker et al. [41], which combined taxonomic, genomic, and metabolic data to determine whether shifts in the vaginal microbiome explain shifts in the vaginal metabolome.) Because GEMs are the foundation of metabolic modeling, we give a concise explanation of how they are created here (Figure 2; but for a more extensive protocol see [42]).
Figure 2. Creating a genome-scale model (GEM) of metabolism involves four sequential steps.
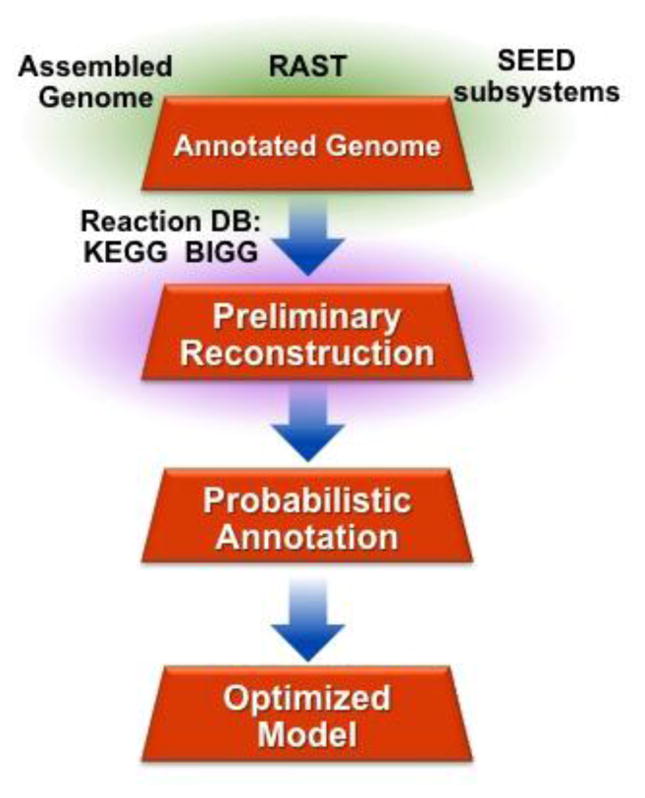
First, an annotated genome is used to produce a preliminary reconstruction of an organism’s metabolism. Typically, a large number of genes in the genome will not be included in this reconstruction, as their function is unknown. To solve this problem, probabilistic annotation, or gap filling, is performed. In this process, gene products are added to an organism’s metabolic network on the basis of a homology-based likelihood maximization algorithm. This results in a final, optimized GEM. The process shown here is based on the ProbModelSEED pipeline [51], but the same essential process is used to create all GEMs.
First, an annotated genome of the organism of interest is required. Databases such as EcoCyc [43,44] and PATRIC [45] contain annotated genomes for a large variety of organisms. However, tools such as RAST [46] and the Prokaryotic Genome Annotation Pipeline [47] allow users to annotate the genomes of organisms that are not present in these databases. Gene annotations, in the form of identified proteins, are then matched to the biochemical reactions that ultimately make up the metabolic network of the organism of interest. Gene-protein-reaction matches are made with databases such as KEGG [48], BRENDA [49], and SEED [50], which contain collections of metabolic reactions shown to exist in a variety of organisms. At this point, the metabolic network of the organism of interest is largely populated with reactions whose existence is supported by genomic information. However, because many genes in a genome have unknown function, further curation is required to complete the model, so that most of the organism’s known metabolic reactions are included. This process is known as gap filling. Finally, metabolic reaction information is converted into a mathematical representation, the GEM, that allows users to investigate the qualitative and quantitative features of an organism’s metabolism.
Reconstructing GEMs once involved a laborious manual process that took at least 6 months per microbial species [42], but today automatic metabolic reconstruction tools such as the RAVEN Toolbox [52], Pathway Tools [53], MicrobesFlux [54], merlin [55], and ModelSEED [56] have significantly reduced the time and effort required. ModelSEED, the most widely used tool, automatically reconstructs metabolic models for bacterial species with full genome sequences, and gap-fills them using a parsimonious approach in which the least number of reactions is added to complete a particular pathway. The first version of this framework created models with an average accuracy of 66% before optimization and 87% after optimization, as determined using experimental validation [56]. However, recent advances in automated metabolic modeling have expanded the ModelSEED algorithm to include a likelihood maximization approach to gap filling that resulted in greater accuracy and the identification of 5–30% additional reactions compared to the original parsimony-based approach[51]. Of the reactions identified by this new approach, 5 to 30% are not identified using the original parsimony-based approach. Given the substantial benefits associated with these automated reconstruction methods, including the ability to analyze high-throughput sequence data, this level of accuracy represents a good avenue into large-scale model creation [57,58].
ANALYZING METABOLIC NETWORKS
Once a GEM is in hand, it can be analyzed either in isolation or together with that of GEMs from other organisms living in the same community. Two of the main avenues for understanding complex microbiomes are topological maps and constraint-based models. Here, we outline each approach.
Topological Metabolic Maps
Topological metabolic maps predict species-species interactions by focusing on metabolic connectivity. An organism’s GEM reveals its potential for carrying out various metabolic functions; for example, based on its genome alone, researchers were recently able to infer that a novel human gut microbe produces the anti-inflammatory short-chain fatty acids butyrate and acetate, and relies on other organisms for metabolically costly molecules such as cobalamin, methionine, and branch-chained amino acids [59]. This illustrates how sequence data can be used to infer a substantial amount of information about an organism’s metabolic niche in the gut. In a topological map, this information is used to draw connections between different organisms based on possible metabolic functions: If one microbe’s genome encodes the cellular machinery to make a sugar and another organism’s genome encodes the machinery to metabolize that sugar, the two are linked in a network.
This network can then be used to make predictions about how a community functions. For example, Zelezniak et al. [57] have used these maps to identify specific metabolites that can promote the survival of metabolically interdependent groups of bacteria under nutritionally challenging conditions; such information might one day prove useful in protecting desirable gut microbes. And Freilich et al. [60] have predicted that metabolic flexibility (measured by the number of metabolic environments in which a species is predicted to grow) is associated with a need to grow rapidly. Interestingly, Levy and Borenstein [61] have used interaction-based topological metabolic maps to investigate the relative importance of cooperation and competition in determining the makeup of the microbiome. They observed a positive association between species co-occurrence and competition, suggesting that species with similar nutrient requirements establish themselves in similar environments; this scenario differs from one in which species are distributed in the gut according to competitive exclusion by other species with similar metabolic requirements. Clearly topological metabolic maps have already generated intriguing hypotheses about community dynamics within the gut microbiome that are worth testing.
Topological metabolic maps have both advantages and drawbacks. One major advantage of these maps is that the metabolic links between species recapitulate a significant portion of interactions identified by correlation coefficients between species [57]. Thus, providing evidence that GEMs can be used to predict species-species interactions and thus community dynamics. Another advantage is that they are easy to produce, as they require no simulations. They are thus well suited to analyzing high-throughput data, and researchers have published several user-friendly tools for creating this type of map, of which NetSeed is the most popular for single species [62] and NetCooperate for pairs of organisms [63]. The main drawback of topological metabolic maps is that they infer species-species interactions based on organisms’ potential to carry out metabolic reactions. In reality, no organism carries out every potential function encoded by its genome; just because a gut microbe can produce a certain sugar transporter does not mean that it does so under normal circumstances. Because the nutritional environment plays an important role in influencing microbial metabolism, topological maps—which ignore context—run the risk of inferring species-species interactions that do not occur in the particular scenarios that researchers wish to study [57].
Constraint-based metabolic models
Constraint-based metabolic models address the major drawback of topological metabolic maps by considering context. The most common approach to describing the metabolism of an organism in constraint-based modeling is flux balance analysis (FBA). FBA incorporates information on the flow of nutrients, or flux, to an organism, allowing researchers to predict which potential metabolic reactions it actually carries out. This information can then be used to generate more accurate species-species interaction networks and even growth and secretion rates, which lead to more accurate predictions about community dynamics. Moreover, dynamic FBA (dFBA) models allow users to simulate conditions in which the level of nutrient flux changes [64], as occurs constantly within the gut, providing an opportunity to improve the accuracy of predictions even further.
Abundant evidence underscores the utility of constraint-based metabolic models. For well-studied organisms, such as E. coli, FBA predictions are fairly consistent with experimental observations in a variety of environments [36,65]. This framework has been successfully used to not only predict bacterial growth rates under different media conditions [66], but to test the effect of gene knockouts on growth [67], and to predict drug targets [68]. In addition, Klitgord and Segrè [69] have used FBA to identify culture media that would induce various types of interactions between pairs of microbes. They predicted and experimentally confirmed that, when cultured with NH3, H2S, and lactate, the sulfate reducer Desulfovibrio vulgaris produces metabolites that are used by Methanococcus maripaludis, and that when NH3 is replaced by alanine, M. maripaludis produces metabolites that can be used by D. vulgaris as well. A model that did not take into account the nutrient environment would not have been able to predict these interactions. Furthermore, Chiu et al. [24] have shown that emergent biosynthetic capacity is common by using dFBA to predict instances among a large collection of two-species communities (30% of 1,400 pairings resulted in at least one emergent metabolite). Intriguingly, they found emergent biosynthesis was most common when two community members were neither too similar nor too different functionally and phylogenetically. Given this array of important findings, constraint-based models represent a significant advance in our ability to model the gut microbiome.
Recently, researchers have begun to make these models even more realistic by considering the role of the host in microbial community dynamics. For example, Ines Thiele and colleagues have combined metabolic models of the gut microbe B. thetaiotaomicron and the mouse to simulate growth of the bacterium in the host lumen under five different diets [58]. This approach generated testable predictions about the metabolites exchanged between microbe and host, the ability of B. thetaiotaomicron to rescue certain lethal enzymopathies in the host, and the ability of the host to do the same for the bacterium. In a more complex study, the same research group modeled pairwise interactions between 11 representative gut microbes in conjunction with human small intestinal enterocytes, under three different diets [70]. They found that the anoxic conditions of the large intestine appear to drive mutualistic crossfeeding, resulting in a more complex ecosystem than is present in the less anoxic small intestine. In addition, this model suggests that nutrients produced by enterocytes induce competition among the microbes. Together, this work establishes the feasibility and promise of using such models to learn about the gut microbiome.
Many constraint-based metabolic modeling tools are available[71], including the COBRA Toolbox [72,73] and COBRApy [74]. In addition, researchers have created hybrid constraint-based modeling tools, including Netlogo, which allow users to create agent-based models that reflect individual GEMs [75,76], and DyMMM to create kinetic-based models that simulate metabolite concentrations through time [77]. Finally, the technical advances packaged in tools such as DFBAlab now permit us to run dFBA faster [78], a prerequisite for analyzing complex communities such as the human gut.
Historically, the drawback of this type of model is that it is time intensive. As mentioned, in the not-so-distant past, a single GEM could take half a year to construct. Fortunately, recent advances have reduced the time and effort involved in this type of modeling. Heinken and Thiele [79] have demonstrated that existing manually curated metabolic models can be combined to systematically predict health-relevant human-microbial co-metabolism, allowing researchers a means of simulating the effects of scenarios such as pathogen introduction and probiotic administration. Manually curating comprehensive metabolic models is a painstaking effort, however, and only a limited number of such models are available. Fortunately, novel computational tools such as MMinte [80] provide users with an automated method to assess pairwise microbial metabolic interactions on a large scale under a variety of metabolic environments for their community of interest. Thus, armed only with genome sequences, researchers can analyze interactions to explore how various conditions might affect community features such as stability and resilience. Unfortunately, even with these methodological advances, metabolic models are not suitable for analyzing large, complex microbial communities such as the gut. Consider what would happen if we wanted to use this type of model to determine the effects of a diet with increased levels of starch on the gut microbiome. The molecular cascade of reactions arising from the breakdown of starch would have to be evaluated across hundreds of thousands to millions of reactions in the gut community. When considered alongside the fact that the vast majority of the reactions are repeated across multiple species, this not only seems computationally challenging, but incredibly inefficient.
FUTURE DIRECTIONS: COMMUNITY METABOLIC MODELS, NON-METABOLIC FACTORS THAT AFFECT INTERACTION, AND MORE SOPHISTICATED VALIDATION METHODS
Above, we discussed the pros and cons of two types of models for predicting gut microbial dynamics: ecological and metabolic. Here, we would like to propose combining the two to create a multiscale community metabolic model that can effectively predict microbial dynamics while giving users the practical, mechanistic data needed to determine the effects of a given intervention (Figure 3). Briefly, this type of modeling starts with metabolic models for each organism within a community. These models are used to generate an interaction matrix that contains a single net term for each pairwise microbe-microbe and microbe-host interaction. This matrix is then used to parameterize an ecological model. When this type of model is combined with novel automated methods of assessing pairwise metabolic interactions, such as MMinte [80], the result should be tractable and easily interpretable calculations that accurately predict community dynamics on the basis of high-throughput microbiome sequencing data.
Figure 3. We argue that community metabolic models, which combine features of metabolic and ecological models, are needed to understand the mechanisms underlying community dynamics in the gut microbiome.

This type of modeling starts with a metabolic model for each organism within the community (left). These models are the used to generate an interaction matrix that contains a single net term for each pairwise microbe-microbe and microbe-host interaction (depiction of pairwise interactions between microbes, middle; green represents cooperative interactions, red represents competitive interactions). This matrix is then used to parameterize an ecological model, which can be used to predict how manipulating specific aspects of the microbiome—with probiotics, prebiotics, or nutritional interventions, for example—will affect properties such as stability and resilience.
To return to our starch example, using a community metabolic model we could take sequence data obtained from the gut microbiome, translate this data into a matrix of pairwise metabolic interactions using an automated tool such as MMinte, and plug these interaction data into an ecological model that predicts how altering the level of starch-related flux affects interactions and thus community dynamics. The beauty of this approach is that we would not need novel empirical data to determine the interaction parameters, as metabolic modeling would provide that information. In addition, the species interaction–based approach reduces the computational challenge of a 101 element stoichiometric matrix into a relatively tractable 104 element interaction matrix by focusing on species instead of biochemical reactions.
Moreover, community models would allow us to integrate important predictions from ecological and metabolic models. Recall from above: Heinken and Theile’s [70] metabolic models predict that host-derived metabolites from enterocytes induce competition within the microbiome. Meanwhile, Coyte et al.’s [31] ecological model indicates that increased levels of competition help stabilize the microbiome. Put together, these findings suggest that increasing levels of host-derived metabolites, such as mucin, could represent one avenue toward increasing gut microbiome stability. By building a community model to simulate this scenario from start to finish, we could better determine whether this is the case. This ability to generate predictions that begin with basic metabolism and end with essential information about community-wide properties is the major advance this type of model can deliver.
Microbe-microbe and microbe-host interactions are composed of more than just metabolism, of course. Although metabolism is a major determinant of cross-feeding interactions, other factors also influence the nature and magnitude of interactions. For example, the gut is full of pockets of distinct microbial communities that constrain the ability of a given microbe to interact with other microbes, and factors such as diet influence patterns of spatial clustering [81]. Even in simple communities with little complex spatial structure, simulations reveal that where a particular microbe resides within a community matters [82]. Harcombe et al. [83] have used constraint-based models to make the surprising prediction that when two collaborative, cross-feeding colonies of the gut microbes E. coli and S. enterica are separated by a competitor colony of S. enterica, growth of the original S. enterica colony increases, and they have confirmed these results experimentally. Although the spatial structure present within the gut is much more complex, this example shows the potential benefits of studying the effects of spatial structure on patterns of microbial growth, and our ability to model the spatial structure of the gut will no doubt improve in the future. Other factors we expect to play a similarly important role in shaping interactions within the microbiome are host immunology and intercellular signaling. Community metabolic models can be expanded to include all of these factors.
We now know that pathogens exploit the oxygen gradient to gain dominance within the gut[19], a fact that underscores the need for models that consider oxygen levels. For some time, researchers have hypothesized that the host inflammatory response in conditions such as inflammatory bowel disease may increase the level of oxygen present in the gut, resulting in overgrowth of aerotolerant organisms and undergrowth of health-promoting, butyrate-producing anaerobic microbes[21]. Recently, a number of studies have provided data that support the general idea that the inflammatory response can benefit pathogens while harming beneficial bacteria. For example, S. typhimurium infection spurs an immune response in mice that reduces levels of butyrate-producing Clostridia; the resulting decrease in butyrate levels increases oxygen levels in the colonocytes lining the intestine, allowing the further expansion of the facultative anaerobe S. typhimurium within the gut[84]. However, not all data support this model. In mice the enteric pathogen Citrobacter rodentium increases oxygenation of the mucosal surface of the intestine, allowing the aerobic C. rodentium to proliferate in the colon, as we might predict—but despite higher levels of oxygen, Clostridia species increase in abundance[85]. Community metabolic models that incorporate the oxygen gradient are needed to help us better understand these perplexing findings.
Finally, all modeling efforts require some form of experimental validation in order to be applicable to reality. The development of experimental platforms for testing community model predictions holds much promise. Bioreactors allow researchers to analyze the dynamics of complex communities growing in physiologically relevant conditions. For example, the Simulator of the Human Intestinal Microbial Ecosystem [86], which comprises five sequentially connected bioreactors to simulate the different gastrointestinal compartments, can be used to understand the dynamics of the communities in each compartment. Community model predictions can also be tested using murine model systems. These are particularly important when researchers want to understand the role of the host in determining community dynamics [70]. As models of murine metabolism improve, it will become easier to confirm or reject predictions using this experimental system. Ultimately, the goal is to generate predictions using community metabolic models that can be tested with data collected before and after interventions, such as the administration of prebiotics or probiotics.
CONCLUSIONS
Here, we have argued that merging ecological and metabolic models to create a novel community metabolic model will allow us to predict community dynamics within the gut in response to specific interventions that target metabolic interactions between microbes and between microbes and host cells. Recent developments have given us the tools to simulate the flux of nutrients within the gut, and we are beginning to explore how factors such as spatial structure determine community dynamics as well. In the near future, we expect to see metabolic models increasingly used as scaffolds for additional layers of complexity, such as gene regulation, intercellular signaling, and the immune system; the groundwork is already being laid [38,87]. Finally, as we develop high-throughput, culture-based experiments, we will be able to test model predictions on a new scale. The past decade has witnessed a remarkable increase in data on the gut microbiome; we hope that the next decade will witness a similar increase in meaningful understanding of these data. Although mechanistically designing interventions such as prebiotics and probiotics based on our understanding of the human gut microbiome appears a daunting task, we hope to have convinced the reader that community metabolic models have the potential to translate sequence data into testable hypotheses, practical knowledge, and novel clinical interventions.
HIGHLIGHTS.
A framework is needed to translate microbiome sequence data into practical knowledge
Ecological models can predict microbiome dynamics but lack mechanistic detail
Metabolic models provide mechanistic detail but are computationally intense
Community metabolic models combine the strengths of ecological and metabolic models
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [grant 1R01CA179243 to Nicholas Chia].
We would like to thank Patricio Jeraldo for discussions that improved this article and the Mayo Clinic Center for Individualized Medicine for its support.
ABBREVIATIONS
- dFBA
dynamic flux balance analysis
- FBA
flux balance analysis
- GEM
Genome-scale model
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Enders G. Gut: The Inside Story of Our Body’s Most Underrated Organ. Vancouver, BC: Greystone Books Ltd; 2015. [Google Scholar]
- 2.Chutkan R. The Microbiome Solution. New York City: Avery; 2016. [Google Scholar]
- 3.Weingarden A, Gonzalez A, Vazquez-Baeza Y, Weiss S, Humphry G, et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome. 2015;3:10. doi: 10.1186/s40168-015-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strogatz SH. Sync: How Order Emerges From Chaos In the Universe, Nature, and Daily Life. Hachette Books; 2004. [Google Scholar]
- 5.Bauer E, Laczny CC, Magnusdottir S, Wilmes P, Thiele I. Phenotypic differentiation of gastrointestinal microbes is reflected in their encoded metabolic repertoires. Microbiome. 2015;3:55. doi: 10.1186/s40168-015-0121-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carraway KL, Ramsauer VP, Haq B, Carothers Carraway CA. Cell signaling through membrane mucins. Bioessays. 2003;25:66–71. doi: 10.1002/bies.10201. [DOI] [PubMed] [Google Scholar]
- 7.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper R, de Vos W, et al. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes. 2010;1:254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergstrom KS, Xia L. Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology. 2013;23:1026–1037. doi: 10.1093/glycob/cwt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 10.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annual Review of Nutrition. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 11.Bradshaw D, Homer K, Marsh P, Beighton D. Metabolic cooperation in oral microbial communities during growth on mucin. Microbiology. 1994;140:3407–3412. doi: 10.1099/13500872-140-12-3407. [DOI] [PubMed] [Google Scholar]
- 12.Ramírez-Puebla ST, Servín-Garcidueñas LE, Jiménez-Marín B, Bolaños LM, Rosenblueth M, et al. Gut and root microbiota commonalities. Applied and Environmental Microbiology. 2013;79:2–9. doi: 10.1128/AEM.02553-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haller D, Colbus H, Gänzle M, Scherenbacher P, Bode C, et al. Metabolic and functional properties of lactic acid bacteria in the gastro-intestinal ecosystem: a comparative in vitro study between bacteria of intestinal and fermented food origin. Systematic and Applied Microbiology. 2001;24:218–226. doi: 10.1078/0723-2020-00023. [DOI] [PubMed] [Google Scholar]
- 14.Nava GM, Carbonero F, Croix JA, Greenberg E, Gaskins HR. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. The ISME Journal. 2012;6:57–70. doi: 10.1038/ismej.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. 2010;285:22082–22090. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 17.Salminen S, Von Wright A. Lactic acid bacteria: microbiological and functional aspects. CRC Press; 2004. [Google Scholar]
- 18.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol. 2012;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espey MG. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radical Biology and Medicine. 2013;55:130–140. doi: 10.1016/j.freeradbiomed.2012.10.554. [DOI] [PubMed] [Google Scholar]
- 20.Wallace N, Zani A, Abrams E, Sun Y. Chapter Four-The Impact of Oxygen on Bacterial Enteric Pathogens. Advances in applied microbiology. 2016;95:179–204. doi: 10.1016/bs.aambs.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147:1055–1063. e1058. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Million M, Alou MT, Khelaifia S, Bachar D, Lagier J-C, et al. Increased Gut Redox and Depletion of Anaerobic and Methanogenic Prokaryotes in Severe Acute Malnutrition. Scientific reports. 2016:6. doi: 10.1038/srep26051. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Chia N, Woese CR, Goldenfeld N. A collective mechanism for phase variation in biofilms. Proceedings of the National Academy of Sciences. 2008;105:14597–14602. doi: 10.1073/pnas.0804962105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiu H-C, Levy R, Borenstein E. Emergent biosynthetic capacity in simple microbial communities. PLoS Comput Biol. 2014;10:18. doi: 10.1371/journal.pcbi.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106:5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The Application of Ecological Theory Toward an Understanding of the Human Microbiome. Science. 2012;336:8. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.May RM, McLean A. Theoretical Ecology - Principles and Applications. 2007. [Google Scholar]
- 28.Murdoch WW. Stabilizing effects of spatial heterogeneity in predator-prey systems. Theoretical population biology. 1977;11:252–273. doi: 10.1016/0040-5809(77)90028-4. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson AJ, Bailey VA. The Balance of Animal Populations—Part I. Wiley Online Library; 1935. pp. 551–598. [Google Scholar]
- 30.Pimm S, Lawton J. On feeding on more than one trophic level. Nature. 1978;275:542–544. [Google Scholar]
- 31.Coyte KZ, Schluter J, Foster KR. The ecology of the microbiome: Networks, competition, and stability (SI) Science. 2015;350:663–666. doi: 10.1126/science.aad2602. [DOI] [PubMed] [Google Scholar]
- 32.Stein RR, Bucci V, Toussaint NC, Buffie CG, Ratsch G, et al. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Comput Biol. 2013;9:e1003388. doi: 10.1371/journal.pcbi.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher CK, Mehta P. Identifying keystone species in the human gut microbiome from metagenomic timeseries using sparse linear regression. PLoS One. 2014;9:e102451. doi: 10.1371/journal.pone.0102451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henry CS, Bernstein HC, Weisenhorn P, Taylor RC, Lee JY, et al. Microbial community metabolic modeling: a community data-driven network reconstruction. Journal of Cellular Physiology. 2016 doi: 10.1002/jcp.25428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feist AM, Palsson BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abram F. Systems-based approaches to unravel multi-species microbial community functioning. Comput Struct Biotechnol J. 2015;13:24–32. doi: 10.1016/j.csbj.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sung J, Hale V, Merkel AC, Kim P-J, Chia N. Metabolic modeling with Big Data and the gut microbiome. Applied & Translational Genomics. 2016 doi: 10.1016/j.atg.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bebek G, Koyuturk M, Price ND, Chance MR. Network biology methods integrating biological data for translational science. Brief Bioinform. 2012;13:446–459. doi: 10.1093/bib/bbr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Covert M, Knight EM, Reed JL, Herrgard MJ, Palsson B. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:5. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 41.Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, et al. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems. 2016;1:e00013–00015. doi: 10.1128/mSystems.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caspi R, Altman T, Billington R, Dreher K, Foerster H, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:D581–591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Angiuoli SV, Gussman A, Klimke W, Cochrane G, Field D, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS. 2008;12:137–141. doi: 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanehisa M, Goto S. KEGG: Kyoto Encyclopetia of Genes and Genomes. Nucleic Acids Res. 2000;28:4. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schomburg I, Chang A, Ebeling C, Gremse M, Heldt C, et al. BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 2004;32:D431–433. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeJongh M, Formsma K, Boillot P, Gould J, Rycenga M, et al. Toward the automated generation of genome-scale metabolic networks in the SEED. BMC Bioinformatics. 2007;8:139. doi: 10.1186/1471-2105-8-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benedict MN, Mundy MB, Henry CS, Chia N, Price ND. Likelihood-based gene annotations for gap filling and quality assessment in genome-scale metabolic models. PLoS Comput Biol. 2014;10:e1003882. doi: 10.1371/journal.pcbi.1003882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, et al. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput Biol. 2013;9:e1002980. doi: 10.1371/journal.pcbi.1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karp P, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18:8. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- 54.Feng X, Xu Y, Chen Y, Tang YJ. MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Systems Biology. 2012;6:9. doi: 10.1186/1752-0509-6-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dias O, Rocha M, Ferreira EC, Rocha I. Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 2015;43:3899–3910. doi: 10.1093/nar/gkv294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 57.Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, et al. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci U S A. 2015;112:6. doi: 10.1073/pnas.1421834112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinken A, Sahoo S, Fleming RM, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013;4:28–40. doi: 10.4161/gmic.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeraldo P, Hernandez A, Nielsen HB, Chen X, White BA, et al. Capturing one of the human gut microbiome’s most wanted: reconstructing the genome of a novel butyrate-producing, clostridial scavenger from metagenomic sequence data. Frontiers in Microbiology. 2016;7:783. doi: 10.3389/fmicb.2016.00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freilich S, Kreimer A, Borenstein E, Yosef N, Sharan R, et al. Metabolic-network-driven analysis of bacterial ecological strategies. Genome Biol. 2009;10:R61. doi: 10.1186/gb-2009-10-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci U S A. 2013;110:9. doi: 10.1073/pnas.1300926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carr R, Borenstein E. NetSeed: a network-based reverse-ecology tool for calculating the metabolic interface of an organism with its environment. Bioinformatics. 2012;28:734–735. doi: 10.1093/bioinformatics/btr721. [DOI] [PubMed] [Google Scholar]
- 63.Levy R, Carr R, Kreimer A, Freilich S, Borenstein E. NetCooperate: a network-based tool for inferring host-microbe and microbe-microbe cooperation. BMC Bioinformatics. 2015;16:164. doi: 10.1186/s12859-015-0588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahadevan R, Edwards JS, Doyle FJI. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophysical Journal. 2002;83:10. doi: 10.1016/S0006-3495(02)73903-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edwards JS, Covert M, Palsson B. Metabolic modelling of microbes: the flux-balance approach. Environmental Microbiology. 2002;4:8. doi: 10.1046/j.1462-2920.2002.00282.x. [DOI] [PubMed] [Google Scholar]
- 66.Varma A, Palsson B. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60:8. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 68.Hamilton JJ, Reed JL. Identification of functional differences in metabolic networks using comparative genomics and constraint-based models. PLoS One. 2012;7:e34670. doi: 10.1371/journal.pone.0034670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klitgord N, Segre D. Ecosystems biology of microbial metabolism. Curr Opin Biotechnol. 2011;22:541–546. doi: 10.1016/j.copbio.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 70.Heinken A, Thiele I. Anoxic Conditions Promote Species-Specific Mutualism between Gut Microbes In Silico. Appl Environ Microbiol. 2015;81:4049–4061. doi: 10.1128/AEM.00101-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biggs MB, Medlock GL, Kolling GL, Papin JA. Metabolic network modeling of microbial communities. Wiley Interdiscip Rev Syst Biol Med. 2015;7:317–334. doi: 10.1002/wsbm.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Becker SA, Feist AM, Mo ML, Hannum G, Palsson BO, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat Protoc. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- 73.Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ebrahim A, Lerman JA, Palsson B, Hyduke D. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst Biol. 2013;7:5. doi: 10.1186/1752-0509-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biggs MB, Papin JA. Metabolic network-guided binning of metagenomic sequence fragments. Bioinformatics. 2015:8. doi: 10.1093/bioinformatics/btv671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tisue S, Wilensky U. Netlogo: A simple environment for modeling complexity. Boston, MA: 2004. pp. 16–21. [Google Scholar]
- 77.Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, et al. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011;5:305–316. doi: 10.1038/ismej.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gomez JA, Hoffner K, Barton P. DFBAlab: a fast and reliable MATLAB code for dynamic flux balance analysis. BCM Bioinformatics. 2014;15:10. doi: 10.1186/s12859-014-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heinken A, Thiele I. Systematic prediction of health-relevant human-microbial co-metabolism through a computational framework. Gut Microbes. 2015;6:120–130. doi: 10.1080/19490976.2015.1023494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mendes-Soares H, Mundy M, Mendes Soares L, Chia N. MMinte: An application for predicting metabolic interactions among the microbial species in a community. 2016 doi: 10.1101/059550. bioRxiv preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, et al. Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe. 2015;18:478–488. doi: 10.1016/j.chom.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cole JA, Kohler L, Hedhli J, Luthey-Schulten Z. Spatially-resolved metabolic cooperativity within dense bacterial colonies. BMC Systems Biology. 2015;9:1. doi: 10.1186/s12918-015-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harcombe WR, Riehl WJ, Dukovski I, Granger BR, Betts A, et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014;7:1104–1115. doi: 10.1016/j.celrep.2014.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rivera-Chávez F, Lopez CA, Bäumler AJ. Oxygen as a driver of gut dysbiosis. Free Radical Biology and Medicine. 2016 doi: 10.1016/j.freeradbiomed.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 85.Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, et al. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science. 2016;353:1249–1253. doi: 10.1126/science.aag3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marzorati M, Pinheiro I, Van den Abbeele P, Van de Wiele T, Possemiers S. An in vitro technology platform to assess host-microbiota interactions in the gastrointestinal tract. AGRO FOOD INDUSTRY HI-TECH. 2012;23:VIII–XI. [Google Scholar]
- 87.Covert MW, Palsson BO. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]